Multiscale Computational Simulation of Amorphous Silicates’ Structural, Dielectric, and Vibrational Spectroscopic Properties



Abstract
1. Introduction
2. Computational Methods
2.1. Classical Molecular Dynamics (MD) Simulations
- (1)
- Nose–Hoover thermostat. A total of 17 different final temperatures were considered, between 400 K and 2000 K, separated by 100 K each. It is worth mentioning that higher temperatures than the melting point of Mg2SiO4 (at about 1800 K) would have been desirable to accelerate the dynamics of the melting. However, we found that with MD simulations at these temperatures (e.g., 2500 K), the system was unstable because the interatomic potential parameters fail. After the heating process, classical MD simulations at the (N,V,T) ensemble at the given temperature were performed, which consisted of a 100 ps equilibration phase and a 200 ps production phase.
- (2)
- Cooling phase: After the production phase of the previous step, the resulting systems were subjected to MD simulations where the temperature was decreased by 0.005 K every time-step to reach the temperature of 300 K. At this temperature, the systems were equilibrated for 50 ps.
- (3)
- Second thermal/production phase: The structures obtained after the cooling simulations were then heated again to the temperature chosen in the first step. Then, MD simulations with an equilibration phase of 100 ps and a production phase of 200 ps production were performed.
2.2. Quantum Mechanical (QM) Calculations
3. Results and Discussion
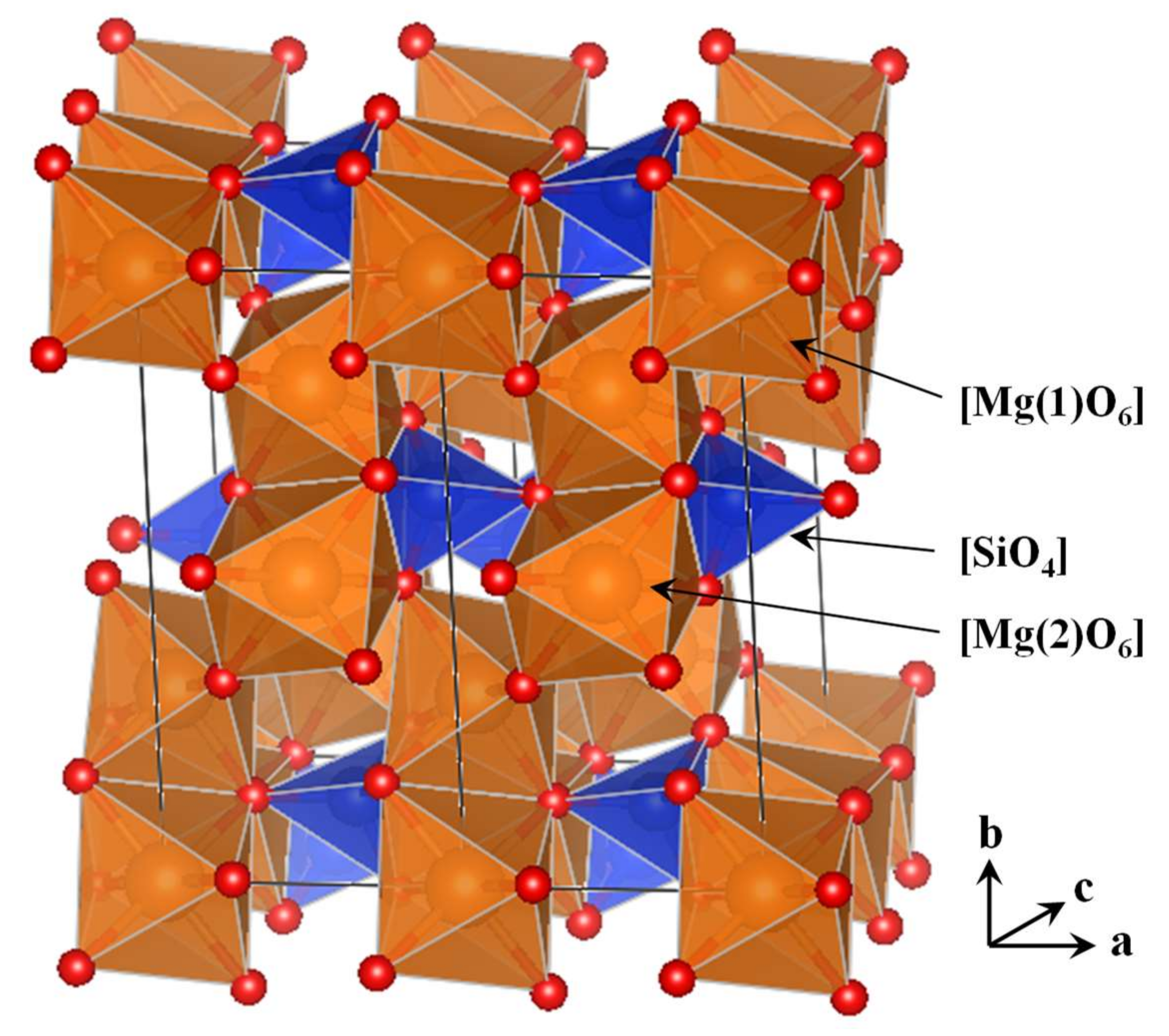
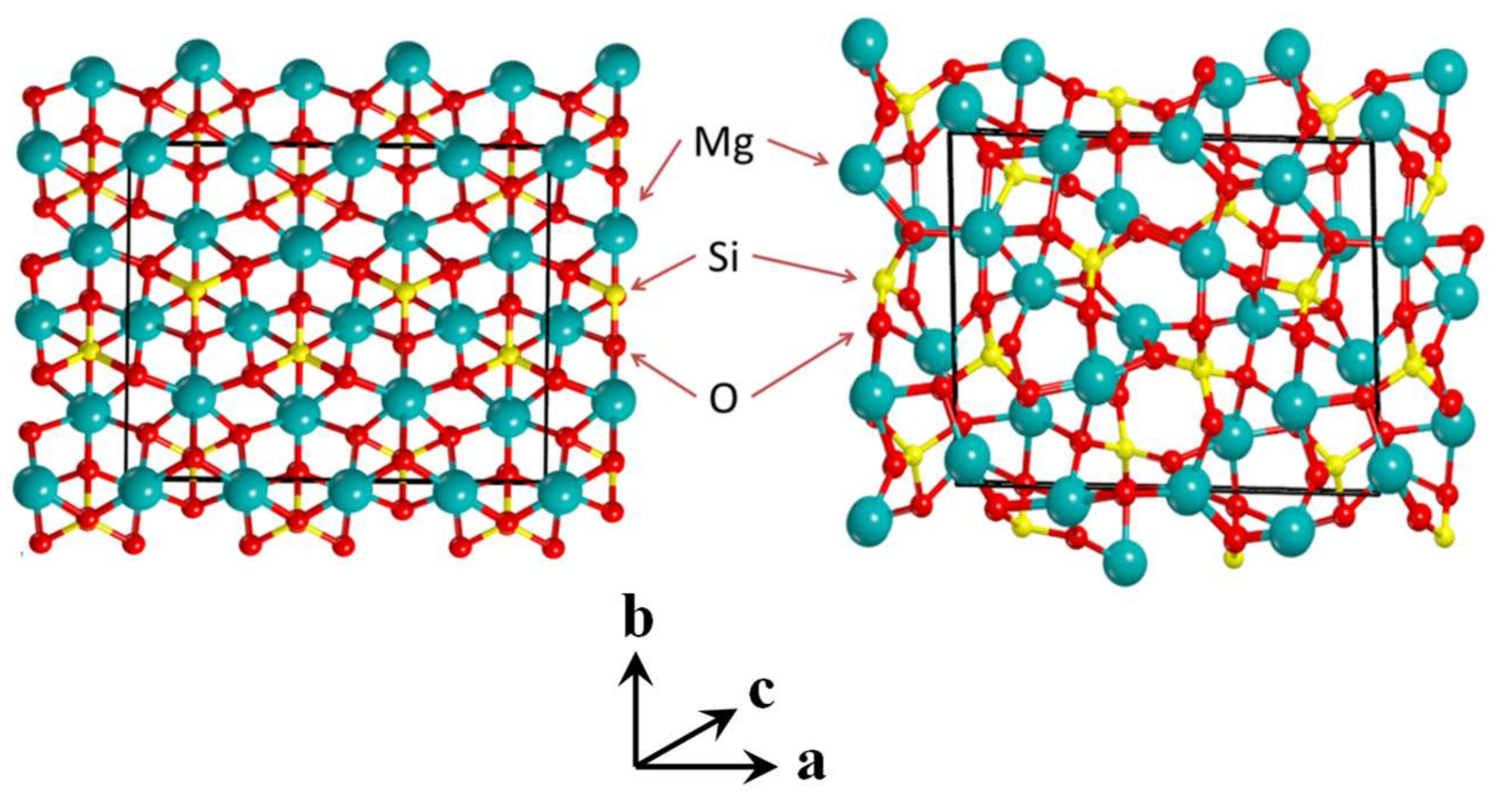
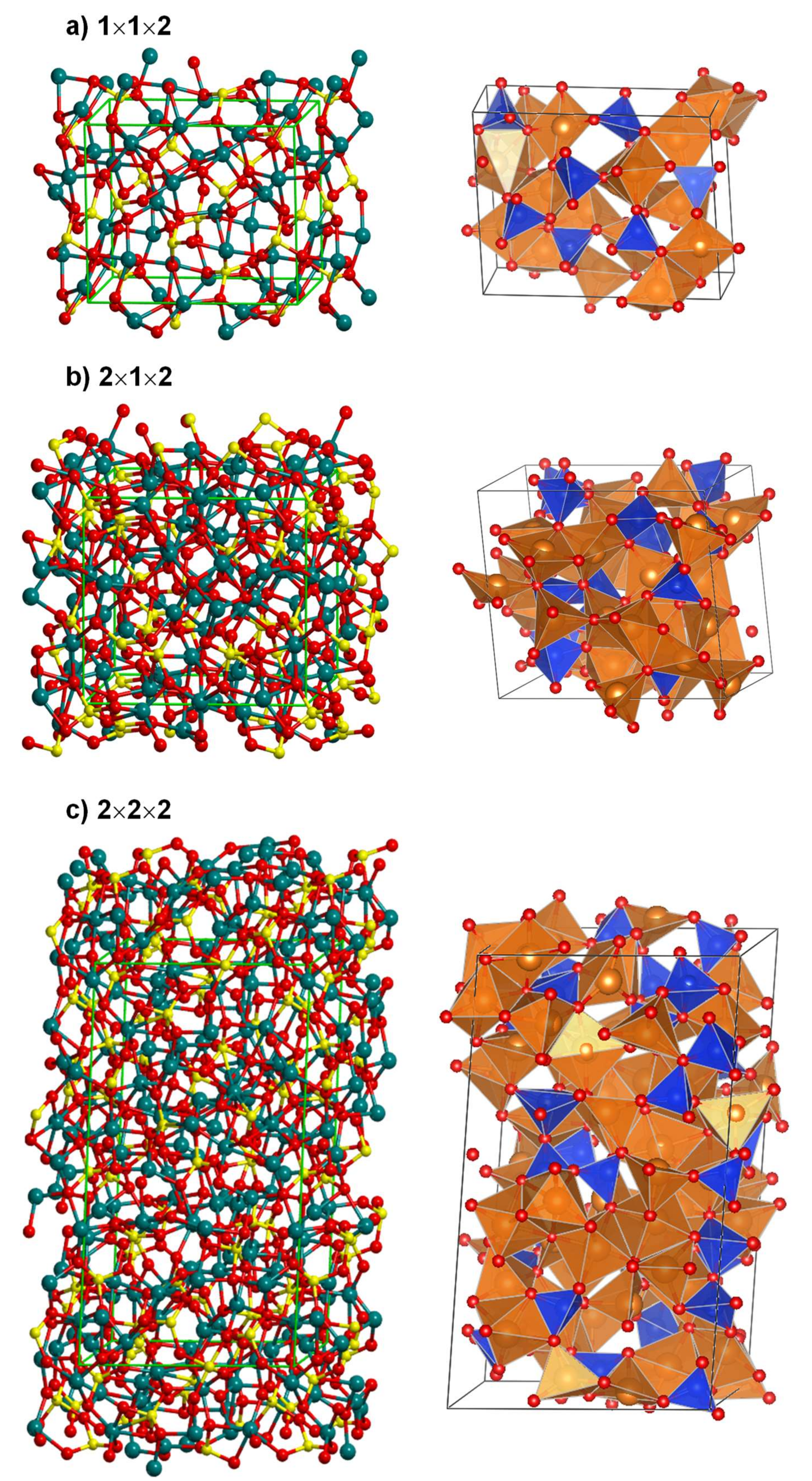
3.1. The Amorphous 1 × 1 × 2 Super-Cell Mg2SiO4 System
3.2. Unit Cell Size Effects in the Amorphisation of the Mg2SiO4 System
4. Conclusions
- The combined approach of classical MD for structure evolution and quantum chemical DFT for electronic structure analysis is a reliable technique to investigate the physicochemical properties of silicates.
