1. Introduction
The Freetown Peninsula, Sierra Leone consists of a thick (7 km) sequence of layered gabbroic rocks. Platinum-group minerals (PGM) have been located in these rocks and have shown to be altered by superficial weathering [
1,
2]. Alluvial PGM have been recovered from the streams draining the intrusion and they are notably different in mineral assemblage, composition, and size from the PGM in the unaltered host rocks [
3,
4]. This difference has created debate concerning whether supergene processes could be responsible for the observed mineralogical differences. It has been suggested [
5,
6,
7] that some of the PGM from the host rocks were altered during weathering and the platinum-group elements (PGE) products transported in solution to a regime of changed Eh and pH where growth of a new PGM suite occurred. Others consider this to be a “scientifically untenable hypothesis” [
8]. There is textural evidence in the alluvial PGM, however, that is consistent with low-temperature growth and re-solution and re-growth. Aware of the abundant organic material and microbial fauna beneath the tropical rain forest cover of the intrusion and the ease with which the PGE react with organic materials [
9,
10], some studies [
11,
12,
13] have examined the possible role of humic and fulvic acid in taking the PGE into solution at 25 °C. It is clearly possible for the PGE to enter solution under these conditions within a short time frame. Importantly, recent work has shown the ability of bacteria to play a part in the growth of PGM [
14,
15]. There is a problem in developing an overall mechanism because there is evidence of different stages in the alteration and re-growth process, but the connections between these stages are circumstantial. A reviewer’s comments on a recent paper describing the mineralogy, geochemistry and genesis of the alluvial Freetown PGM [
4] can be paraphrased as: “OK we accept there is a question to be answered but if organic acids are involved how could that work?” This paper is a novel attempt to answer that question and to suggest an alternative mechanism to a conventional organic geochemical approach. The question to be asked is less “how does humic acid permit alteration of the PGM or transport of the PGE?”, but rather “how is humic acid broken down by the presence of the PGE in soils?”
2. Humic Acids in Soils
Humic acids are products of the breakdown of dead organic matter derived from the biosphere (e.g., leaf litter). In the living cell, the organic molecular structures are enzymatically controlled, either as independent molecules (leaf waxes) or biopolymers (proteins, carbohydrates, and lipids). The molecules comprising biopolymers have a reactive functional group (e.g., acid, alcohol) through which they are bound to the biopolymer. Upon death of a cell, the biopolymers are broken down (hydrolyzed) in to smaller polymers and free molecules (e.g., protein → peptide → amino acid). The free molecules and biopolymer breakdown products are overwhelmingly (>99.9%) degraded by microbial processes, ultimately to CO
2 and H
2O [
16].
The molecules comprising biopolymers have reactive functional groups that can recombine non-enzymatically to form unorganized polymers, which become geopolymers. Molecules without reactive functional groups can escape degradation by adsorption and clathration within the randomly structured geopolymers. Initially, relatively low molecular weight geopolymers are formed (fulvic acids) which become humic acids as random polymerization continues to increase the geopolymer molecular weight. Further polymerization yields humins (protokerogen), and ultimately kerogen, the quantitatively dominant geopolymer. As polymerization increases molecular weight, the geopolymers become more difficult to isolate and characterize. Fulvic and humic acids are both soluble in a sodium hydroxide solution, whereas humin and kerogen are insoluble. Fulvic acids are soluble in hydrochloric acid, whereas humic acids and humins are insoluble [
16,
17].
The transition from fulvic acid to humic acid and to kerogens is a continuum, with definition dependent only on solubility. The geoploymers however retain in part the molecular structures that are inherited from the living sources. To deduce the original biological sources, bulk methods such as carbon and nitrogen stable isotope analyses can be used. If the original molecular structures are sought, a common technique is to thermally fragment the polymer (pyrolysis), followed by quantitative identification of the individual molecules by gas chromatography-mass spectrometry (GC-MS) [
18]. Pyrolysis can produce artifacts. For example, a C
16 straight chain hydrocarbon with a terminal acid or alcohol can become thermally broken adjacent to the functional group to yield the C
15 hydrocarbon in the final GC-MS analysis, as well as any free C
15. Among the many compounds detected from pyrolysis of humic acids is an important group, the n (normal) -alkanes, where n is the number of carbons. These are straight chains, with no branches or cyclics, and have the general formula C
nH
(2n+2), where n ranges from 1 to 100 or more and their melting points increase with n. The n-alkanes with low values of n form gases, such as methane (CH
4, i.e., n = 1). In the range n = 5 to 16 the alkanes are liquids at 20 °C (e.g., octane, C
8H
18, n = 8) and for higher values of n they are solid and waxy.
