
Reduction-Driven Mobilization of Structural Fe in Clay Minerals with High Fe Content

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Clay Mineral Preparation
2.2. Clay Mineral Reduction and Re-Oxidation
2.3. Sequential Extraction
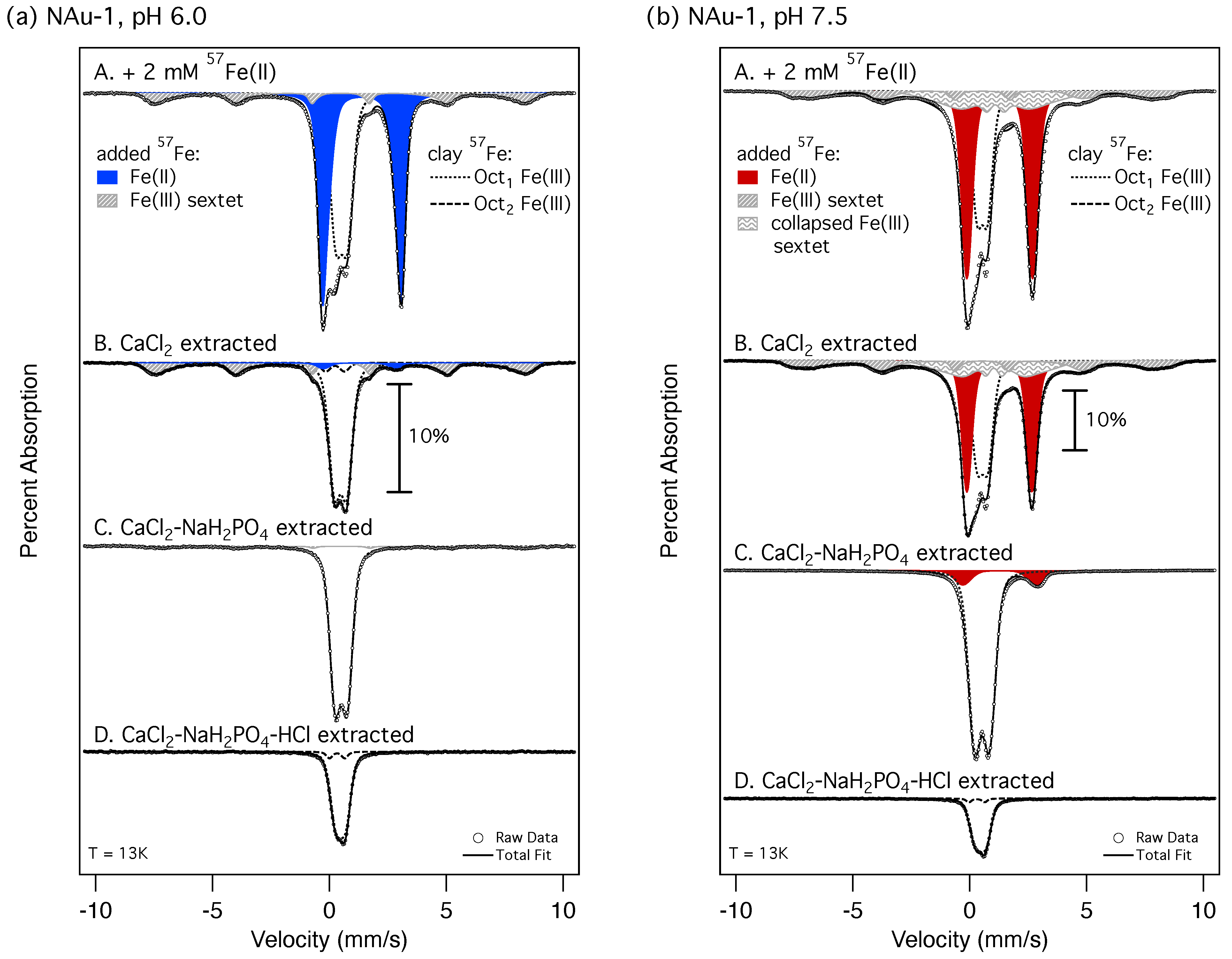
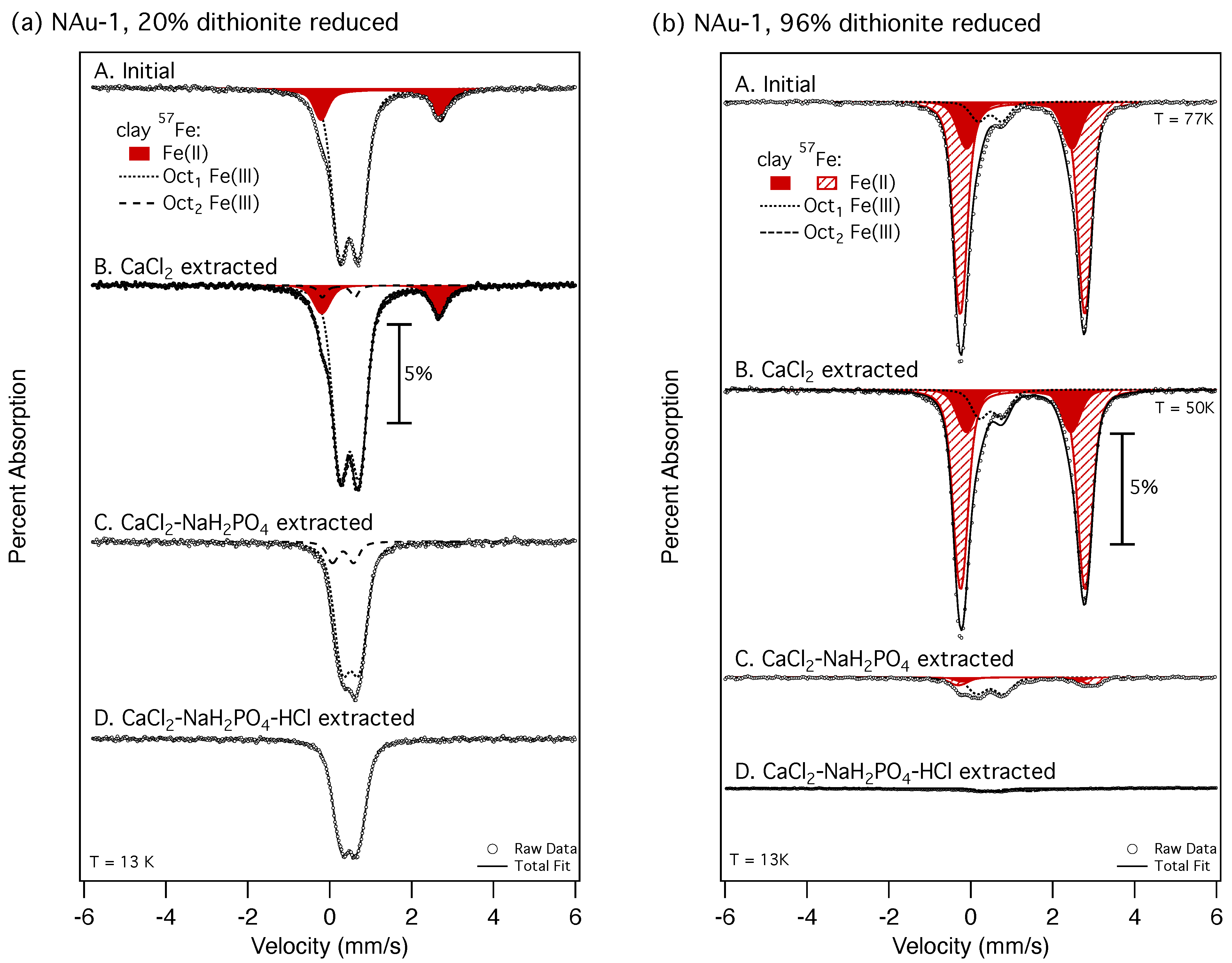
2.4. Mössbauer Spectroscopy
3. Results and Discussion
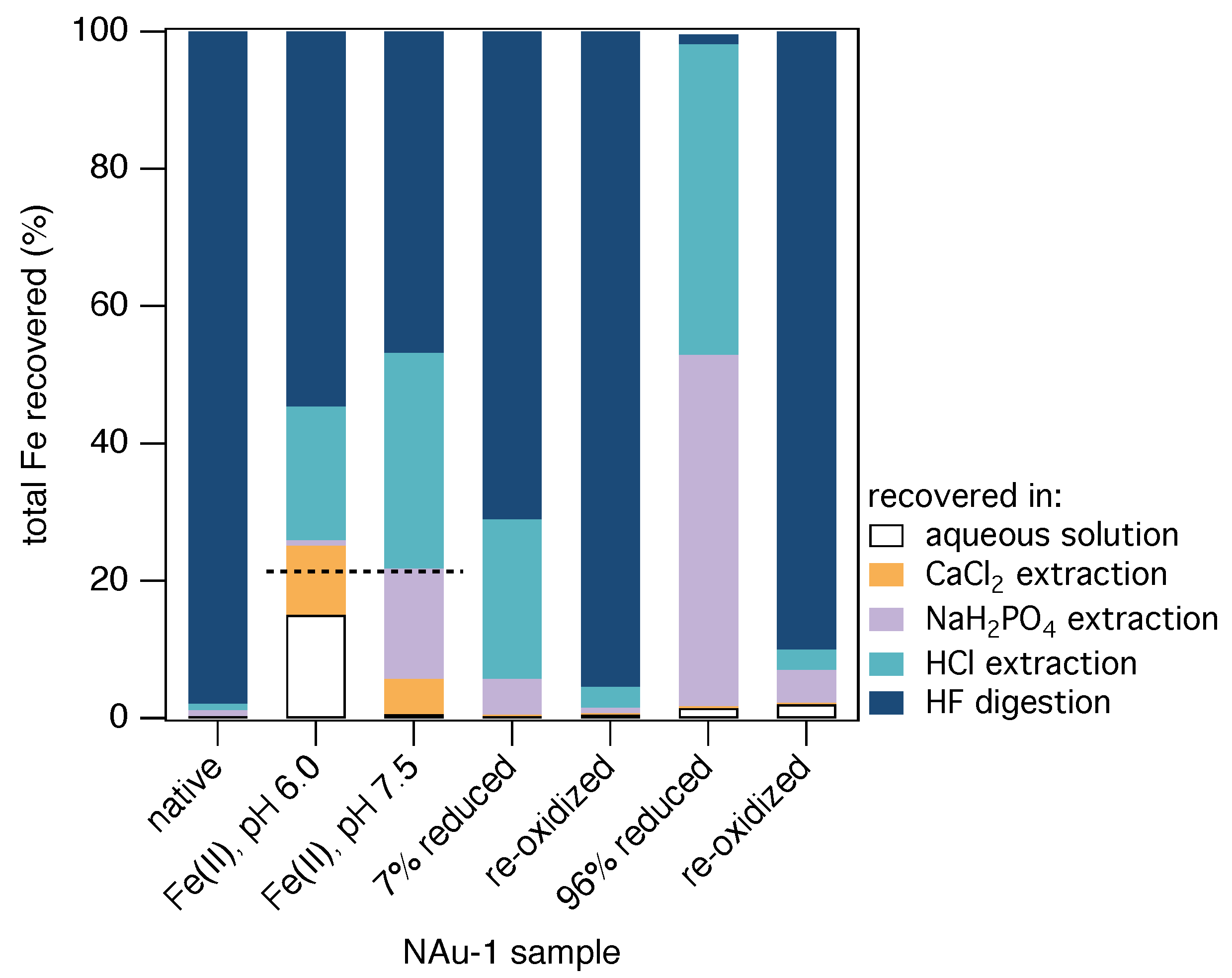
3.1. Quantification of Fe Pools in Fe(II)-Reacted Clay Minerals

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aqueous Fe(II) | Extracted Fe | Extracted Fe | HCl Extracted Fe | HF Extracted Fe | Total Fe | Fe(II) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NAu-1 Sample | Initial | of Total a | Final | of Recovered b | Sorbed c | Fe(II)/ Fe(tot) | Fe(tot) | of Recovered b | Fe(II)/ Fe(tot) | Fe(tot) | of Recovered b | Fe(II)/ Fe(tot) | Fe(tot) | of Recovered b | Fe(II)/ Fe(tot) | Fe(tot) | of Recovered b | Intial d | Recov- ered | of Initial | of initial |
| μmol | % | μmol | % | μmol | % | μmol | % | % | μmol | % | % | μmol | % | % | μmol | % | μmol | μmol | % | % | |
| untreated | |||||||||||||||||||||
| NAu-1 | – | – | 0.04(0.02) | <1 | – | 100 | 0.05(0.02) | <1 | 15 | 0.61(0.04) | 1 | 11 | 0.55(0.16) | 1 | 2 | 58.1(6.4) | 98 | 129.4(0.6) | 59.4(6.6) | 46 | – |
| NAu-1 e,f | – | – | – | – | – | 100 | 0.01(0.01) | <1 | 5 | 0.77(0.14) | 1 | 29 | 0.14(0.07) | <1 | 112.3(5.7) | – | |||||
| Fe(II)-reacted, pH 6.0 | |||||||||||||||||||||
| NAu-1 | 32.3(0.2) | 21 | 17.5(0.3) | 15 | 14.8(0.5) | 96 | 11.8(0.2) | 10 | 14 | 0.90(0.01) | 1 | 7 | 22.7(0.3) | 19 | 7 | 63.8(4.2) | 55 | 152.0(0.7) | 116.7(4.9) | 77 | 109 |
| NAu-1 e | 29.9(3.1) | 20 | 20.5(0.4) | 21 | 9.61(3.50) | 100 | 7.40(0.14) | 8 | 24 | 8.26(0.08) | 8 | 3 | 20.7(0.2) | 21 | 1 | 42.3 | 43 | 148.4(1.3) | 99.2(0.7) | 67 | 106 |
| Fe(II)-reacted, pH 7.5 | |||||||||||||||||||||