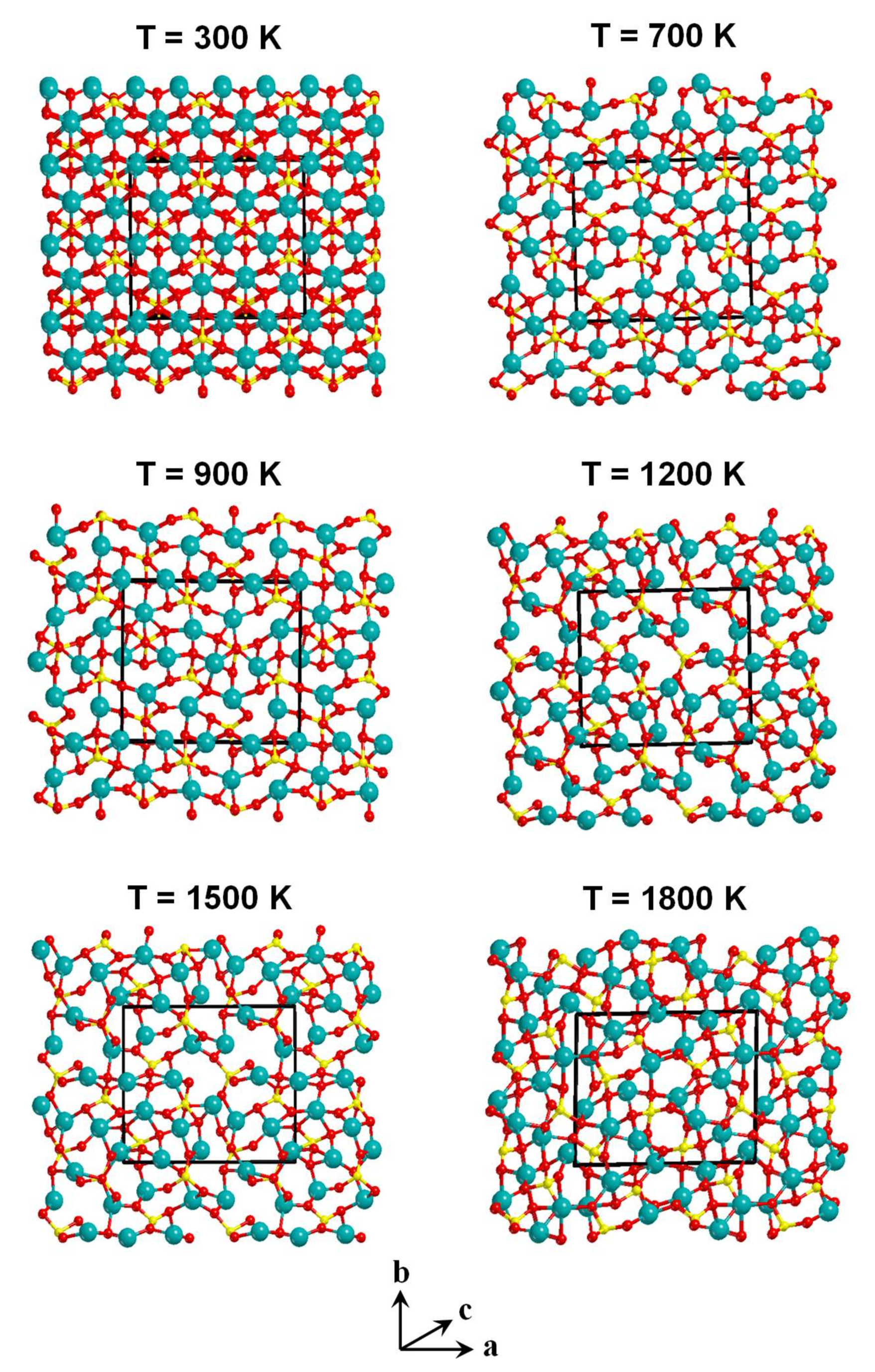
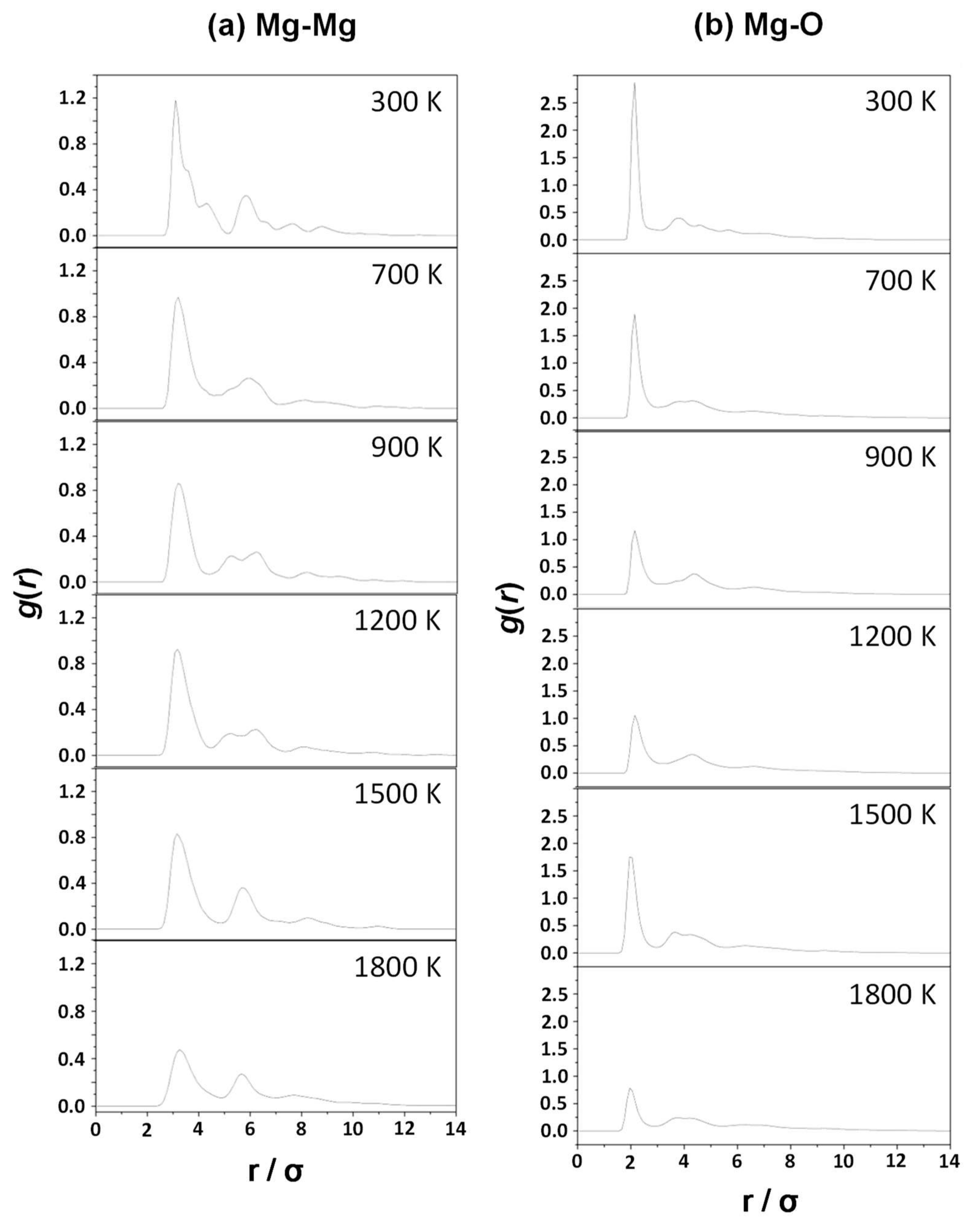
- The Mg–Mg and Mg–O pair correlation functions, g(r), derived from the classical MD simulations indicate that an increase of the temperature produces broader profiles of g(r), indicating a progressive loss of crystallinity with temperature.