The carbon number, n, can be indicative of the original biological source. Aquatic plants (e.g., algae and phytoplankton) yield high concentrations of n-alkanes in the nC
15–17 range. Sub-aerial plants synthesize high boiling point waxes (nC
27–31) as protection from desiccation by the sun and wind, to cover leaves and fruit. Odd-carbon numbered n-alkanes usually dominate [
19]. The reason is that the cell polymerizes acetic acid (C
2H
4O
2) to synthesize fatty (carboxylic) acids, which containing multiples of two carbon atoms, have even carbon numbers. The cell then removes the terminal acid functional group (loosing 1 carbon), resulting in the odd carbon numbered predominance in the n-alkanes.
It is normal alkanes with n = 27, 29, and 31 that are of particular interest here and they are abbreviated to nC
27, nC
29, and nC
31. In their simplest form the alkanes consist of a long straight chain of carbon atoms with the hydrogen atoms attached along the length. The waxy, non-water soluble alkanes form in the leaves of vegetation [
20,
21,
22], especially in the leaves of broad-leaved trees [
23]. They can be found to be preserved in the soil as their melting points range from 59.5 to 67.9 °C.
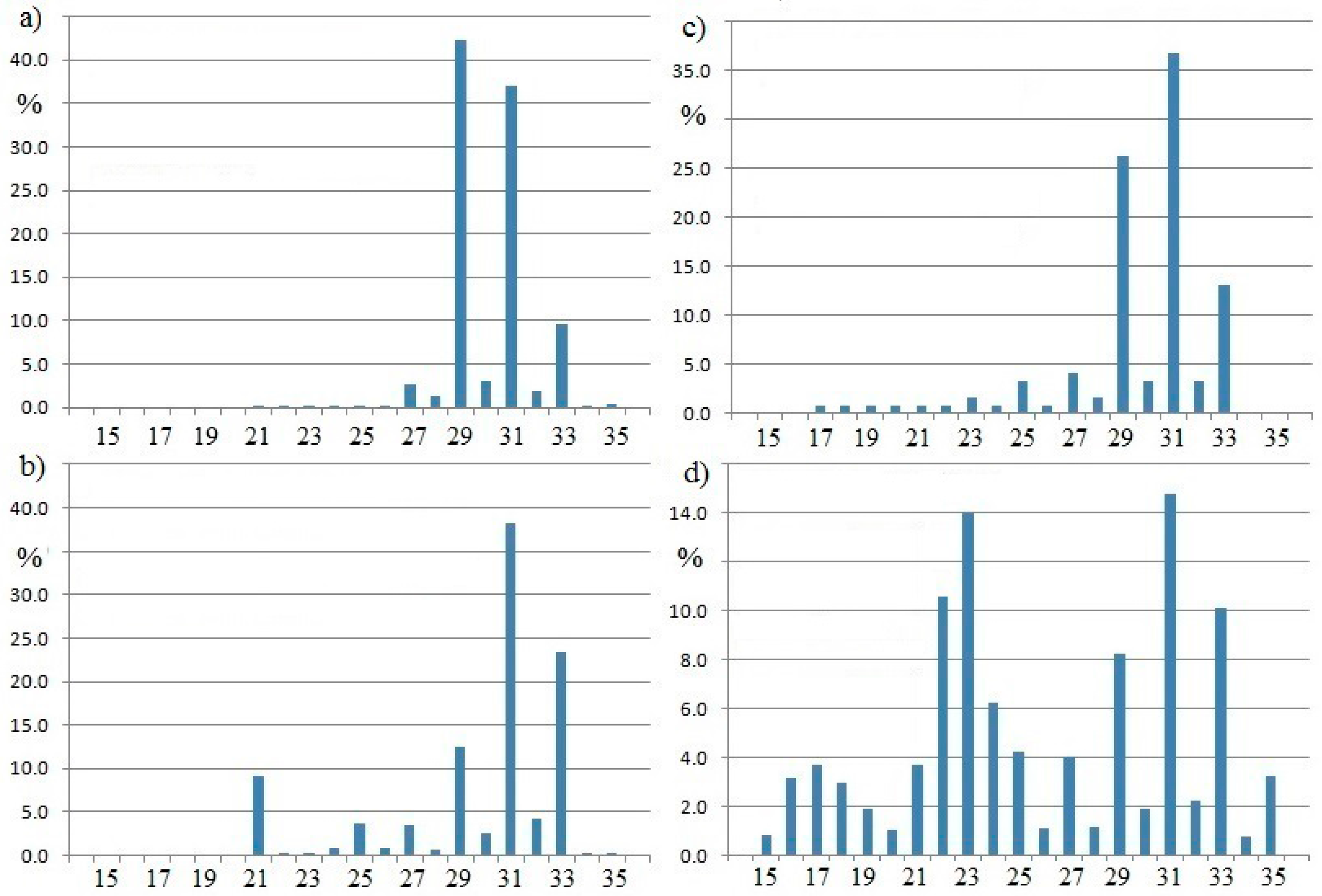
Histograms showing the distribution of n-alkanes in tree leaves and soils are shown in
Figure 1. The samples chosen for this illustration deliberately come from a wide geographic area and are from moist tropical forests. In all cases, the alkane distribution consists of the nC
27, nC
29, nC
31 alkanes to the virtual exclusion of lighter alkanes, especially those lighter than nC
22. The lighter alkanes (nC
12 to nC
19) are indicative of derivation from algae in marine or lacustrine environments. This summary includes recent studies that correspond with the established view of this subject [
24,
25,
26,
27].
