Desiccator Volume: A Vital Yet Ignored Parameter in CaCO3 Crystallization by the Ammonium Carbonate Diffusion Method


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Carbonate Precipitation
2.3. Characterization
2.4. Carbonate Dissolution Quantification
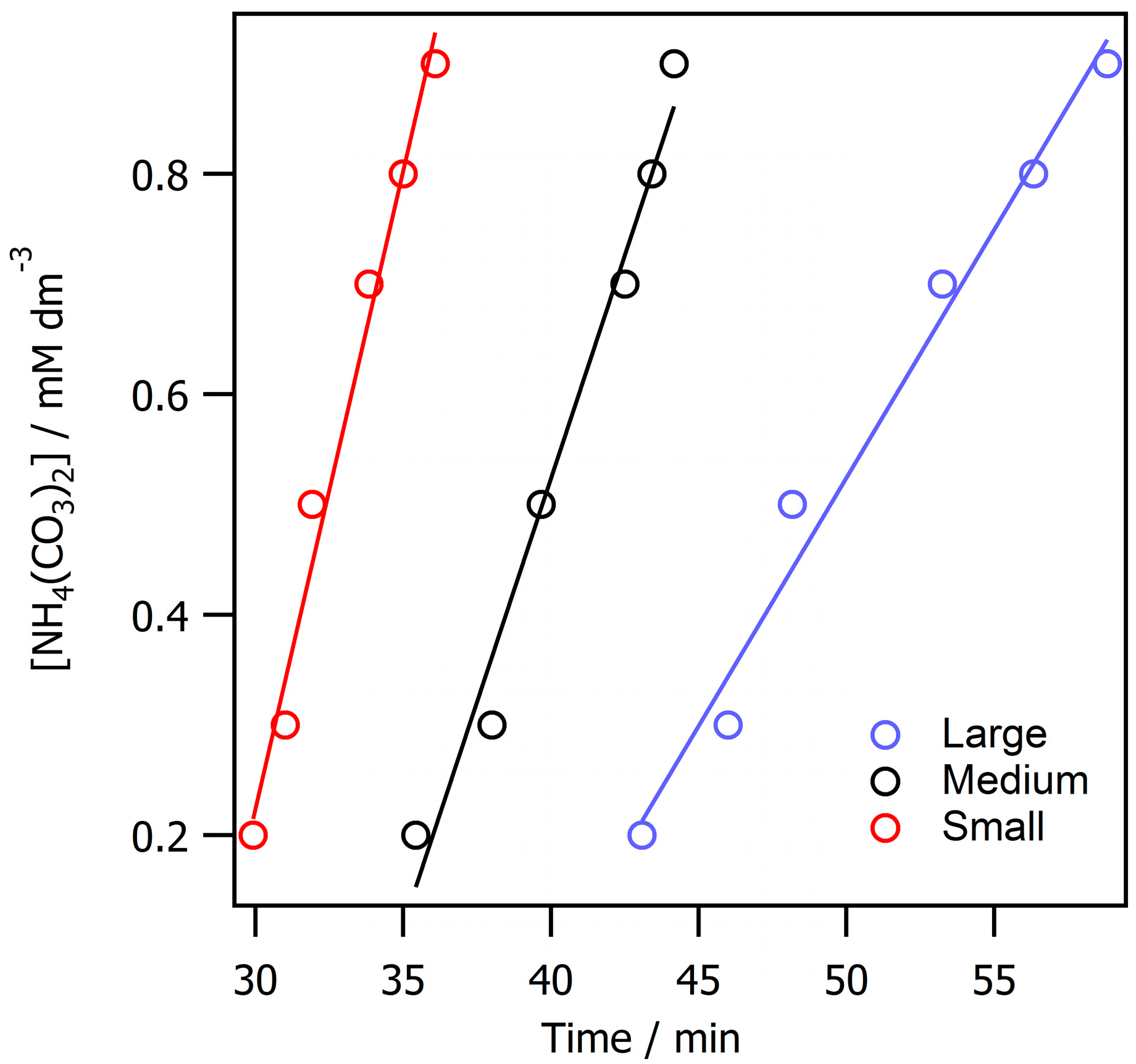
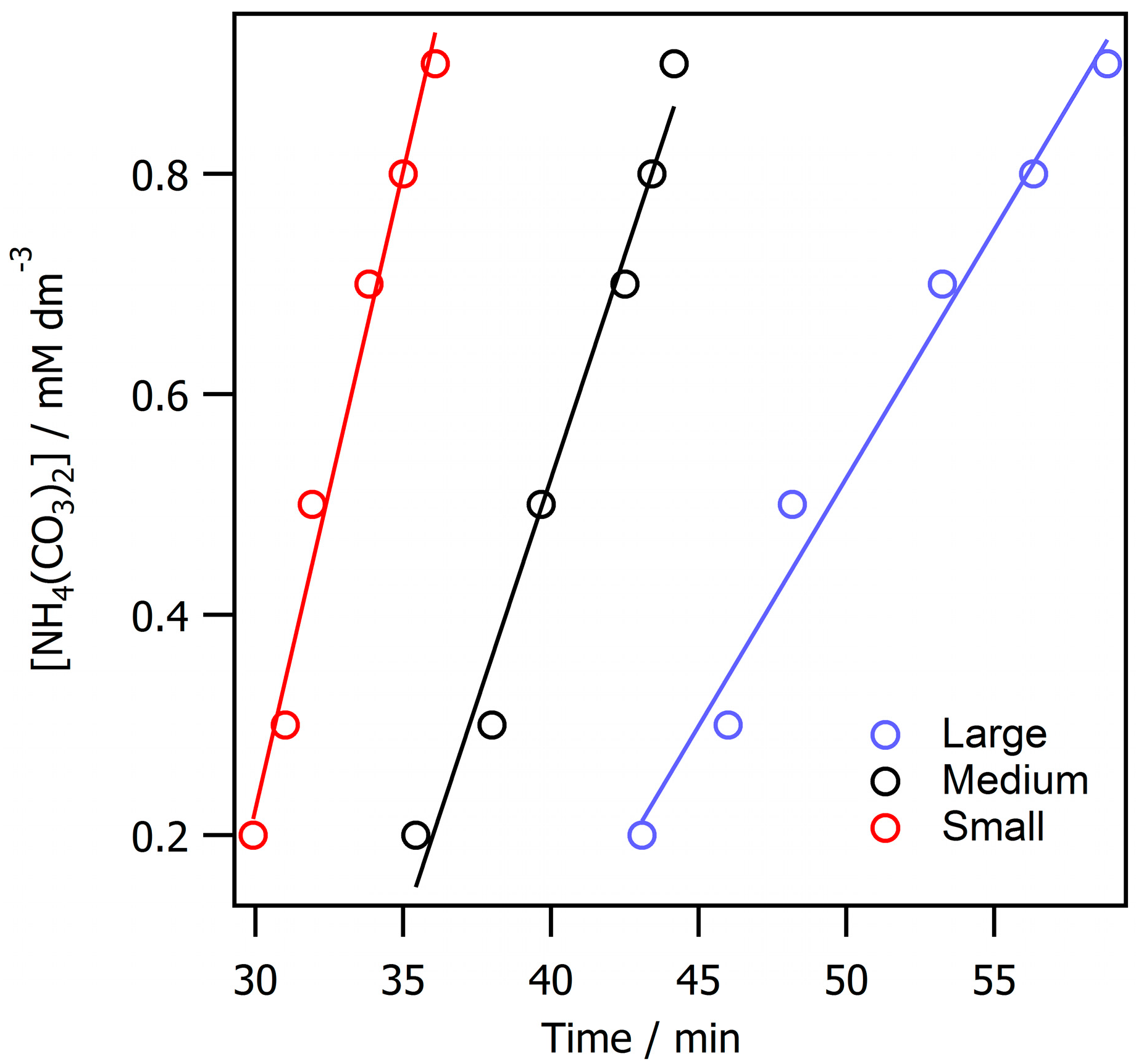
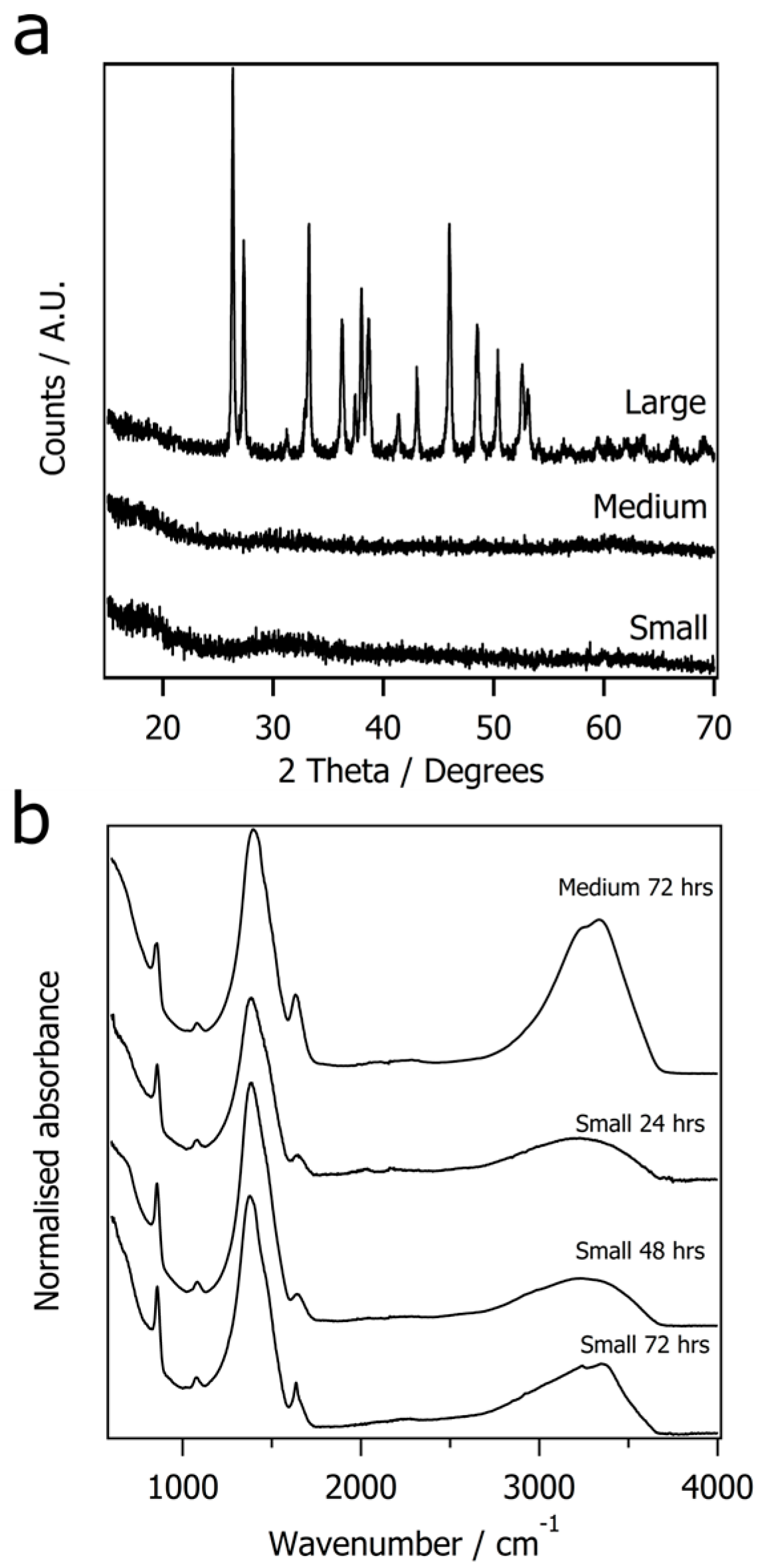
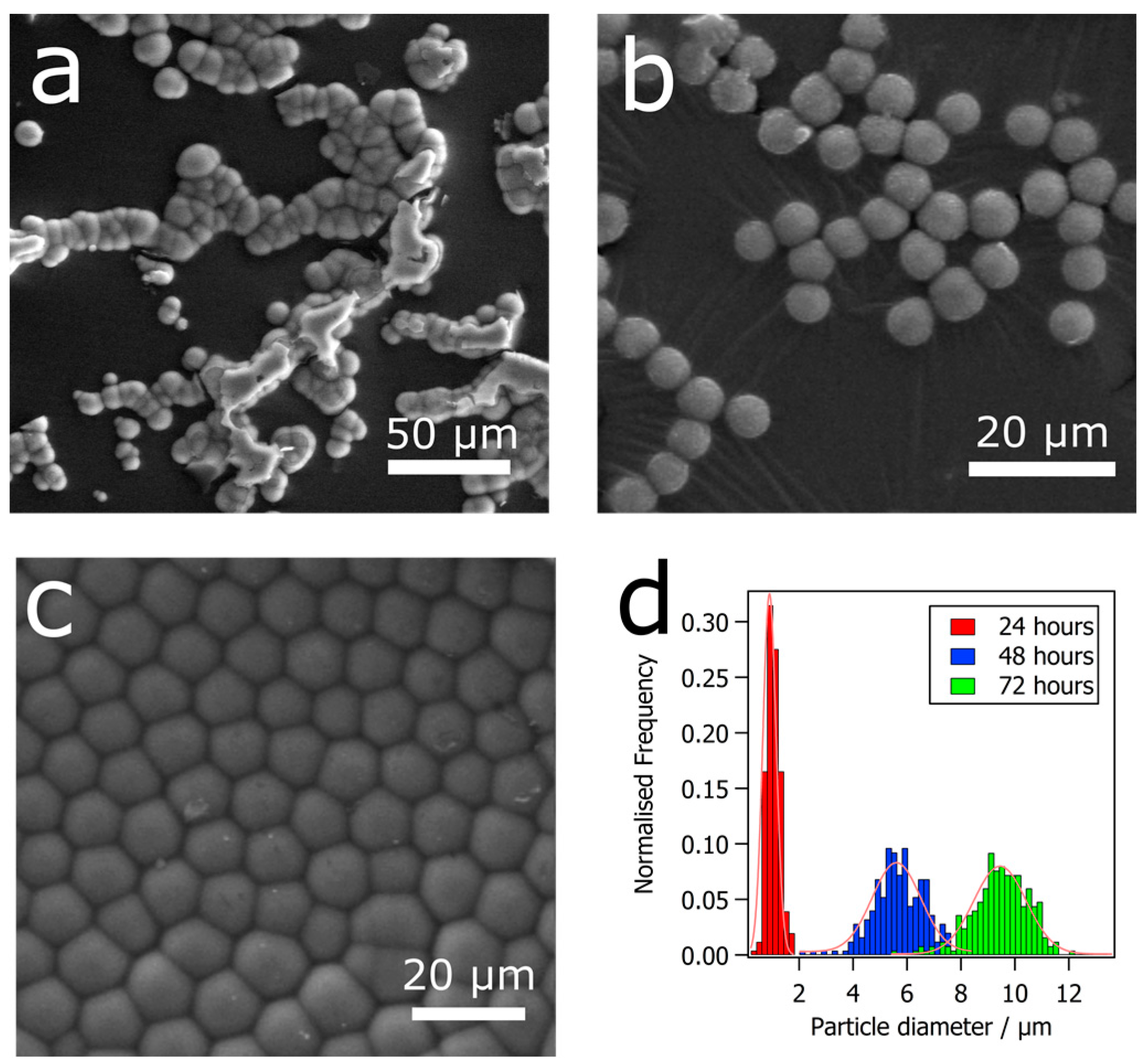
3. Results
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, Y.; Aichmayer, B.; Paris, O.; Fratzl, P.; Meibom, A.; Metzler, R.A.; Politi, Y.; Addadi, L.; Gilbert, P.U.P.A.; Weiner, S. The grinding tip of the sea urchin tooth exhibits exquisite control over calcite crystal orientation and Mg distribution. Proc. Natl. Acad. Sci. USA 2009, 106, 6048–6053. [Google Scholar] [CrossRef] [PubMed]