- B3LYP-optimized geometries of the amorphous bulk systems present larger distributions of bond lengths and angles than the crystal analogues (for instance, the Si–O distance ranges, 1.618–1.693 Å vs 1.628–1.673 Å) due to the significant disorganization of the internal atomic positions. Consequently, the tetrahedral SiO4 and octahedral MgO6 motifs become largely distorted, which is characteristic of amorphous materials. Calculated densities of the amorphous bulks are lower than the crystalline ones; that is, 3.373 (1 × 1 × 2), 3.214 (2 × 1 × 2), 3.172 (2 × 2 × 2) versus 3.34 (crystalline), due to the presence of internal cavities produced during the amorphization process.
- Related to the high-frequency dielectric constants along the x, y, and z directions (ε∞,x, ε∞,y, and ε∞,z), while in the crystalline Mg2SiO4 they are clearly different (2.571, 2.424, and 2.475, respectively), in agreement with the direction dependency caused by the inherently crystallinity of the system, in the amorphous phases, they are more similar (e.g., for the 2 × 2 × 2 system, 2.148, 2.135, and 2.132, respectively), showing a convergence between the calculated ε∞,x, ε∞,y, and ε∞,z values. Accordingly, ε∞ values do not depend on a particular direction because the system is likewise disordered in all the directions.