3. The Sample Site
The rocks beneath the sample site consist of a layered gabbroic sequence within which there is a PGE-enriched horizon containing small, weathered PGM [
1,
2,
26]. The PGM are disseminated with rare minute Cu-sulfides. Pyrite and pentlandite are absent. Whole rock assays typically show the sulfur content below 0.04% with no known sulfide-rich layer. Weathering of these rocks does not produce a gossan. The rainfall is high (3000 to 5000 mm/year) [
27], most falling in July to September. A thin soil, normally about 10 cm in thickness, overlies saprolite that is derived from weathering of the layered rocks. The humic and fulvic acid contents of a sample from that soil have been examined previously [
12,
13]. A comparison of the geochemistry of the rocks, saprolite, and soil has indicated that Pt, Pd and Au are mobile within the saprolite and soils [
2]. The saprolite that covers the PGE-enriched horizon contains Pt, Pd, and Au. The saprolite retains details of the igneous layering and concentric weathering textures so it is in place and directly derived from the rocks that it covers. The saprolite that lies down slope of the PGE-enriched horizon also contains Pt, Pd and Au. This is not due to mechanical down slope movement, but it is the result of movement of the metals in solution. There is an additional weathering feature; the upper levels of the saprolite contain less Pt, Pd, and Au than the lower levels and this is likely to have been caused by leaching under the heavy rainfall. The whole area is covered by closed canopy humid tropical forest [
28]. The tree species mentioned in the older literature (
Khaya anthotheca,
Guared cedrata,
Mimusops heckelii,
Entandrophragma utile,
Entandrophragma macrophyllum,
Oldfieldia africana, and
Cordia platythrsa, [
29] are mainly the hardwoods, such as mahogany and teak of commercial interest. The forest covering the sample area now consists mainly of secondary forest consisting of smaller trees with waxy, drip-tip leaves that are designed to cope with the high rainfall.
The sample site is on a well drained hilltop rising to 290 m above present sea level. The hill is a ridge (roughly N-S) which slopes steeply dropping by about 200 m on either side. This topography has existed for a long time. There are Eocene wave cut platforms and hard pan deposits at the base of the ridge (up to 50 m), and there is a lateritized raised beach at a height of about 200 m above present sea level. The sample site is at about 250 m above sea level and some 50 m above the lateritized raised beach (
Figure 2). Field work is normally practical only outside of the rainy season, but at this time, the topography and freely draining saprolite mean that soil samples are dry. An attempt to sample ground water to determine the PGE contents and speciation was not successful.
4. Methods
The soil sample was sieved to produce −6 mm and −177 μm (−80 mesh) fractions. Both of the fractions were assayed for Pt, Pd, and Au by Genalysis (Australia) using fire assay with a Pb collector using ICP-MS (
Table 1). Assays of the reference material SARM 7 in the same run gave results that were close to the certified values.
The humic acids were separated following the established method [
30] and the dried humic acid analyzed by on-line pyrolysis gas chromatography-mass spectroscopy. The sample treatment and analysis have already been described [
12,
13].