- Hovden, R.; Wolf, S.E.; Holtz, M.E.; Marin, F.; Muller, D.A.; Estroff, L.A. Nanoscale assembly processes revealed in the nacroprismatic transition zone of Pinna nobilis mollusc shells. Nat. Commun. 2015, 6, 10097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, J.C.; Milliron, G.W.; Miserez, A.; Evans-Lutterodt, K.; Herrera, S.; Gallana, I.; Mershon, W.J.; Swanson, B.; Zavattieri, P.; DiMasi, E.; et al. The stomatopod dactyl club: A formidable damage-tolerant biological hammer. Science 2012, 336, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Wagermaier, W.; Masic, A.; Kommareddy, K.P.; Bennet, M.; Manjubala, I.; Lee, S.-W.; Park, S.B.; Cölfen, H.; Fratzl, P. Self-assembly of amorphous calcium carbonate microlens arrays. Nat. Commun. 2012, 3, 725. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Mey, I.; Hajir, M.; Mondeshki, M.; Wolf, S.E. Pseudomorphic transformation of amorphous calcium carbonate films follows spherulitic growth mechanisms and can give rise to crystal lattice tilting. CrystEngComm 2015, 17, 6831–6837. [Google Scholar] [CrossRef]
- Marie, B.; Joubert, C.; Tayalé, A.; Zanella-Cléon, I.; Belliard, C.; Piquemal, D.; Cochennec-Laureau, N.; Marin, F.; Gueguen, Y.; Montagnani, C. Different secretory repertoires control the biomineralization processes of prism and nacre deposition of the pearl oyster shell. Proc. Natl. Acad. Sci. USA 2012, 109, 20986–20991. [Google Scholar] [CrossRef] [PubMed]