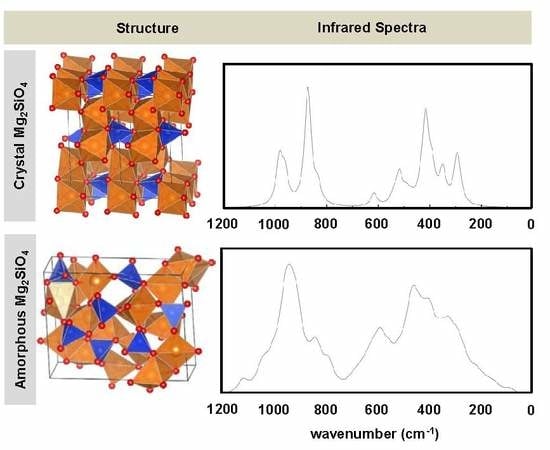
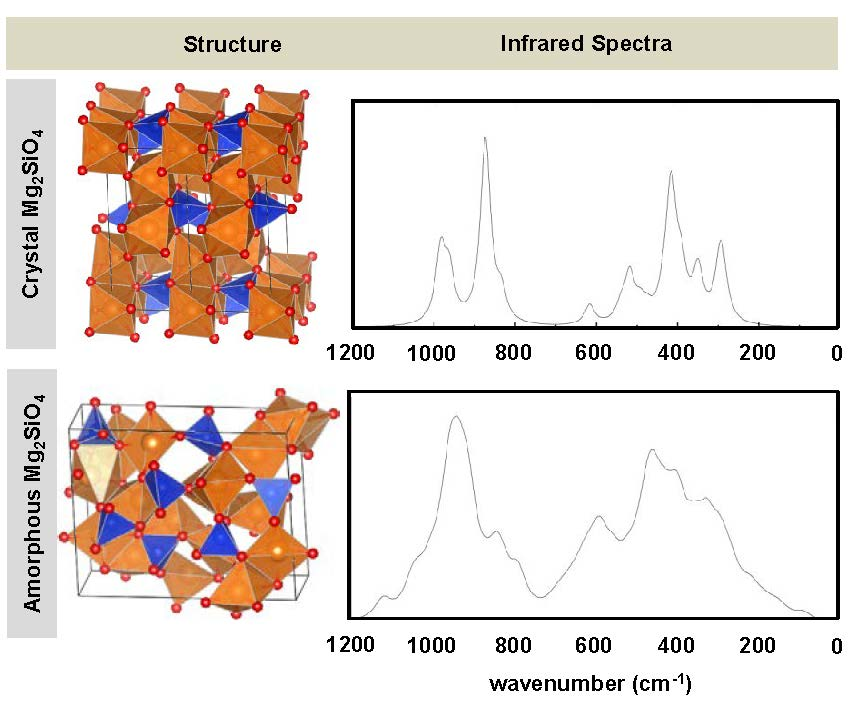
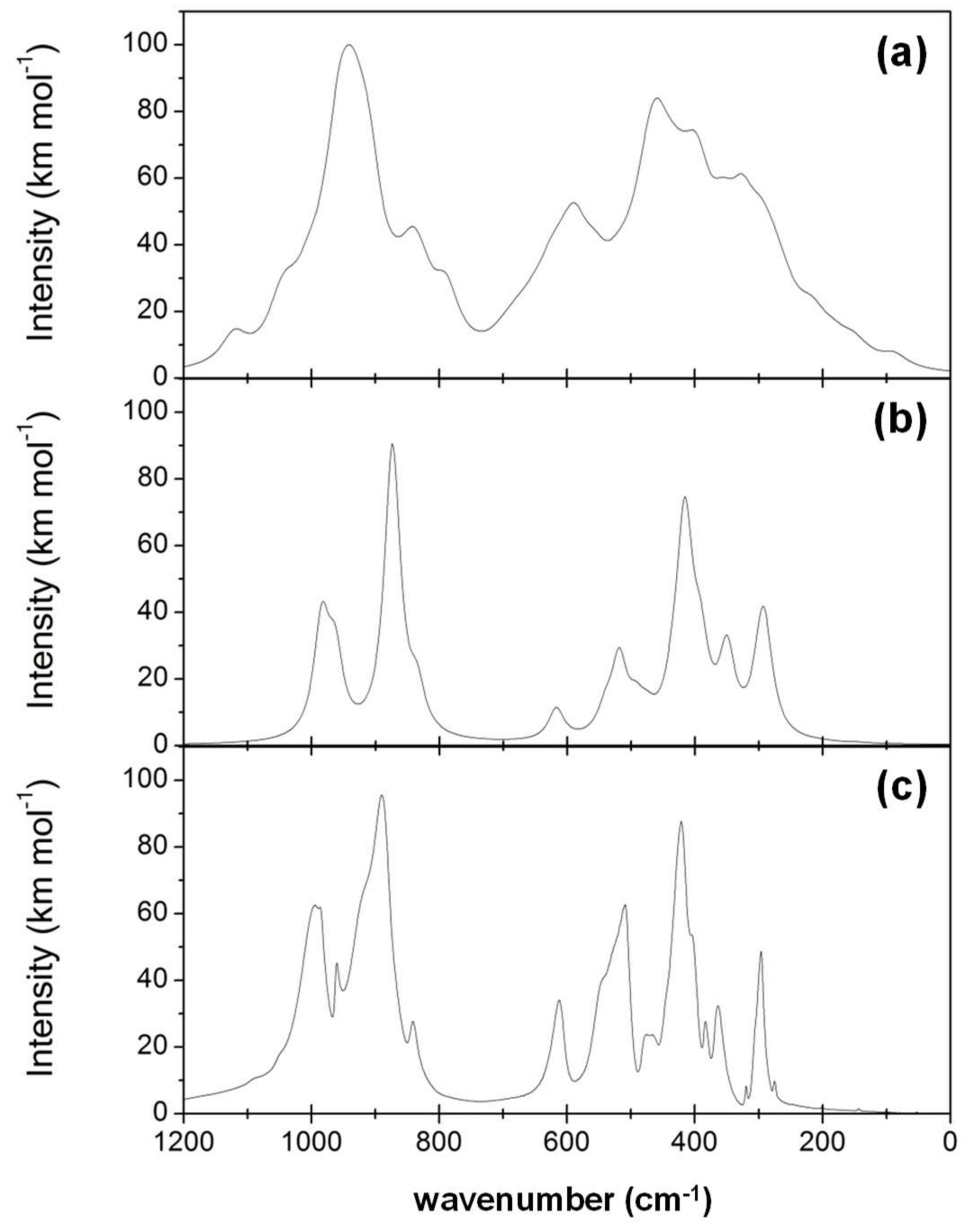
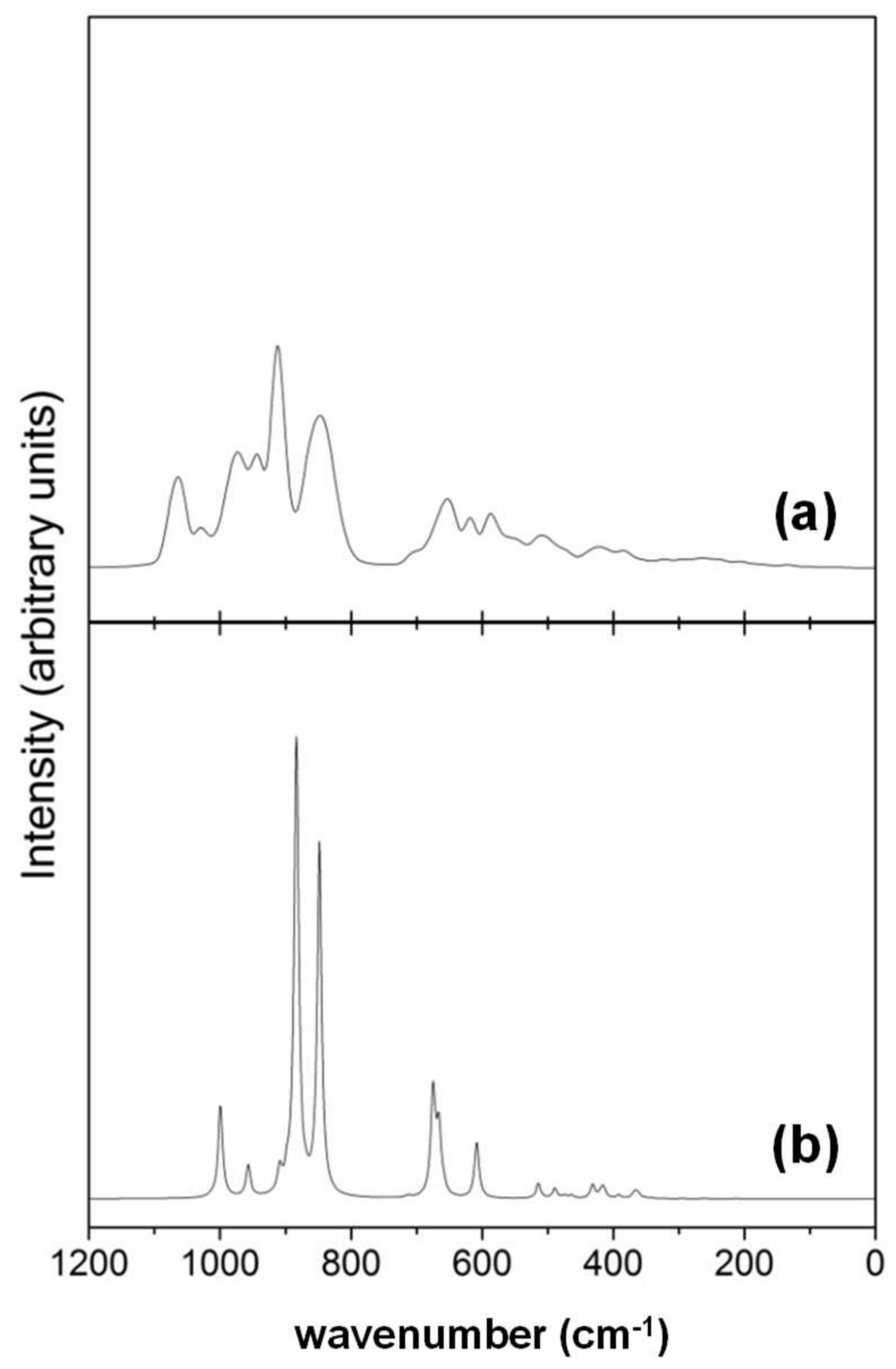
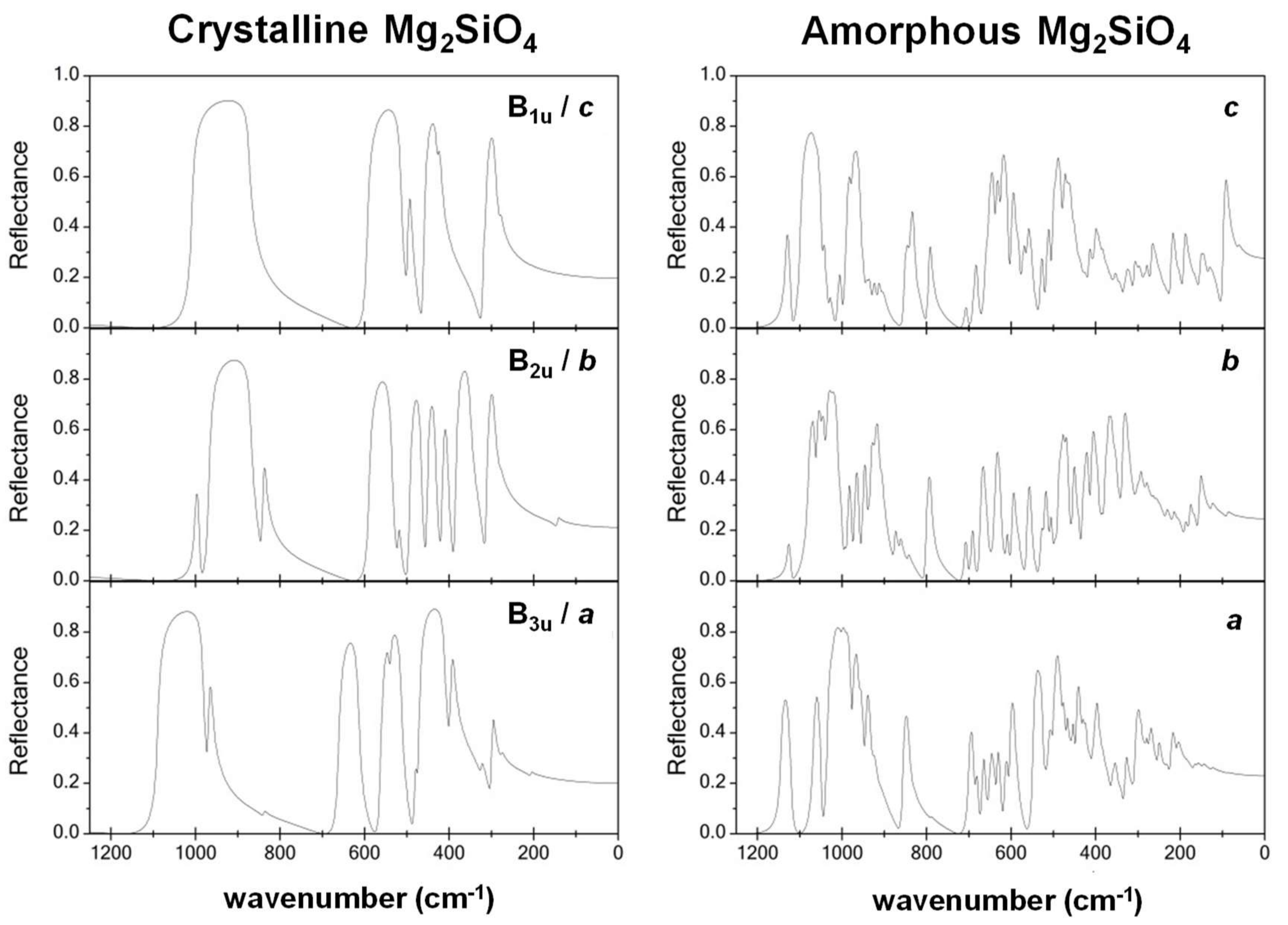
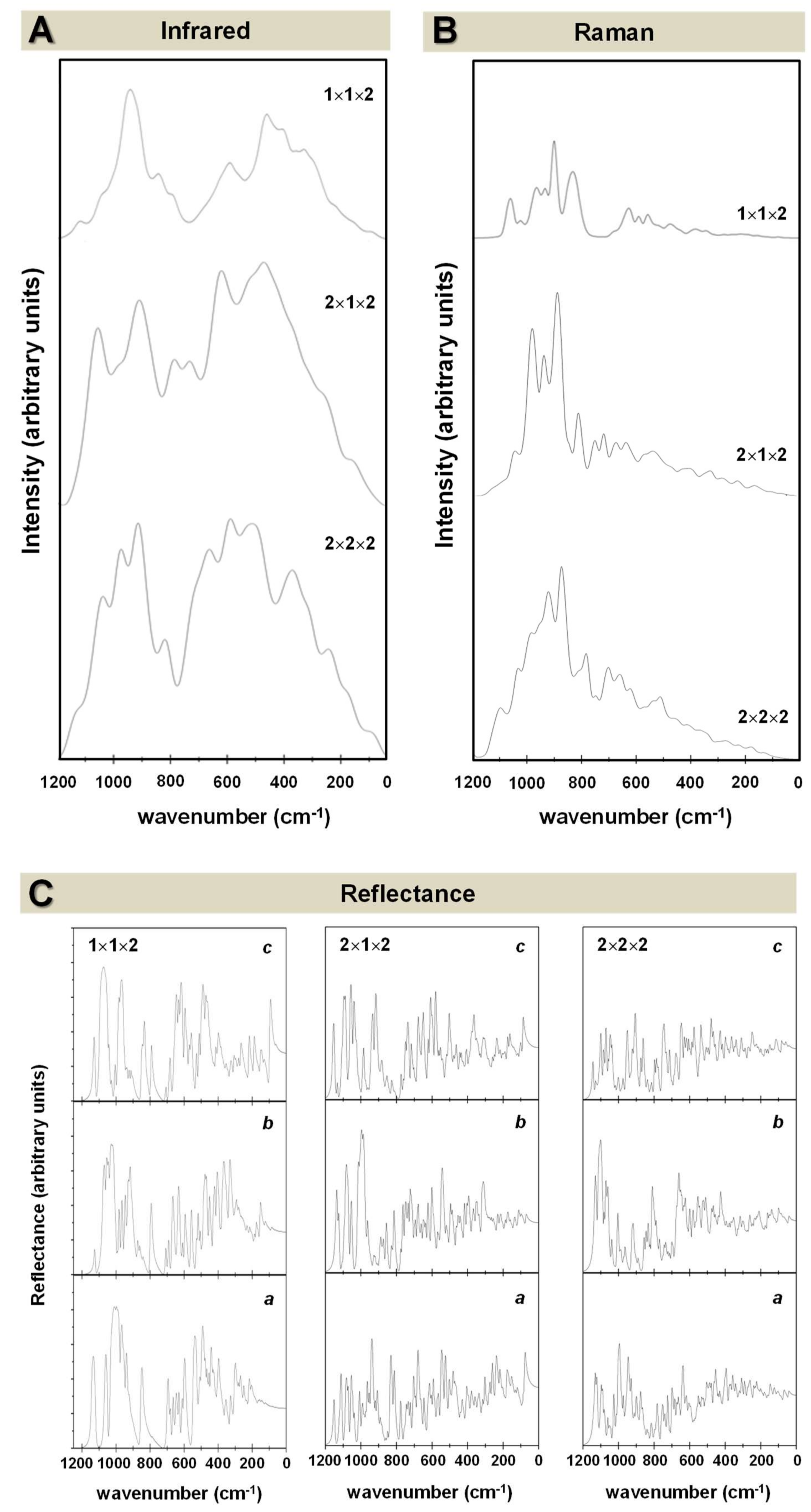
- While the IR and Raman spectra of the crystalline phase present well-defined narrow bands covering the 1000–200 cm−1 region, the spectra of the amorphous analogues, due to the wider and more diverse distribution of lengths and angles, are dominated by broader bands caused by multiple bands overlapping; namely, for IR, three broad bands are observed (between 1000–800 cm−1, a peak at 600 cm−1, and between 500–300 cm−1), while for Raman, two broad bands are observed (between 1000–800 cm−1 and between 700–400 cm−1). The simulated spectra agree fairly well with those recorded for Mg2SiO4 in astrophysical environments, with the IR broad bands being centered at 1000 cm−1 and 500 cm−1. In the case of reflectance spectra, the broad reflectance curves of the crystalline system change with bands increasing in sharpness and number for the amorphous systems, due to the presence of more active modes.