| NAu-1 | 32.5(0.5) | 21 | 0.65(0.00) | 1 | 31.9(0.5) | 93 | 5.42(0.09) | 5 | 87 | 17.2(0.5) | 16 | 13 | 33.5(0.8) | 31 | 7 | 50.0(7.6) | 47 | 152.5(0.7) | 106.7(9.0) | 70 | 88 |
| NAu-1 e | 27.9(0.1) | 19 | 2.48(0.06) | 2 | 25.5(0.2) | 100 | 2.30(0.11) | 2 | 96 | 18.6(0.5) | 16 | 9 | 27.2(1.6) | 24 | 1 | 64.1 | 56 | 147.7(0.1) | 114.5(2.3) | 78 | 88 |
| NAu-1 g | 32.6(0.8) | 21 | 0.88(0.01) | 1 | 31.7(0.4) | 100 | 1.28(0.04) | 1 | 96 | 29.3(0.2) | 22 | 11 | 30.8(0.3) | 23 | 1 | 69.9 | 53 | 155.0(0.8) | 132.1(0.6) | 85 | 101 |
| partly dithionite-reduced h | |||||||||||||||||||||
| NAu-1 | – | – | 0.02(0.02) | <1 | – | 83 | 0.12(0.08) | <1 | 48 | 4.15(2.42) | 5 | 9 | 18.1(0.5) | 23 | 1 | 55.5(2.6) | 71 | 103.9(1.3) | 77.9(5.6) | 75 | 60 |
| NAu-1 e | – | – | 0.00(0.00) | 0 | – | 93 | 0.28(0.01) | 2 | 79 | 9.74(0.56) | 59 | 19 | 4.32(0.15) | 26 | 4 | 2.24 | 14 | 72.7(0.3) | 16.59(0.73) | 23 | 65 |
| partly dithionite-reduced, re-oxidized i | |||||||||||||||||||||
| NAu-1 | – | – | 0.01(0.01) | <1 | – | 100 | 0.03(0.03) | <1 | 7 | 0.61(0.49) | 1 | 4 | 1.79(0.74) | 3 | 1 | 57.7(0.8) | 96 | 102.0(7.2) | 60.1(2.1) | 59 | – |
| completely dithionite-reduced j | |||||||||||||||||||||
| NAu-1 | – | – | 3.32(1.27) | 1 | – | 100 | 1.27(0.03) | <1 | 100 | 124.8(4.3) | 51 | 95 | 109.7(16.3) | 45 | 7 | 3.55(0.46) | 1 | 238.2(2.4) | 242.6(22.4) | 102 | 104 |
| NAu-1 e | – | – | 0.28(0.05) | 0 | – | 100 | 0.69(0.02) | 0 | 100 | 123.7(0.5) | 87 | 72 | 14.9(4.3) | 10 | 4 | 2.66 | 2 | 158.8 | 142.2(4.9) | 90 | 92 |
| completely dithionite-reduced, re-oxidized k | |||||||||||||||||||||
| NAu-1 j | – | – | 0.02(0.00) | 2 | – | – | n.d. l | 0 | <1 | 2.51(0.02) | 6 | 3 | 1.40(0.16) | 3 | 1 | 36.62 | 88 | 67.6(0.4) | 41.45(0.55) | 61 | – |
3.2. Source of Extractable Fe Pools in Fe(II)-Reduced Clay Minerals
3.3. Iron Mobilization in Dithionite-Reduced Clay Minerals
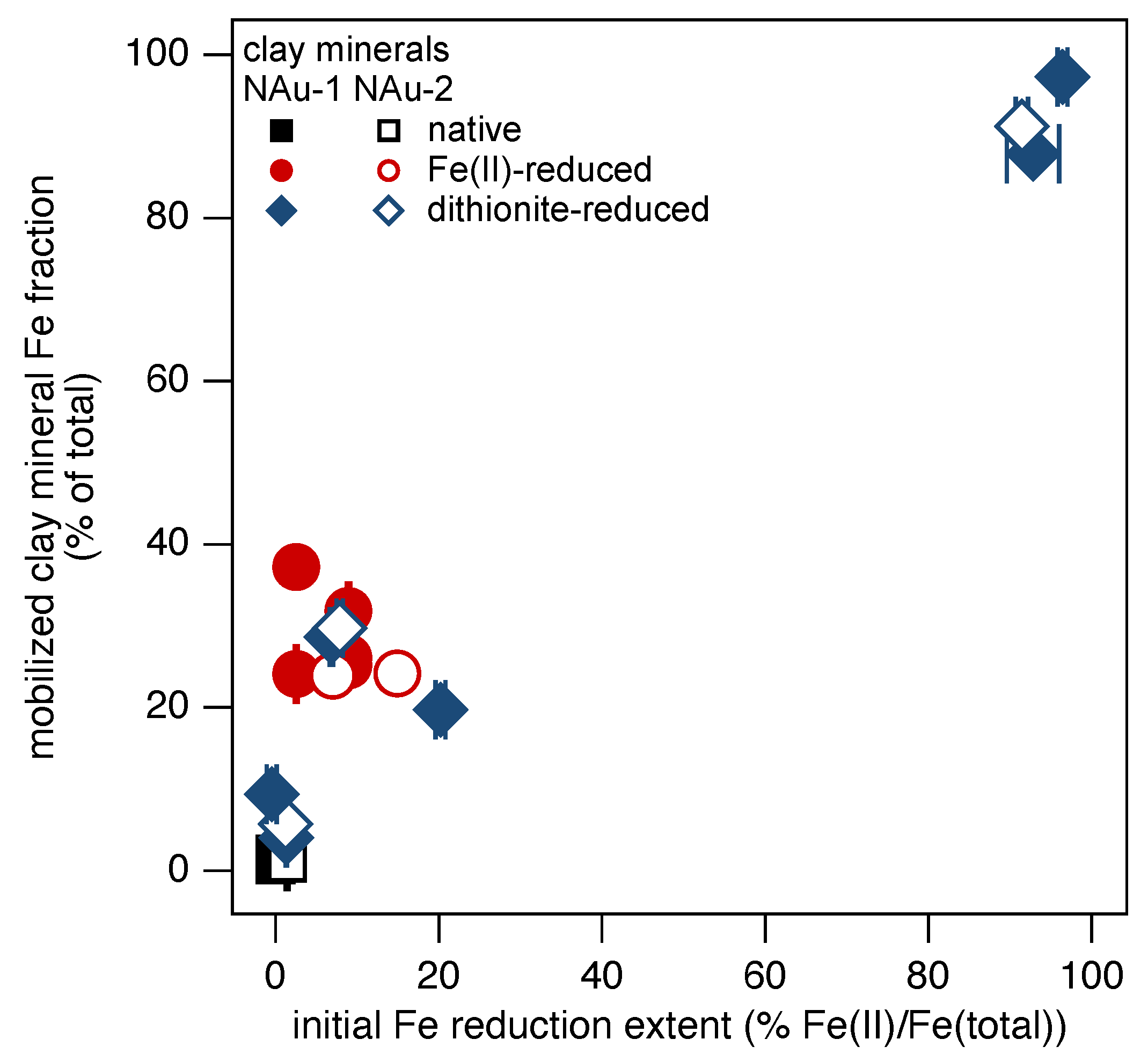