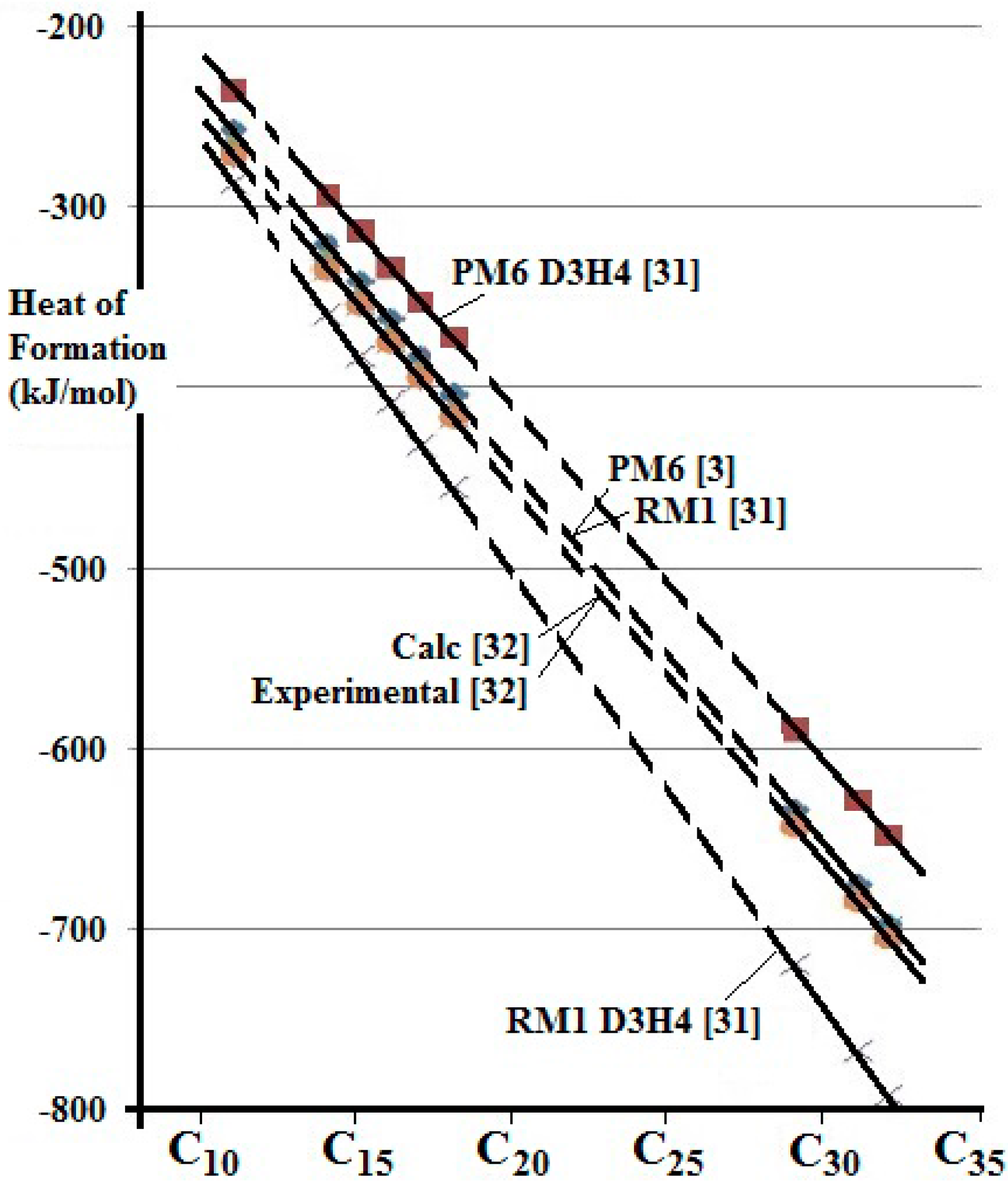
The thermodynamic properties of some straight chain alkanes and their possible reactions with Pt- bearing species were studied by computational chemistry using the AMPAC 10 program [
31]. This program offers several models corresponding to progressive development of the method. Two semi-empirical models (PM6 and RM1) from the AMPAC 10 program have been used here. Of these, PM6 is the most helpful for the present study because it can include calculations involving the PGE and Au, having been designed with biological molecules and catalysis in mind. There is a modification (D3H4) that is available to both of these models, which is intended improve the modelling of hydrogen bonding. Calculation of the Heat of Formation (Formation Enthalpy) of some n-alkanes from C
11 to C
32 indicates that the RM1 and PM6 models provide a good fit to values published previously [
32], as shown in
Figure 3. The D3H4 option does not offer an improvement in these examples. The Heat of Formation has previously been calculated [
32] using a density functional model and when compared with experimental values derived from the NIST Chemistry Web Book (
http://webbook.nist.gov/chemistry).