- Regarding the unit cell size effects, results clearly provide evidence that an increase of the unit cell size infers a larger degree of amorphization due to the presence of more internal degrees of freedom to be disordered.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.-F.; Ugliengo, P. Silica surface features and their role in the adsorption of biomolecules: Computational modeling and experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, I.; Prandi, L.; Tomatis, M.; Fubini, B. Free radical generation in the toxicity of inhaled mineral particles: The role of iron speciation at the surface of asbestos and silica. Redox Rep. 2001, 6, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Fubini, B.; Otero Arean, C. Chemical aspects of the toxicity of inhaled mineral dusts. Chem. Soc. Rev. 1999, 28, 373–381. [Google Scholar] [CrossRef]
- Otero Areán, C.; Barceló, F.; Fenoglio, I.; Fubini, B.; Llabrés i Xamena, F.X.; Tomatis, M. Free radical activity of natural and heat treated amphibole asbestos. J. Inorg. Chem. 2001, 83, 211–216. [Google Scholar] [CrossRef]
- Turci, F.; Colonna, M.; Tomatis, M.; Mantegna, S.; Cravotto, G.; Gulino, G.; Aldieri, E.; Ghigo, D.; Fubini, B. Surface reactivity and cell responses to chrysotile asbestos nanofibers. Chem. Res. Toxicol. 2012, 25, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Merget, R.; Bauer, T.; Küpper, H.; Philippou, S.; Bauer, H.; Breitstadt, R.; Bruening, T. Health hazards due to the inhalation of amorphous silica. Arch. Toxicol. 2002, 75, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L.; Polak, J.M. Third-generation biomedical materials. Science 2002, 295, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Edit. 2007, 46, 7548–7558. [Google Scholar] [CrossRef] [PubMed]
- Ceresoli, D.; Vanderbilt, D. Structural and dielectric properties of amorphous ZrO2 and HfO2. Phys. Rev. B 2006, 74, 125108. [Google Scholar] [CrossRef]
- Wang, Y.; Zahid, F.; Wang, J.; Guo, H. Structure and dielectric properties of amorphous high-k oxides: HfO2, ZrO2, and their alloys. Phys. Rev. B 2012, 85, 224110. [Google Scholar] [CrossRef]
- Zhao, X.; Ceresoli, D.; Vanderbilt, D. Structural, electronic, and dielectric properties of amorphous ZrO2 from ab initio molecular dynamics. Phys. Rev. B 2005, 71, 085107. [Google Scholar] [CrossRef]
- Hofmeister, A.M. Single-crystal absorption and reflection infrared spectroscopy of forsterite and fayalite. Phys. Chem. Miner. 1987, 14, 499–513. [Google Scholar] [CrossRef]
- Madon, M.; Price, G.D. Infrared spectroscopy of the polymorphic series (enstatite, ilmenite, and perovskite) of MgSiO3, MgGeO3, and MnGeO3. J. Geophys. Res. Solid Earth 1989, 94, 15687–15701. [Google Scholar] [CrossRef]
- Fabian, D.; Jäger, C.; Henning, T.; Dorschner, J.; Mutschke, H. Steps toward interstellar silicate mineralogy V. Thermal evolution of amorphous magnesium silicates and silica. Astron. Astrophys. 2000, 364, 282–292. [Google Scholar]
- Henning, T.; Mutschke, H. Low-temperature infrared properties of cosmic dust analogues. Astron. Astrophys. 1997, 327, 743–754. [Google Scholar]
- Jäger, C.; Molster, F.J.; Dorschner, J.; Henning, T.; Mutschke, H.; Waters, L.B.F.M. Steps toward interstellar silicate mineralogy. IV. The crystalline revolution. Astron. Astrophys. 1998, 339, 904–916. [Google Scholar]
- Billinge, S.J.L.; Levin, I. The problem with determining atomic structure at the nanoscale. Science 2007, 316, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Tilocca, A. Current challenges in atomistic simulations of glasses for biomedical applications. Phys. Chem. Chem. Phys. 2014, 16, 3874–3880. [Google Scholar] [CrossRef] [PubMed]
- Berardo, E.; Pedone, A.; Ugliengo, P.; Corno, M. DFT Modeling of 45S5 and 77S soda-lime phospho-silicate glass surfaces: Clues on different bioactivity mechanism. Langmuir 2013, 29, 5749–5759. [Google Scholar] [CrossRef] [PubMed]
- Bondi, R.J.; Lee, S.; Hwang, G.S. First-principles study of the mechanical and optical properties of amorphous hydrogenated silicon and silicon-rich silicon oxide. Phys. Rev. B 2010, 81, 195207. [Google Scholar] [CrossRef]
- Corno, M.; Pedone, A.; Dovesi, R.; Ugliengo, P. B3LYP simulation of the full vibrational spectrum of 45S5 bioactive silicate glass compared to v-silica. Chem. Mater. 2008, 20, 5610–5621. [Google Scholar] [CrossRef]
- Karazhanov, S.Z.; Kroll, P.; Holt, A.; Bentzen, A.; Ulyashin, A. Comparative analysis of electronic structure and optical properties of crystalline and amorphous silicon nitrides. J. Appl. Phys. 2009, 106, 053717. [Google Scholar] [CrossRef]
- Kroll, P. A DFT study of amorphous silicon oxynitride. J. Non-Cryst. Solids 2005, 351, 1127–1132. [Google Scholar] [CrossRef]
- Landmann, M.; Köhler, T.; Rauls, E.; Frauenheim, T.; Schmidt, W.G. The atomic structure of ternary amorphous TixSi1−xO2 hybrid oxides. J. Phys. Condens. Matter 2014, 26, 253201. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Bondi, R.J.; Hwang, G.S. Ab initio parameterized valence force field for the structure and energetics of amorphous SiOx (0 <x <2) materials. Phys. Rev. B 2011, 84, 045202. [Google Scholar]
- Umari, P.; Pasquarello, A. Infrared and raman spectra of disordered materials from first principles. Diam. Relat. Mater. 2005, 14, 1255–1261. [Google Scholar] [CrossRef]
- Marquardt, K.; Faul, U.H. The structure and composition of olivine grain boundaries: 40 years of studies, status and current developments. Phys. Chem. Miner. 2018, 45, 139–172. [Google Scholar] [CrossRef]
- Henning, T. Cosmic silicates. Annu. Rev. Astron. Astrophys. 2010, 48, 21–46. [Google Scholar] [CrossRef]
- Mumma, M.J.; Charnley, S.B. The chemical composition of comets—Emerging taxonomies and natal heritage. Annu. Rev. Astron. Astrophys. 2011, 49, 471–524. [Google Scholar] [CrossRef]
- Rubin, A.E. Mineralogy of meteorite groups. Meteorit. Planet. Sci. 1997, 32, 231–247. [Google Scholar] [CrossRef]
- Molster, F.; Kemper, C. Crystalline silicates. Space Sci. Rev. 2005, 119, 3–28. [Google Scholar] [CrossRef]
- Vidali, G. H2 Formation on interstellar grains. Chem. Rev. 2013, 113, 8762–8782. [Google Scholar] [CrossRef] [PubMed]
- Hama, T.; Watanabe, N. Surface processes on interstellar amorphous solid water: Adsorption, diffusion, tunneling reactions, and nuclear-spin conversion. Chem. Rev. 2013, 113, 8783–8839. [Google Scholar] [CrossRef] [PubMed]
- Van Dishoeck, E.F.; Herbst, E.; Neufeld, D.A. Interstellar water chemistry: From laboratory to observations. Chem. Rev. 2013, 113, 9043–9085. [Google Scholar] [CrossRef] [PubMed]
- Shankland, T.J.; Hemmenway, K. Synthesis of forsterite crystals. Am. Mineral. 1963, 48, 200. [Google Scholar]
- Brindley, G.W.; Hayami, R. Kinetics and mechanism of formation of forsterite (Mg2SiO4) by solid state reaction of MgO and SiO2. Phil. Mag. Lett. 1965, 12, 505–514. [Google Scholar] [CrossRef]
- Shankland, T.J. Synthetic forsterite. Ceram. Bull. 1967, 46, 1160–1162. [Google Scholar]
- Cunha-Duncan Flavia, N.; Bradt Richard, C. Synthesis of magnesium aluminate spinels from bauxites and magnesias. J. Am. Ceram. Soc. 2004, 85, 2995–3003. [Google Scholar] [CrossRef]
- Kazakos, A.; Komarneni, S.; Roy, R. Preparation and densification of forsterite (Mg2SiO4) by nanocomposite sol-gel processing. Mater. Lett. 1990, 9, 405–409. [Google Scholar] [CrossRef]
- Park, D.G.; Duchamp, J.C.; Duncan, T.M.; Burlitch, J.M. Preparation of forsterite by pyrolysis of a xerogel: The effect of water. Chem. Mater. 1994, 6, 1990–1995. [Google Scholar] [CrossRef]
- Ban, T.; Ohya, Y.; Takahashi, Y. Low-temperature crystallization of forsterite and orthoenstatite. J. Am. Ceram. Soc. 2004, 82, 22–26. [Google Scholar] [CrossRef]
- Finch, C.B.; Clark, G.W. Czochralski growth of single-crystal Mg2SiO4 (forsterite). J. Cryst. Growth 1971, 8, 307–308. [Google Scholar] [CrossRef]
- Takei, H.; Hosoya, S.; Ozima, M. Synthesis of large single crystals of silicates and titanates. In Materials Science of the Earth’s Interior; Sunagawa, I., Ed.; Terra Scientific Publishing: Tokyo, Japan, 1984; pp. 107–130. [Google Scholar]