- Loste, E.; Wilson, R.M.; Seshadri, R.; Meldrum, F.C. The role of magnesium in stabilising amorphous calcium carbonate and controlling calcite morphologies. J. Cryst. Growth 2003, 254, 206–218. [Google Scholar] [CrossRef]
- Ihli, J.; Bots, P.; Kulak, A.; Benning, L.G.; Meldrum, F.C. Elucidating mechanisms of diffusion-based calcium carbonate synthesis leads to controlled mesocrystal formation. Adv. Funct. Mater. 2013, 23, 1965–1973. [Google Scholar] [CrossRef]
- Davis, K.J.; Dove, P.M.; De Yoreo, J.J. The role of Mg2+ as an impurity in calcite growth. Science 2000, 290, 1134–1138. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harris, J.; Wolf, S.E. Desiccator Volume: A Vital Yet Ignored Parameter in CaCO3 Crystallization by the Ammonium Carbonate Diffusion Method. Minerals 2017, 7, 122. https://doi.org/10.3390/min7070122
Harris J, Wolf SE. Desiccator Volume: A Vital Yet Ignored Parameter in CaCO3 Crystallization by the Ammonium Carbonate Diffusion Method. Minerals. 2017; 7(7):122. https://doi.org/10.3390/min7070122
Chicago/Turabian StyleHarris, Joe, and Stephan E. Wolf. 2017. "Desiccator Volume: A Vital Yet Ignored Parameter in CaCO3 Crystallization by the Ammonium Carbonate Diffusion Method" Minerals 7, no. 7: 122. https://doi.org/10.3390/min7070122
APA StyleHarris, J., & Wolf, S. E. (2017). Desiccator Volume: A Vital Yet Ignored Parameter in CaCO3 Crystallization by the Ammonium Carbonate Diffusion Method. Minerals, 7(7), 122. https://doi.org/10.3390/min7070122