5. Results
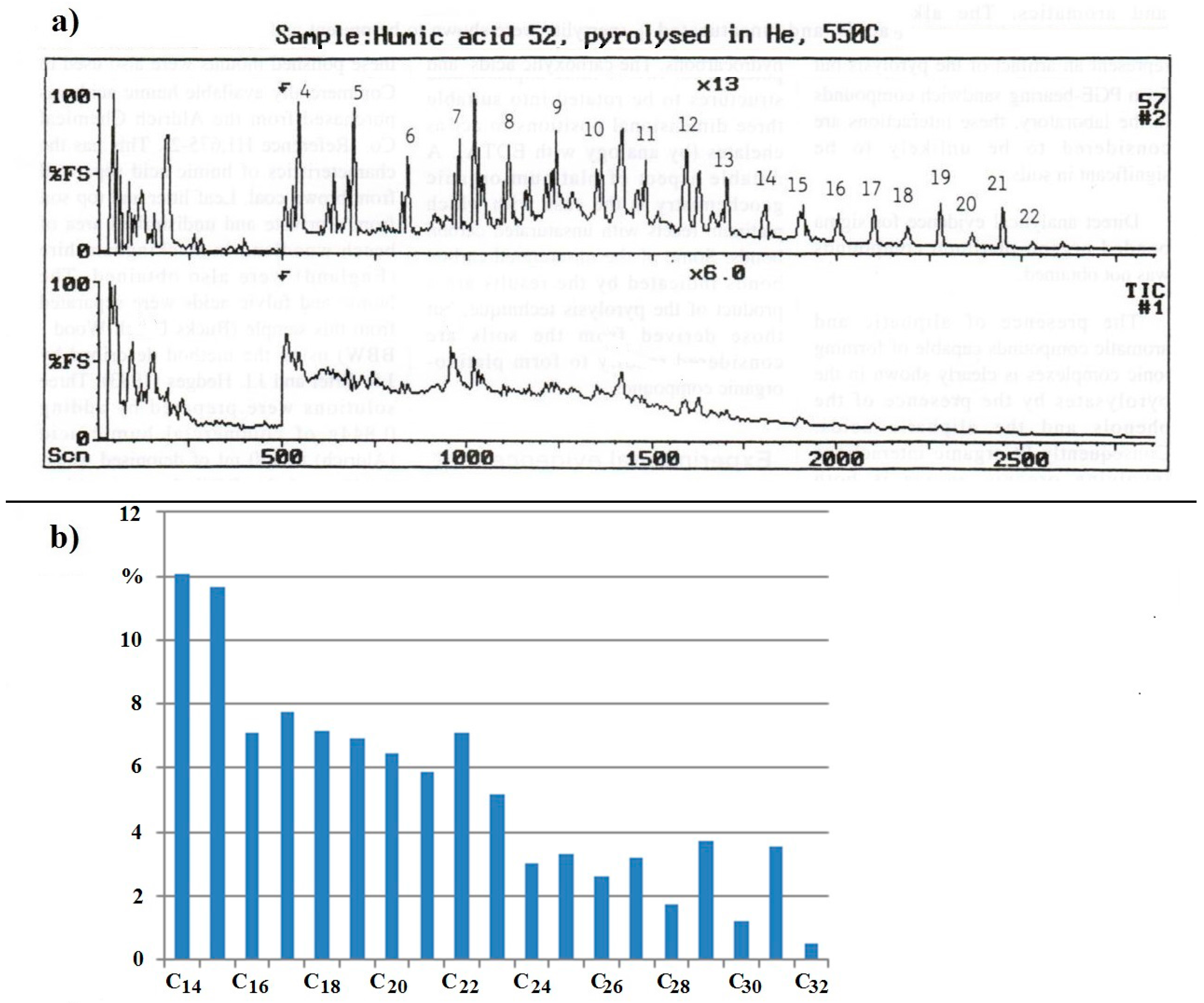
The Total Ion Chromatograph (TIC) trace for the humic acid from soil sample from the Freetown Peninsula, Sierra Leone, is reproduced as
Figure 4a. The alkanes from the central trace in
Figure 4a are represented as a histogram in
Figure 4b for ease of comparison with
Figure 1. nC
27, nC
29, and nC
31 are present in larger proportions than their neighbours (nC
28, nC
30, nC
32), as is to be expected from the data in
Figure 1. Surprisingly, the n-alkanes C
14 to C
24 are present in higher proportions than nC
27, nC
29, and nC
31, which is unusual. The high proportions of nC
14 and nC
15 are particularly noticeable given that their melting points are 5.9 and 9.9 °C, respectively. nC
16 and nC
17 with melting points of 18.2 and 21 °C are also present. As a measure for comparison with the n-alkanes in other broad leaved trees and tropical soils the ratio: average proportion of C
14–C
26/average proportion of C
27–C
35 can be calculated from earlier results [
20,
21,
22,
23]. In the leaves of broad leafed trees, this ratio is 0.006 to 0.092 and it is 0.07 to 0.68 in tropical soils. For the sample from the Freetown Intrusion, this ratio is 3.34.