- Ito, K.; Sato, H.; Takei, H.; Tamada, O.; Kitazawa, T. Synthesis of large high-quality forsterite single crystals to 200 mm length and its significance. Geochem. Geophys. Geosyst. 2003, 4. [Google Scholar] [CrossRef]
- Williams, Q.; McMillan, P.; Cooney, T.F. Vibrational spectra of olivine composition glasses: The Mg-Mn join. Phys. Chem. Miner. 1989, 16, 352–359. [Google Scholar] [CrossRef]
- Cooney, T.F.; Sharma, S.K. Structure of glasses in the systems Mg2SiO4-Fe2SiO4, Mn2SiO4-Fe2SiO4, Mg2SiO4-CaMgSiO4, and Mn2SiO4-CaMnSiO4. J. Non-Cryst. Solids 1990, 122, 10–32. [Google Scholar] [CrossRef]
- Tangeman, J.A.; Phillips, B.L.; Navrotsky, A.; Weber, J.K.R.; Hixson, A.D.; Key, T.S. Vitreous forsterite (Mg2SiO4): Synthesis, structure, and thermochemistry. Geophys. Res. Lett. 2001, 28, 2517–2520. [Google Scholar] [CrossRef]
- Thompson, S.P.; Evans, A.; Jones, A.P. Structural evolution in thermally processed silicates. Astron. Astrophys. 1996, 308, 309–320. [Google Scholar]
- Hanada, T.; Soga, N.; Tachibana, T. Coordination state of magnesium ions in rf-sputtered amorphous films in the system MgO-SiO2. J. Non-Cryst. Solids 1988, 105, 39–44. [Google Scholar] [CrossRef]
- Jeanloz, R.; Ahrens, T.J.; Lally, J.S.; Nord, G.L.; Christie, J.M.; Heuer, A.H. Shock-produced olivine glass: First observation. Science 1977, 197, 457. [Google Scholar] [CrossRef] [PubMed]
- Kosanović, C.; Stubičar, N.; Tomašić, N.; Bermanec, V.; Stubičar, M. Synthesis of a forsterite powder by combined ball milling and thermal treatment. J. Alloys Compd. 2005, 389, 306–309. [Google Scholar] [CrossRef]
- Mirhadi, S.M.; Forghani, A.; Tavangarian, F. A modified method to synthesize single-phase forsterite nanoparticles at low temperature. Ceram. Int. 2016, 42, 7974–7979. [Google Scholar] [CrossRef]
- Mennella, V.; Brucato, J.R.; Colangeli, L. Carbon and silicate grains in the laboratory as analogues of cosmic dust. Spectrochim. Acta Part A 2001, 57, 787–795. [Google Scholar] [CrossRef]
- Colangeli, L.; Henning, T.; Brucato, J.R.; Clément, D.; Fabian, D.; Guillois, O.; Huisken, F.; Jäger, C.; Jessberger, E.K.; Jones, A.; et al. The role of laboratory experiments in the characterisation of silicon–based cosmic material. Astron. Astrophys. Rev. 2003, 11, 97–152. [Google Scholar] [CrossRef]
- Brunetto, R.; Roush, T.L.; Marra, A.C.; Orofino, V. Optical characterization of laser ablated silicates. Icarus 2007, 191, 381–393. [Google Scholar] [CrossRef]
- Tavangarian, F.; Emadi, R. Synthesis of nanocrystalline forsterite (Mg2SiO4) powder by combined mechanical activation and thermal treatment. Mater. Res. Bull. 2010, 45, 388–391. [Google Scholar] [CrossRef]
- Tavangarian, F.; Emadi, R. Mechanochemical synthesis of single phase nanocrystalline forsterite powder. Int. J. Mod. Phys. B 2010, 24, 343–350. [Google Scholar] [CrossRef]
- Nurbaiti, U.; Zainuri, T.M.; Pratapa, S. Synthesis of microforsterite using derived-amorphous-silica of silica sands. AIP Conf. Proc. 2016, 1725, 020056. [Google Scholar]
- Servoin, J.L.; Piriou, B. Infrared reflectivity and raman scattering of Mg2SiO4 single crystal. Phys. Status Solidi B 1973, 55, 677–686. [Google Scholar] [CrossRef]
- Iishi, K. Lattice dynamics of forsterite. Am. Mineral. 1978, 63, 1198–1208. [Google Scholar]
- Gervais, F.; Blin, A.; Massiot, D.; Coutures, J.P.; Chopinet, M.H.; Naudin, F. Infrared reflectivity spectroscopy of silicate glasses. J. Non-Cryst. Solids 1987, 89, 384–401. [Google Scholar] [CrossRef]
- Brucato, J.R.; Colangeli, L.; Mennella, V.; Palumbo, P.; Bussoletti, E. Mid-infrared spectral evolution of thermally annealed amorphous pyroxene. Astron. Astrophys. 1999, 348, 1012–1019. [Google Scholar]
- Abdi Maghsoudlou, M.S.; Ebadzadeh, T.; Sharafi, Z.; Arabi, M.; Zahabi, K.R. Synthesis and sintering of nano-sized forsterite prepared by short mechanochemical activation process. J. Alloys Compd. 2016, 678, 290–296. [Google Scholar] [CrossRef]
- Suzuki, N.; Kimura, S.; Nakada, T.; Kaito, C.; Saito, Y.; Koike, C. Correlation between crystallographic structure and infrared spectra of silicon oxide films containing iron or magnesium atoms. Meteorit. Planet. Sci. 2010, 35, 1269–1273. [Google Scholar] [CrossRef]
- Tsuchiyama, A. Condensation experments using a forsterite evaporation source in H2 at low pressures. Mineral. J. 1998, 20, 59–80. [Google Scholar] [CrossRef]
- Black, J.R.; Carroll, S.A.; Haese, R.R. Rates of mineral dissolution under CO2 storage conditions. Chem. Geol. 2015, 399, 134–144. [Google Scholar] [CrossRef]
- Meysman, F.J.R.; Montserrat, F. Negative CO2 emissions via enhanced silicate weathering in coastal environments. Biol. Lett. 2017, 13, 20160905. [Google Scholar] [CrossRef] [PubMed]
- Montserrat, F.; Renforth, P.; Hartmann, J.; Leermakers, M.; Knops, P.; Meysman, F.J.R. Olivine dissolution in seawater: Implications for CO2 sequestration through enhanced weathering in coastal environments. Environ. Sci. Technol. 2017, 51, 3960–3972. [Google Scholar] [CrossRef] [PubMed]
- Quadery, A.H.; Pacheco, S.; Au, A.; Rizzacasa, N.; Nichols, J.; Le, T.; Glasscock, C.; Schelling, P.K. Atomic-scale simulation of space weathering in olivine and orthopyroxene. J. Geophys. Res. Planets 2015, 120, 643–661. [Google Scholar] [CrossRef]
- Strefler, J.; Amann, T.; Bauer, N.; Kriegler, E.; Hartmann, J. Potential and costs of carbon dioxide removal by enhanced weathering of rocks. Environ. Res. Lett. 2018, 13. [Google Scholar] [CrossRef]
- Ramesh, S.; Yaghoubi, A.; Sara Lee, K.Y.; Christopher Chin, K.M.; Purbolaksono, J.; Hamdi, M.; Hassan, M.A. Nanocrystalline forsterite for biomedical applications: Synthesis, microstructure and mechanical properties. J. Mech. Behav. Biomed. Mater. 2013, 25, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Samadikuchaksaraei, A.; Gholipourmalekabadi, M.; Farhadihosseinabadi, B.; Rezvani, Z.; Mozafari, M. Carboxymethyl chitosan/forsterite bone tissue engineering scaffolds: Correlations between composition and physico-chemical characteristics. Biointerf. Res. Appl. Chem. 2016, 6, 1222–1228. [Google Scholar]
- Kharaziha, M.; Fathi, M.H.; Edris, H. Development of novel aligned nanofibrous composite membranes for guided bone regeneration. J. Mech. Behav. Biomed. Mater. 2013, 24, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yazdanpanah, A.; Kamalian, R.; Moztarzadeh, F.; Mozafari, M.; Ravarian, R.; Tayebi, L. Enhancement of fracture toughness in bioactive glass-based nanocomposites with nanocrystalline forsterite as advanced biomaterials for bone tissue engineering applications. Ceram. Int. 2012, 38, 5007–5014. [Google Scholar] [CrossRef]
- Diba, M.; Fathi, M.H.; Kharaziha, M. Novel forsterite/polycaprolactone nanocomposite scaffold for tissue engineering applications. Mater. Lett. 2011, 65, 1931–1934. [Google Scholar] [CrossRef]
- Yang, H.M.; Shi, J.X.; Gong, M.L.; Cheah, K.W. Synthesis and photo luminescence of Eu3+- or Tb3+-doped Mg2SiO4 nanoparticles prepared by a combined novel approach. J. Lumin. 2006, 118, 257–264. [Google Scholar] [CrossRef]
- Naik, R.; Prashantha, S.C.; Nagabhushana, H.; Sharma, S.C.; Nagabhushana, B.M.; Nagaswarupa, H.P.; Premkumar, H.B. Low temperature synthesis and photoluminescence properties of red emitting Mg2SiO4:Eu3+ nanophosphor for near UV light emitting diodes. Sens. Actuators B 2014, 195, 140–149. [Google Scholar] [CrossRef]
- Zampiva, R.Y.S.; Acauan, L.H.; Venturini, J.; Garcia, J.A.M.; da Silva, D.S.; Han, Z.H.; Kassab, L.R.P.; Wetter, N.U.; Agarwal, A.; Alves, A.K.; et al. Tunable green/red luminescence by infrared upconversion in biocompatible forsterite nanoparticles with high erbium doping uptake. Opt. Mater. 2018, 76, 407–415. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Erba, A.; Zicovich-Wilson, C.M.; Civalleri, B.; Casassa, S.; Maschio, L.; Ferrabone, M.; De La Pierre, M.; D’Arco, P.; et al. CRYSTAL14: A program for the ab initio investigation of crystalline solids. Int. J. Quantum Chem. 2014, 114, 1287–1317. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: Oxford, UK, 1954; 420p. [Google Scholar]