3.4. Reversibility of Iron Mobilization in Clay Minerals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amonette, J.E. Iron redox chemistry of clays and oxides: Environmental applications. In Electrochemical Properties of Clays; CMS Workshop Lectures; Fitch, A., Ed.; The Clay Minerals Society: Aurora, CO, USA, 2002; Volume 10, pp. 89–146. [Google Scholar]
- Murad, E.; Fischer, W.R. The Geobiochemical Cycle of Iron. In Iron in Soils and Clay Minerals; Stucki, J.W., Goodman, B.A., Schwertmann, U., Eds.; Springer: Dordrecht, The Netherlands, 1988; pp. 1–18. [Google Scholar] [CrossRef]
- Stucki, J.W. Structural Iron in Smectites. In Iron in Soils and Clay Minerals; Stucki, J.W., Goodman, B.A., Schwertmann, U., Eds.; Springer: Dordrecht, The Netherlands, 1988; pp. 625–675. [Google Scholar] [CrossRef]
- Stucki, J.W. Properties and Behavior of Iron in Clay Minerals. In Handbook of Clay Science; Bergaya, F., Theng, B., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 423–477. [Google Scholar]
- Stucki, J.W.; Komadel, P.; Wilkinson, H.T. Microbial reduction of structural Iron(III) in smectites. Soil Sci. Soc. Am. J. 1987, 51, 1663–1665. [Google Scholar] [CrossRef]
- Dong, H.; Jaisi, D.P.; Kim, J.; Zhang, G. Microbe-clay mineral interactions. Am. Mineral. 2009, 94, 1505–1519. [Google Scholar] [CrossRef]
- Kostka, J.E.; Dalton, D.D.; Skelton, H.; Dollhopf, S.; Stucki, J.W. Growth of iron(III)-reducing bacteria on clay minerals as the sole electron acceptor and comparison of growth yields on a variety of oxidized iron forms. Appl. Environ. Microbiol. 2002, 68, 6256–6262. [Google Scholar] [CrossRef]
- Kukkadapu, R.K.; Zachara, J.M.; Fredrickson, J.K.; Kennedy, D.W. Biotransformation of two-line silica-ferrihydrite by a dissimilatory Fe(III)-reducing bacterium: Formation of carbonate green rust in the presence of phosphate. Geochim. Cosmochim. Acta 2004, 68, 2799–2814. [Google Scholar] [CrossRef]