6. Discussion
6.1. Are nC14–C25 Breakdown Products of nC27–C31?
Tree leaves and their humic products normally contain the n-alkanes C23–C31 and the range C27–C31 is usually dominant. The C14–C21 content of dry land vegetation is often negligible and C22–C28 spasmodic in occurrence, depending upon the type of vegetation, but they are always lower in proportion when compared with C27–C31. The soil from the Freetown Peninsula is anomalous in that it contains three times as much C14–C15 as C27–C31, and about twice as much C16–C22 when compared with C27–C31. The sample site on a 200 m high ridge makes it unlikely that the presence of the lighter alkanes can be due to lacustrine algae. Although there are the remnants of raised beaches in the area these are at a lower level and of significant age in an area of very heavy rainfall so that lighter alkanes due to marine algae should not now be present.
Could the presence of C
14 and C
15 be explained as a breakdown product of C
27–C
31 by reactions such as:
It would be more realistic to present these as oxidizing or reducing reactions:
Similarly, the presence of C
16 and above might be explained in terms of a breakdown into two unequal parts, for example:
For this we would assume that the lighter alkanes <C14 are sufficiently liquid to drain through the soil and that the alkanes C14–C17 are sufficiently viscous that they do not drain away or are being produced at a sufficient rate to compensate for any loss.
There is a huge problem with this suggestion. The alkanes, such as C
27–C
31, do not normally breakdown spontaneously because a large energy input is necessary to enable their destruction. Typically, this involves combustion. Calculation of the Heat of Formation of the components (
Table 2) illustrates this problem. The reactions above can be shown with the calculated Heat of Formation (in square brackets and in kJ/mol).
For these reactions, there is an energy gain provided by breakdown but it is small.
The oxidizing reactions with the addition of oxygen produce CO
2 and provide a heat gain. They are energetically favourable and the heat gain [−358 kJ/mol] is almost entirely due to the heat of formation of the CO
2. However, in practice, these reactions are only found to proceed if heat is added to trigger combustion. The reducing reactions are not energetically favourable and would require an additional +344 kJ/mol to proceed. The failure of these reactions to proceed without heat input explains why the non-oxidizing reactions with a smaller heat gain also fail to proceed. In these examples, different starting materials (C
29 or C
31) breakdown to equal or unequal components appears to make little difference. The oxidizing reactions above correspond only to partial combustion. Full combustion:
provides a very much greater heat gain.
The situation can be illustrated by the classic activation energy diagram (
Figure 5), in which reactions proceed from left to right. The oxidation reaction leading to production of CO
2 is energetically more favourable but the reaction does not progress due to the energy requirements of the transition state. Only with the addition of heat (by combustion for example) can the transition state be reached. However, in the presence of a catalyst, the energy requirement of the transition state is reduced and the reaction could proceed without additional heat input.
One distinguishing feature of the soil sample that is studied here is the Pt, Pd, and Au content. These elements are not likely to be present in significant amounts in most other soils, such as those illustrated in
Figure 1. Oxidized hydrous Pt species have a much lower heat of formation of than the un-oxidized species. For instance, [with Heat of Formation in kJ/mol and in square brackets]:
offers a gain of −452 kJ/mol. Thus, a linked Pt species-alkane reaction such:
has a much greater heat gain compared with the simpler alkane oxidation reactions described above. If such a reaction could occur, then it could provide the incentive for the breakdown of the alkane and overcome the energy of the transition state.
6.2. Can Catalysis Involving the PGE Facilitate Breakdown of the Heavier Alkanes?
Oxidation and ignition of four light alkanes in the presence of a Pt catalyst [
33] has shown that the oxidation temperatures are lowered to: C
1 540 °C, C
2 320 °C, C
3 250 °C, C
4 220 °C, whereas these alkanes normally oxidize above 1000 °C. The trend is expected continue so that the longer chain alkanes should oxidize at even lower temperatures. A straight line on a plot of temperature against C–H bond energy predicts a very low oxidation temperature for the longer chain alkanes [
33]. The data suggest that a curve would be more appropriate, resulting in a gentler change of oxidation temperature with alkane number. A curve drawn through the data points by eye or using a Lagrange polynomial extrapolation suggests that C
29, C
31 might oxidize at 195 °C, but such an extrapolation is highly speculative and ignores both the large error bars on the data and the phase change from gas to solid. Somewhere between the straight line interpretation and a curve it appears likely that longer chain alkanes, such as C
29 and C
31, could oxidize at room temperature in the presence of a Pt catalyst. Platinum was shown to be a more effective than Pd, Rh or Ir catalysts. At a slightly higher alkane/air mixture, the Pt catalyst oxidized C
2 at 265 °C, whilst oxidation temperatures of 350 °C (Pd), 410 °C (Rh), and 450 °C (Ir) were reported [
33].