- Gale, J.D.; Rohl, A.L. The general utility lattice program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Flikkema, E.; Bromley, S.T. A new interatomic potential for nanoscale silica. Chem. Phys. Lett. 2003, 378, 622–629. [Google Scholar] [CrossRef]
- Hassanali, A.A.; Singer, S.J. Model for the water—Amorphous silica interface: The undissociated surface. J. Phys. Chem. B 2007, 111, 11181–11193. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; Johnston, R.L. Investigation of the structures of MgO clusters using a genetic algorithm. Phys. Chem. Chem. Phys. 2001, 3, 5024–5034. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Civalleri, B.; D’Arco, P.; Orlando, R.; Saunders, V.R.; Dovesi, R. Hartree–Fock geometry optimisation of periodic systems with the CRYSTAL code. Chem. Phys. Lett. 2001, 348, 131–138. [Google Scholar] [CrossRef]
- De La Pierre, M.; Carteret, C.; Orlando, R.; Dovesi, R. Use of Ab initio methods for the interpretation of the experimental IR reflectance spectra of crystalline compounds. J. Comput. Chem. 2013, 34, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- De La Pierre, M.; Orlando, R.; Maschio, L.; Doll, K.; Ugliengo, P.; Dovesi, R. Performance of six functionals (LDA, PBE, PBESOL, B3LYP, PBE0, and WC1LYP) in the simulation of vibrational and dielectric properties of crystalline compounds. The case of forsterite Mg2SiO4. J. Comput. Chem. 2011, 32, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Noel, Y.; Catti, M.; D’Arco, P.; Dovesi, R. The vibrational frequencies of forsterite Mg2SiO4: An all-electron ab initio study with the CRYSTAL code. Phys. Chem. Miner. 2006, 33, 383–393. [Google Scholar] [CrossRef]
- Noel, Y.; De La Pierre, M.; Maschio, L.; Rérat, M.; Zicovich-Wilson, C.M.; Dovesi, R. Electronic structure, dielectric properties and infrared vibrational spectrum of fayalite: An ab initio simulation with an all-electron gaussian basis set and the B3LYP functional. Int. J. Quantum Chem. 2012, 112, 2098–2108. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Zicovich-Wilson, C.M.; Bert, A.; Roetti, C.; Dovesi, R.; Saunders, V.R. Characterization of the electronic structure of crystalline compounds through their localized wannier functions. J. Chem. Phys. 2002, 116, 1120–1127. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Dovesi, R.; Saunders, V.R. A General method to obtain well localized wannier functions for composite energy bands in linear combination of atomic orbital periodic calculations. J. Chem. Phys. 2001, 115, 9708–9719. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Torres, F.J.; Pascale, F.; Valenzano, L.; Orlando, R.; Dovesi, R. Ab initio simulation of the IR spectra of pyrope, grossular, and andradite. J. Comput. Chem. 2008, 29, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Civalleri, B.; Rérat, M.; Orlando, R.; Dovesi, R. The calculation of the static first and second susceptibilities of crystalline urea: A comparison of Hartree-Fock and density functional theory results obtained with the periodic coupled perturbed Hartree-Fock/Kohn-Sham scheme. J. Chem. Phys. 2009, 131, 214704. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R. The calculation of static polarizabilities of 1-3D periodic compounds. The implementation in the crystal code. J. Comput. Chem. 2008, 29, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Carteret, C.; De La Pierre, M.; Dossot, M.; Pascale, F.; Erba, A.; Dovesi, R. The vibrational spectrum of CaCO3 aragonite: A combined experimental and quantum-mechanical investigation. J. Chem. Phys. 2013, 138, 014201. [Google Scholar] [CrossRef] [PubMed]
- De La Pierre, M.; Carteret, C.; Maschio, L.; André, E.; Orlando, R.; Dovesi, R. The raman spectrum of CaCO3 polymorphs calcite and aragonite: A combined experimental and computational study. J. Chem. Phys. 2014, 140, 164509. [Google Scholar] [CrossRef] [PubMed]
- Lacivita, V.; Erba, A.; Noël, Y.; Orlando, R.; D’Arco, P.; Dovesi, R. Zinc oxide nanotubes: An ab initio investigation of their structural, vibrational, elastic, and dielectric properties. J. Chem. Phys. 2013, 138, 214706. [Google Scholar] [CrossRef] [PubMed]
- Maschio, L.; Demichelis, R.; Orlando, R.; Pierre, M.D.L.; Mahmoud, A.; Dovesi, R. The raman spectrum of grossular garnet: A quantum mechanical simulation of wavenumbers and intensities. J. Raman Spectrosc. 2014, 45, 710–715. [Google Scholar] [CrossRef]
- Maschio, L.; Kirtman, B.; Salustro, S.; Zicovich-Wilson, C.M.; Orlando, R.; Dovesi, R. Raman spectrum of pyrope garnet. A quantum mechanical simulation of frequencies, intensities, and isotope shifts. J. Phys. Chem. A 2013, 117, 11464–11471. [Google Scholar] [CrossRef] [PubMed]
- Maul, J.; Erba, A.; Santos, I.M.G.; Sambrano, J.R.; Dovesi, R. In silico infrared and raman spectroscopy under pressure: The case of CaSnO3 perovskite. J. Chem. Phys. 2015, 142, 014505. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, M.; Maschio, L.; Kirtman, B.; Salustro, S.; Erba, A.; Dovesi, R. Raman spectrum of NaAlSi2O6 jadeite. A quantum mechanical simulation. J. Raman Spectrosc. 2014, 45, 703–709. [Google Scholar] [CrossRef]
- Ricca, C.; Grishin, A.; Ringuede, A.; Cassir, M.; Adamo, C.; Labat, F. Modeling composite electrolytes for low-temperature solid oxide fuel cell application: Structural, vibrational and electronic features of carbonate-oxide interfaces. J. Mater. Chem. A 2016, 4, 17473–17482. [Google Scholar] [CrossRef]
- Schneider, A.G.; Schomborg, L.; Ulpe, A.C.; Rüscher, C.H.; Bredow, T. Structure, vibrational spectra and 11B-NMR chemical shift of Na8[AlSiO4]6(B(OH)4)2: Comparison of theory and experiment. J. Phys. Chem. A 2016, 120, 7503–7509. [Google Scholar] [CrossRef] [PubMed]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the vibration frequencies of α-Quartz: The effect of hamiltonian and basis set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Rérat, M.; Kirtman, B.; Dovesi, R. Calculation of first and second static hyperpolarizabilities of one- to three-dimensional periodic compounds. Implementation in the CRYSTAL code. J. Chem. Phys. 2008, 129, 244110. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R. Coupled perturbed hartree-fock for periodic systems: The role of symmetry and related computational aspects. J. Chem. Phys. 2008, 128, 014110. [Google Scholar] [CrossRef] [PubMed]
- Baranek, P.; Zicovich-Wilson, C.M.; Roetti, C.; Orlando, R.; Dovesi, R. Well localized crystalline orbitals obtained from bloch functions: The case of KNbO3. Phys. Rev. B 2001, 64, 125102. [Google Scholar] [CrossRef]
- Dall’Olio, S.; Dovesi, R.; Resta, R. Spontaneous polarization as a berry phase of the hartree-fock wave function: The case of KNbO3. Phys. Rev. B 1997, 56, 10105–10114. [Google Scholar] [CrossRef]
- Noel, Y.; Zicovich-Wilson, C.M.; Civalleri, B.; D’Arco, P.; Dovesi, R. Polarization properties of ZnO and BeO: An ab initio study through the berry phase and wannier functions approaches. Phys. Rev. B 2001, 65, 014111. [Google Scholar] [CrossRef]
- Dovesi, R.; Pierre, M.D.L.; Ferrari, A.M.; Pascale, F.; Maschio, L.; Zicovich-Wilson, C.M. The IR vibrational properties of six members of the garnet family: A quantum mechanical ab initio study. Am. Mineral. 2011, 96, 1787–1798. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Valenzano, L.; Meyer, A.; Orlando, R.; Dovesi, R. Quantum-mechanical ab initio simulation of the raman and IR spectra of Fe3Al2Si3O12 almandine. J. Phys. Chem. A 2009, 113, 11289–11294. [Google Scholar] [CrossRef] [PubMed]
- Maschio, L.; Ferrabone, M.; Meyer, A.; Garza, J.; Dovesi, R. The infrared spectrum of spessartine: An ab initio all electron simulation with five different functionals (LDA, PBE, PBESOL, B3LYP and PBE0). Chem. Phys. Lett. 2011, 501, 612–618. [Google Scholar] [CrossRef]
- Deer, W.A.; Howie, R.A.; Zussman, J. An Introduction to the Rock-Forming Minerals, 2nd ed.; Longman: London, UK, 1992; 724p. [Google Scholar]