- Neumann, A.; Hofstetter, T.B.; Skarpeli-Liati, M.; Schwarzenbach, R.P. Reduction of Polychlorinated Ethanes and Carbon Tetrachloride by Structural Fe(II) in Smectites. Environ. Sci. Technol. 2009, 43, 4082–4089. [Google Scholar] [CrossRef]
- Hofstetter, T.B.; Neumann, A.; Schwarzenbach, R.P. Reduction of nitroaromatic compounds by Fe(II) species associated with iron-rich smectites. Environ. Sci. Technol. 2006, 40, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.; Hofstetter, T.B.; Lussi, M.; Cirpka, O.A.; Petit, S.; Schwarzenbach, R.P. Assessing the redox reactivity of structural iron in smectites using nitroaromatic compounds as kinetic probes. Environ. Sci. Technol. 2008, 42, 8381–8387. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Boyanov, M.I.; Kemner, K.M.; Thalhammer, K.O. Reduction of Hg(II) by Fe(II)-Bearing Smectite Clay Minerals. Minerals 2020, 10, 1079. [Google Scholar] [CrossRef]
- Brigatti, M.F.; Franchini, G.; Lugli, C.; Medici, L.; Poppi, L.; Turci, E. Interaction between aqueous chromium solutions and layer silicates. Appl. Geochem. 2000, 15, 1307–1316. [Google Scholar] [CrossRef]
- Peretyazhko, T.; Zachara, J.M.; Heald, S.M.; Jeon, B.H.; Kukkadapu, R.K.; Liu, C.; Moore, D.; Resch, C.T. Heterogeneous reduction of Tc(VII) by Fe(II) at the solid-water interface. Geochim. Cosmochim. Acta 2008, 72, 1521–1539. [Google Scholar] [CrossRef]
- Qian, Y.; Scheinost, A.C.; Grangeon, S.; Hoving, A.; Churakov, S.V.; Marques Fernandes, M. Influence of structural Fe content in clay minerals on selenite redox reactions: Kinetics and structural transformations. Geochim. Cosmochim. Acta 2024, 377, 19–33. [Google Scholar] [CrossRef]
- Ernstsen, V. Reduction of nitrate by Fe2+ in clay minerals. Clays Clay Miner. 1996, 44, 599–608. [Google Scholar] [CrossRef]