There are reactions available both in Nature and are used as commercial processes that involve Pt (or the other PGE) as catalysts. Some of these reactions are summarized below. A small quantity of PGE acting as a catalyst is not consumed, so it can alter a very much larger quantity of humic acid.
6.3. Industrial Catalytic Reactions Used to Modify the Alkanes
Hydrogenation breaks a carbon-carbon double bond using a catalyst, such as Pt, Pd, or Ni in the presence of excess H
2. The organic molecule and the H
2 are adsorbed onto the catalyst surface and the H
2 dissociates, which allows the two hydrogen atoms to bond to different carbon atoms in the organic molecule, which is then released from the surface. Ni offers a slower reaction than Pt or Pd and requires a higher temperature. Industrial applications mainly involve the modification of the alkenes and there are variations that are adapted to create particular products, converting unsaturated compounds into saturated compounds (margarine, paraffin, napththene), generally with better storage properties [
34,
35].
Dehydrogenation removes hydrogen from an organic molecule and is used to convert alkanes into more reactive products. Industrial processes use Ag, Fe + Mo or vanadium oxide as a catalyst, but require higher temperatures (250–400 °C) to perform [
36].
Hydrogenolysis breaks a single bond, typically a carbon-carbon single bond but C–O, C–N or C–S bonds can also be broken. Industrially, it is used to remove sulphur, but in the laboratory it is used for organic synthesis [
37].
Alkane metathesis acts on a carbon-carbon single bonds and can be used both to generate a longer or a shorter alkane. A TaH (tantalum hydride) catalyst acts at 25–200°C and catalysts using either Pt in conjunction with tungsten oxide or Ir with Rh are used in industry. In all cases, the presence of SiO
2 appears to be essential. The industrial processes are usually designed to produce longer chain alkanes, modifying C
9H
20 to produce C
19H
40, for instance [
38,
39].
6.4. Possible Pt-Alkane Reactions in a Soil Environment
The physical situation envisaged in many of the industrial catalytic applications is a Pt foil with n-alkanes mobile in solution and the ends of the n-alkanes that are able to touch the Pt foil. In a humic-rich soil with the Pt mobile as a result of weathering, the Pt can be envisaged as free to move and able interact with an n-alkane at any point along its length.
The AMPAC 10 program [
31] has been used to construct the n-alkane molecules and calculate the Heats of Formation C
11 to C
32 listed in
Table 2. The C
31 molecule is shown in
Figure 6a and it has a Heat of Formation of −674.54 kJ/mol.
Allowing for a Pt atom to replace an H raises the Heat of Formation. If the Pt is added at the end (at the 31st C atom indicated here as 31C) the Heat of Formation becomes −347.80 kJ/mol (
Figure 6b). At the centre (16C) of the alkane (
Figure 6c), a Pt atom changes the Heat of Formation to −359.73 kJ/mol. In either case, there is little difference to the structure. If a Pt atom is added ¾ of the way along the alkane (24C) then the effect is to bend and twist the alkane (
Figure 6d) and the Heat of Formation becomes −378.61 kJ/mol.
A tetrahedrally coordinated Pt atom at the end of the alkane (31C) makes little difference to the structure (
Figure 6e, Heat of Formation −385.5 kJ/mol) but, in the centre of the alkane (16C), the alkane is both bent and twisted (
Figure 6f, Heat of Formation −394.52 kJ/mol).
PtOH placed at the end of the alkane also has little effect on the structure (
Figure 6g) with a similar Heat of Formation (−341.22 kJ/mol) to that given by Pt alone.
Platinum may occur in solution as Pt
2+(H
2O)
2(OH)
2 or Pt
4+(H
2O)
2(OH)
4. If Pt
2+(H
2O)
2(OH)
2 is placed at the ¾ position (24C,
Figure 6h) or at the centre (31C,
Figure 6i) of the C
31 alkane the result is a bending and twisting of the alkane and the Heats of Formation are −1277.85 and −1294.56 kJ/mol respectively. Pt
2+(H
2O)
2(OH)
4 at the centre (
Figure 6j) results in bending and twisting of the alkane and a significantly lower Heat of Formation (−1681.71 kJ/mol).