- Chopelas, A. Single crystal raman spectra of forsterite, fayalite, and monticellite. Am. Mineral. 1991, 76, 1100–1109. [Google Scholar]
- Hofmeister, A.M. Thermal conductivity of spinels and olivines from vibrational spectroscopy: Ambient conditions. Am. Mineral. 2001, 86, 1188–1208. [Google Scholar] [CrossRef]
- Kolesov, B.A.; Geiger, C.A. A raman spectroscopic study of Fe–Mg olivines. Phys. Chem. Miner. 2004, 31, 142–154. [Google Scholar] [CrossRef]
- Reynard, B. Single-crystal infrared reflectivity of pure Mg2SiO2 forsterite and (Mg0.86, Fe0.14)2SiO4 olivine. Phys. Chem. Miner. 1991, 18, 19–25. [Google Scholar] [CrossRef]
- Sogawa, H.; Koike, C.; Chihara, H.; Suto, H.; Tachibana, S.; Tsuchiyama, A.; Kozasa, T. Infrared reflection spectra of forsterite crystal. Astron. Astrophys. 2006, 451, 357–361. [Google Scholar] [CrossRef]
- Suto, H.; Sogawa, H.; Tachibana, S.; Koike, C.; Karoji, H.; Tsuchiyama, A.; Chihara, H.; Mizutani, K.; Akedo, J.; Ogiso, K.; et al. Low-temperature single crystal reflection spectra of forsterite. Mon. Not. R. Astron. Soc. 2006, 370, 1599–1606. [Google Scholar] [CrossRef]
- Navarro-Ruiz, J.; Ugliengo, P.; Rimola, A.; Sodupe, M. B3LYP periodic study of the physicochemical properties of the nonpolar (010) Mg-pure and Fe-containing olivine surfaces. J. Phys. Chem. A 2014, 118, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Wunder, B.; Jahn, S.; Koch-Müller, M.; Speziale, S. The 3.65 Å phase, MgSi(OH)6: Structural insights from DFT-calculations and T-dependent IR spectroscopy. Am. Mineral. 2012, 97, 1043–1048. [Google Scholar] [CrossRef]
- Yang, H.; Prewitt, C.T.; Frost, D.J. Crystal structure of the dense hydrous magnesium silicate, phase D. Am. Mineral. 1997, 82, 651–654. [Google Scholar] [CrossRef]
- Frost, D.J.; Fei, Y. Stability of phase D at high pressure and high temperature. J. Geophys. Res. Solid Earth 1998, 103, 7463–7474. [Google Scholar] [CrossRef]
- Hajilar, S.; Shafei, B.; Cheng, T.; Jaramillo-Botero, A. Reactive molecular dynamics simulations to understand mechanical response of thaumasite under temperature and strain rate effects. J. Phys. Chem. A 2017, 121, 4688–4697. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Charges | |||
|---|---|---|---|
| Ions | Core | Shell | Core–shell interactions (eV Å−2) |
| Mg | +2.00000 | ||
| Si | +4.00000 | ||
| O | +0.84819 | −2.84819 | 74.92 |
| Buckingham Potential | A (eV) | ρ (Å) | C (eV Å6) |
| Mgc–Os | 1428.500 | 0.31813 | 0.00000 |
| Sic–Os | 1283.907 | 0.32052 | 10.66158 |
| Oc–Os | 22,764.300 | 0.14900 | 27.88000 |
| Three-Body Potential | k (eV rad−2) | Θ0 (degree) | |
| Os–Sic–Os | 2.097240 | 109.47000 |
| Experimental | Crystal | Amorphous | |
|---|---|---|---|
| Si–O (Å) | 1.616–1.649 | 1.628–1.673 | 1.618–1.693 |
| Mg1–O (Å) | 2.069–2.126 | 2.073–2.131 | 1.929–2.226 |
| Mg2–O (Å) | 2.040–2.166 | 2.062–2.222 | |
| a (Å) | 4.746 | 4.789 | 4.93 |
| b (Å) | 10.18 | 10.25 | 9.37 |
| c (Å) | 5.976 | 6.009 | 5.97 (11.94) 1 |
| α (degrees) | 90.0 | 90.0 | 87.4 |
| β (degrees) | 90.0 | 90.0 | 90.6 |
| γ (degrees) | 90.0 | 90.0 | 88.6 |
| Volume (Å3) | 288.73 | 295.10 | 275.78 (551.00) 1 |
| Density (g·cm−3) | 3.22 | 3.34 | 3.37 |
| Angle (Degrees) | Ideal Value | Mean Value ± Standard Deviation (Average Deviation) | ||
|---|---|---|---|---|
| 1 × 1 × 2 | 2 × 1 × 2 | 2 × 2 × 2 | ||
| O–Si–O | 110.0 | 109.5 ± 7.2(6.53%) | 108.9 ± 12.4(11.3%) | 104.9 ± 12.8(11.6%) |
| O–Mg–O | 90.0 | 94.7 ± 13.8(15.3%) | 100.9 ± 22.1(24.6%) | 104.1 ± 16.5(18.3%) |
| Experimental 1 | Crystal | Amorphous | |
|---|---|---|---|
| ε∞,x | 2.789 | 2.571 | 2.284 |
| ε∞,y | 2.673 | 2.424 | 2.247 |
| ε∞,z | 2.726 | 2.475 | 2.218 |
| N | This Work | [121] | [119] | [60] | [59] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ν | I (%) | ν | Δν | ν | Δν | ν | Δν | ν | Δν | ||
| Ag | 1 | 212 | 0.05 | 184 | 28 | 183 | 29 | 183 | 29 | 183 | 29 |
| 2 | 261 | 0.08 | 227 | 34 | 226 | 35 | 227 | 34 | 227 | 34 | |
| 3 | 351 | 0.11 | 304 | 47 | 304 | 47 | 305 | 46 | 305 | 46 | |
| 4 | 367 | 1.20 | 329 | 38 | 329 | 38 | 329 | 38 | 329 | 38 | |
| 5 | 391 | 0.78 | 339 | 52 | 332 | 59 | 340 | 51 | 340 | 51 | |
| 6 | 474 | 0.62 | 421 | 53 | 422 | 52 | 424 | 50 | 424 | 50 | |
| 7 | 608 | 12.4 | 544 | 64 | 545 | 63 | 546 | 62 | 545 | 63 | |
| 8 | 675 | 22.2 | 608 | 67 | 608 | 67 | 609 | 66 | 609 | 66 | |
| 9 | 848 | 81.1 | 824 | 24 | 824 | 24 | 826 | 22 | 826 | 22 | |
| 10 | 883 | 100 | 856 | 27 | 856 | 27 | 856 | 27 | 856 | 27 | |
| 11 | 999 | 20.6 | 966 | 33 | 965 | 34 | 966 | 33 | 966 | 33 | |
| B1g | 12 | 253 | 0.00 | - | - | 220 | 33 | 224 | 29 | 197 | 56 |
| 13 | 294 | 0.06 | - | - | 274 | 20 | 260 | 34 | 265 | 29 | |
| 14 | 361 | 1.05 | 316 | 45 | 318 | 43 | 318 | 43 | 317 | 44 | |
| 15 | 417 | 1.68 | - | - | 351 | 66 | - | - | - | - | |
| 16 | 420 | 0.38 | - | - | 383 | 37 | 418 | 2 | 418 | 2 | |
| 17 | 489 | 2.21 | 434 | 55 | 434 | 55 | 434 | 55 | 434 | 55 | |
| 18 | 658 | 1.39 | 588 | 70 | 582 | 76 | 585 | 73 | 583 | 75 | |
| 19 | 713 | 0.43 | - | - | 632 | 81 | 632 | 81 | 632 | 81 | |
| 20 | 869 | 1.28 | 838 | 31 | 838 | 31 | 839 | 30 | 839 | 30 | |
| 21 | 897 | 3.37 | 866 | 31 | 866 | 31 | 866 | 31 | 866 | 31 | |
| 22 | 1012 | 0.37 | - | - | 975 | 37 | 976 | 36 | 976 | 36 | |
| B2g | 23 | - | - | - | 175 | - | 142 | - | - | - | |
| 24 | 229 | 0.01 | 243 | −14 | 242 | −13 | 244 | −15 | 244 | −15 | |
| 25 | 343 | 0.17 | - | - | 323 | 20 | 324 | 19 | 324 | 19 | |
| 26 | 365 | 0.08 | - | - | 365 | 0 | 368 | −3 | 365 | 0 | |
| 27 | 431 | 2.93 | - | - | - | - | - | - | 407 | 24 | |
| 28 | 463 | 0.69 | 434 | 29 | 439 | 24 | 441 | 22 | 438 | 25 | |
| 29 | 665 | 14.3 | 588 | 77 | 586 | 79 | 588 | 77 | 585 | 80 | |
| 30 | 956 | 6.98 | 882 | 74 | 881 | 75 | 884 | 72 | 881 | 75 | |
| B3g | 31 | 203 | 0.00 | 243 | −40 | - | - | 226 | −23 | - | - |
| 32 | 296 | 0.00 | - | - | 286 | 10 | 272 | 24 | - | - | |
| 33 | - | 316 | - | 315 | - | 318 | - | 314 | - | ||
| 34 | 369 | 0.31 | 373 | −4 | 374 | −5 | 376 | −7 | 374 | −5 | |
| 35 | 413 | 1.35 | 409 | 4 | 410 | 3 | 412 | 1 | 406 | 7 | |
| 36 | - | - | - | 435 | - | - | - | 466 | - | ||
| 37 | 514 | 3.34 | - | - | - | - | - | - | 484 | 30 | |
| 38 | 670 | 2.82 | 588 | 82 | 592 | 78 | 595 | 75 | 591 | 79 | |
| 39 | 908 | 5.14 | 920 | −12 | 920 | −12 | 922 | −14 | 920 | −12 | |
| 1 × 1 × 2 | 2 × 1 × 2 | 2 × 2 × 2 | |
|---|---|---|---|
| Si–O (Å) | 1.618–1.693 | 1.592–1.906 | 1.579–1.983 |
| Mg–O (Å) | 1.929–2.226 | 1.860–2.211 | 1.800–2.209 |
| a (Å) | 4.93 | 10.168 | 9.589 |
| b (Å) | 9.37 | 10.177 | 20.574 |
| c (Å) | 11.9 | 11.329 | 11.937 |
| α (degrees) | 87.4 | 89.6 | 85.7 |
| β (degrees) | 90.6 | 83.6 | 93.4 |
| γ (degrees) | 88.6 | 97.1 | 91.7 |
| Volume (Å3) | 551.00 | 1156.77 | 2343.91 |
| Density (g·cm−3) | 3.373 | 3.214 | 3.172 |
| 1 × 1 × 2 | 2 × 1 × 2 | 2 × 2 × 2 | |
|---|---|---|---|
| ε∞,x | 2.284 | 2.153 | 2.148 |
| ε∞,y | 2.247 | 2.161 | 2.135 |
| ε∞,z | 2.218 | 2.168 | 2.132 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-González, J.Á.; Navarro-Ruiz, J.; Rimola, A. Multiscale Computational Simulation of Amorphous Silicates’ Structural, Dielectric, and Vibrational Spectroscopic Properties. Minerals 2018, 8, 353. https://doi.org/10.3390/min8080353
Martínez-González JÁ, Navarro-Ruiz J, Rimola A. Multiscale Computational Simulation of Amorphous Silicates’ Structural, Dielectric, and Vibrational Spectroscopic Properties. Minerals. 2018; 8(8):353. https://doi.org/10.3390/min8080353
Chicago/Turabian StyleMartínez-González, José Ángel, Javier Navarro-Ruiz, and Albert Rimola. 2018. "Multiscale Computational Simulation of Amorphous Silicates’ Structural, Dielectric, and Vibrational Spectroscopic Properties" Minerals 8, no. 8: 353. https://doi.org/10.3390/min8080353
APA StyleMartínez-González, J. Á., Navarro-Ruiz, J., & Rimola, A. (2018). Multiscale Computational Simulation of Amorphous Silicates’ Structural, Dielectric, and Vibrational Spectroscopic Properties. Minerals, 8(8), 353. https://doi.org/10.3390/min8080353