- Zhao, L.; Dong, H.; Kukkadapu, R.K.; Zeng, Q.; Edelmann, R.E.; Pentrák, M.; Agrawal, A. Biological Redox Cycling of Iron in Nontronite and Its Potential Application in Nitrate Removal. Environ. Sci. Technol. 2015, 49, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Hadi, J.; Greneche, J.M.; Wersin, P.; Koho, P.; Pastina, B. Determination of Sulfide Consumption by Fe-bearing Components of Bentonites. Clays Clay Miner. 2023, 71, 577–599. [Google Scholar] [CrossRef]
- Stucki, J.W. A review of the effects of iron redox cycles on smectite properties. Comptes Rendus Geosci. 2011, 343, 199–209. [Google Scholar] [CrossRef]
- Ernstsen, V.; Gates, W.P.; Stucki, J.W. Microbial reduction of structural iron in clays—A renewable source of reduction capacity. J. Environ. Qual. 1998, 27, 761–766. [Google Scholar] [CrossRef]
- Peiffer, S.; Kappler, A.; Haderlein, S.B.; Schmidt, C.; Byrne, J.M.; Kleindienst, S.; Vogt, C.; Richnow, H.H.; Obst, M.; Angenent, L.T.; et al. A biogeochemical–hydrological framework for the role of redox-active compounds in aquatic systems. Nat. Geosci. 2021, 14, 264–272. [Google Scholar] [CrossRef]
- Lee, K.; Kostka, J.E.; Stucki, J.W. Comparisons of structural Fe reduction in smectites by bacteria and dithionite: An infrared spectroscopic study. Clays Clay Miner. 2006, 54, 195–208. [Google Scholar] [CrossRef]
- Ribeiro, F.R.; Fabris, J.D.; Kostka, J.E.; Komadel, P.; Stucki, J.W. Comparisons of structural iron reduction in smectites by bacteria and dithionite: II. A variable-temperature Mossbauer spectroscopic study of Garfield nontronite. Pure Appl. Chem. 2009, 81, 1499–1509. [Google Scholar] [CrossRef]
- Fialips, C.I.; Huo, D.; Yan, L.B.; Wu, J.; Stucki, J.W. Infrared study of reduced and reduced-reoxidized ferruginous smectite. Clays Clay Miner. 2002, 50, 455–469. [Google Scholar] [CrossRef]
- Fialips, C.I.; Huo, D.F.; Yan, L.B.; Wu, J.; Stucki, J.W. Effect of Fe oxidation state on the IR spectra of Garfield nontronite. Am. Mineral. 2002, 87, 630–641. [Google Scholar] [CrossRef]