It seems that the addition of Pt species to the alkane can damage the structure, and that for Pt species, such as Pt2+(H2O)2(OH)2 or Pt4+(H2O)2(OH)4, the Heat of Formation is significantly lower. With such a low Heat of Formation, there is an increased likelihood of a reaction that is able to overcome the energy of the transition state and cause a breakdown of the n-alkane into smaller n-alkanes.
7. Conclusions
The soil above a PGE-bearing horizon within the Freetown Layered Intrusion contains high and anomalous concentrations of n-alkanes (CnH2n+2) that are in the range C14 to C22. The presence of these n-alkanes cannot reasonably be attributed to an algal or lacustrine origin. Longer chain n-alkanes (C23 to C31) in the soil are likely to have been derived from the breakdown of organic matter from leaf litter beneath the closed canopy humid tropical forest. These longer chain n-alkanes are typical of many other tropical to temperature soils.
Unassisted breakdown of the heavier n-alkanes to provide a source for the C14 to C22 alkanes is not likely. The alkanes are normally stable, and only breakdown when additional energy is added to the system, such as by ignition. Although there is an energy gain that is offered by breakdown, the stability of the heavier alkanes can be explained by the energy of a transition state, which is too high to be overcome without the input of additional energy.
In the Freetown soil, the catalytic properties of the PGE (and Pt in particular) may permit breakdown of the heavier alkanes by lowering the temperature at which oxidation can occur. Reaction between the heavier alkanes and Pt species, such as Pt
2+(H
2O)
2(OH)
2 and Pt
4+(H
2O)
2(OH)
4, can bend and twist the alkanes and significantly lower the Heat of Formation. Together, these effects render the breakdown of the heavier alkanes into smaller components more likely. It is possible, therefore, to offer an explanation for the presence of the C
14 to C
22 alkanes as a breakdown product of the natural, longer chain n-alkanes through the catalytic action of Pt, and provide an indication that Pt is present in solution in the soils, as predicted by the geochemical evidence [
2].
The objective of this paper is to offer a preliminary assessment of the potential effects of solid and aqueous PGE (specifically Pt) on humic acids. Our ab initio approach was stimulated by the difficulty of identifying potential sources of biogenic matter for the enhanced concentration of n-alkanes that are centred on nC
15. Although future detailed work might identify biogenic sources, the present study shows that the nC
15 enhancement can be formed abiogenically. The same argument could be applied to methyl alkanes and alkenes in the source leaf waxes. Fragmentation adjacent to weak bonds in long chain leaf waxes may occur, but we show that Pt
2+(H
2O)
2(OH)
2 and Pt
4+(H
2O)
2(OH)
4 can promote scission at the central carbon in nC
31. Other Pt species may occur in natural systems. Evidence for the conditions under which inorganic ligands, such as Pt(OH), Pt(OH)
2, and Pd(OH)
2, and organic siderophiles can occur have been provided by experimental studies [
40,
41,
42,
43,
44,
45]. The initial study reported here did not find Pt(OH) or Pt(OH)
2 to have a strong effect on the structure of nC
31 (e.g.,
Figure 6g). Pt
4+(H
2O)
2(OH)
4, appears to be more effective and was selected for the ab initio calculations on the basis of a low desorption energy, thermodynamic stability, and the ability to dissolve platinum nanoparticles [
46], a situation compatible with the platinum assays for the Freetown studies.
The calculated Heats of Formation assume a linear nC31 structure. Given the ease of C-C rotation in any alkane, it is possible that a linear nC31 molecule only exists in crystalline forms, such as leaf waxes, and not in randomly structured humic acids. Consequently, steric energy changes due to C-C rotation have not been included in our modelling. Our ab initio approach indicates abiogenic catalysis of long chain n-alkanes is energetically viable and fits the analytical data, which was our initial aim. Future work will need to include:
- (a)
the relative importance of the abiogenic, sterile processes presented in this paper compared with the non-sterile biogenic mechanisms;
- (b)
the relative efficiency of a range of oxide and hydroxide species, including especially Pt(OH), and Pt(OH)2;
- (c)
a comparison of Pt oxide and hydroxide species with those of the other PGE, especially Pd;
- (d)
an examination of the role of position along the alkane on the degree of distortion of the molecule;
- (e)
a comparison of the distortion of nC31 with nC29 and nC27; and,
- (f)
the effect of alkane structures other than simple linear chains.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}