- Neumann, A.; Petit, S.; Hofstetter, T.B. Evaluation of redox-active iron sites in smectites using middle and near infrared spectroscopy. Geochim. Cosmochim. Acta 2011, 75, 2336–2355. [Google Scholar] [CrossRef]
- Gorski, C.A.; Klüpfel, L.E.; Voegelin, A.; Sander, M.; Hofstetter, T.B. Redox Properties of Structural Fe in Clay Minerals: 3. Relationships between Smectite Redox and Structural Properties. Environ. Sci. Technol. 2013, 47, 13477–13485. [Google Scholar] [CrossRef]
- Komadel, P.; Madejova, J.; Stucki, J.W. Reduction and Reoxidation of Nontronite-Questions of Reversibility. Clays Clay Miner. 1995, 43, 105–110. [Google Scholar] [CrossRef]
- Rothwell, K.A.; Pentrak, M.P.; Pentrak, L.A.; Stucki, J.W.; Neumann, A. Reduction Pathway-Dependent Formation of Reactive Fe(II) Sites in Clay Minerals. Environ. Sci. Technol. 2023, 57, 10231–10241. [Google Scholar] [CrossRef]
- Kostka, J.E.; Haefele, E.; Viehweger, R.; Stucki, J. Respiration and dissolution of iron(III)-containing clay minerals by bacteria. Environ. Sci. Technol. 1999, 33, 3127–3133. [Google Scholar] [CrossRef]
- Dong, H.L.; Kostka, J.E.; Kim, J. Microscopic evidence for microbial dissolution of smectite. Clays Clay Miner. 2003, 51, 502–512. [Google Scholar] [CrossRef]
- Kim, J.; Dong, H.L.; Seabaugh, J.; Newell, S.W.; Eberl, D.D. Role of microbes in the smectite-to-illite reaction. Science 2004, 303, 830–832. [Google Scholar] [CrossRef]
- Jaisi, D.P.; Kukkadapu, R.K.; Eberl, D.D.; Dong, H.L. Control of Fe(III) site occupancy on the rate and extent of microbial reduction of Fe(III) in nontronite. Geochim. Cosmochim. Acta 2005, 69, 5429–5440. [Google Scholar] [CrossRef]
- Jaisi, D.P.; Dong, H.L.; Morton, J.P. Partitioning of Fe(II) in reduced nontronite (NAu-2) to reactive sites: Reactivity in terms of Tc(VII) reduction. Clays Clay Miner. 2008, 56, 175–189. [Google Scholar] [CrossRef]
- Yang, J.; Kukkadapu, R.K.; Dong, H.; Shelobolina, E.S.; Zhang, J.; Kim, J. Effects of redox cycling of iron in nontronite on reduction of technetium. Chem. Geol. 2012, 291, 206–216. [Google Scholar] [CrossRef]
- Bishop, M.E.; Dong, H.; Glasser, P.; Briggs, B.R.; Pentrak, M.; Stucki, J.W. Microbially mediated iron redox cycling of subsurface sediments from Hanford Site, Washington State, USA. Chem. Geol. 2020, 546, 119643. [Google Scholar] [CrossRef]
- Zhang, G.; Kim, J.; Dong, H.; Sommer, A.J. Microbial effects in promoting the smectite to illite reaction: Role of organic matter intercalated in the interlayer. Am. Mineral. 2007, 92, 1401–1410. [Google Scholar] [CrossRef]
- Stucki, J.W.; Golden, D.C.; Roth, C.B. Effects of reduction and reoxidation of structural iron on the surface charge and dissolution of dioctahedral smectites. Clays Clay Miner. 1984, 32, 350–356. [Google Scholar] [CrossRef]
- Stucki, J.W.; Golden, D.C.; Roth, C.B. Preparation and handling of dithionite-reduced smectite suspensions. Clays Clay Miner. 1984, 32, 191–197. [Google Scholar] [CrossRef]
- Hepburn, L.E.; Butler, I.B.; Boyce, A.; Schröder, C. The use of operationally-defined sequential Fe extraction methods for mineralogical applications: A cautionary tale from Mössbauer spectroscopy. Chem. Geol. 2020, 543, 119584. [Google Scholar] [CrossRef]
- Qian, Y.; Scheinost, A.C.; Grangeon, S.; Greneche, J.M.; Hoving, A.; Bourhis, E.; Maubec, N.; Churakov, S.V.; Fernandes, M.M. Oxidation State and Structure of Fe in Nontronite: From Oxidizing to Reducing Conditions. ACS Earth Space Chem. 2023, 7, 1868–1881. [Google Scholar] [CrossRef]
- Schaefer, M.V.; Gorski, C.A.; Scherer, M.M. Spectroscopic Evidence for Interfacial Fe(II)-Fe(III) Electron Transfer in a Clay Mineral. Environ. Sci. Technol. 2011, 45, 540–545. [Google Scholar] [CrossRef]
- Neumann, A.; Olson, T.L.; Scherer, M.M. Spectroscopic evidence for Fe(II)-Fe(III) electron transfer at clay mineral edge and basal sites. Environ. Sci. Technol. 2013, 47, 6969–6977. [Google Scholar] [CrossRef]
- Latta, D.E.; Neumann, A.; Premaratne, W.A.P.J.; Scherer, M.M. Fe(II)-Fe(III) Electron Transfer in a Clay Mineral with Low Fe Content. ACS Earth Space Chem. 2017, 1, 197–208. [Google Scholar] [CrossRef]
- Entwistle, J.; Latta, D.E.; Scherer, M.M.; Neumann, A. Abiotic Degradation of Chlorinated Solvents by Clay Minerals and Fe(II): Evidence for Reactive Mineral Intermediates. Environ. Sci. Technol. 2019, 53, 14308–14318. [Google Scholar] [CrossRef] [PubMed]
- Luan, F.; Gorski, C.A.; Burgos, W.D. Thermodynamic Controls on the Microbial Reduction of Iron-Bearing Nontronite and Uranium. Environ. Sci. Technol. 2014, 48, 2750–2758. [Google Scholar] [CrossRef] [PubMed]
- Komadel, P.; Madejova, J.; Stucki, J.W. Structural Fe(III) reduction in smectites. Appl. Clay Sci. 2006, 34, 88–94. [Google Scholar] [CrossRef]
- Neumann, A.; Wu, L.; Li, W.; Beard, B.L.; Johnson, C.M.; Rosso, K.M.; Frierdich, A.J.; Scherer, M.M. Atom Exchange between Aqueous Fe(II) and Structural Fe in Clay Minerals. Environ. Sci. Technol. 2015, 49, 2786–2795. [Google Scholar] [CrossRef]
- Keeling, J.L.; Raven, M.D.; Gates, W.P. Geology and characterization of two hydrothermal nontronites from weathered metamorphic rocks at the Uley Graphite Mine, South Australia. Clays Clay Miner. 2000, 48, 537–548. [Google Scholar] [CrossRef]
- Gates, W.P.; Slade, P.G.; Manceau, A.; Lanson, B. Site occupancies by iron in nontronites. Clays Clay Miner. 2002, 50, 223–239. [Google Scholar] [CrossRef]
- Kandegedara, A.; Rorabacher, D.B. Noncomplexing tertiary amines as "better" buffers covering the range of pH 3-11. Temperature dependence of their acid dissociation constants. Anal. Chem. 1999, 71, 3140–3144. [Google Scholar] [CrossRef]
- Good, N.E.; Winget, G.D.; Winter, W.; Connolly, T.N.; Izawa, S.; Singh, R.M.M. Hydrogen Ion buffers for biological research. Biochemistry 1966, 5, 467. [Google Scholar] [CrossRef]
- Schilt, A.A. Applications of 1,10-Phenanthroline and Related Compounds, 1st ed.; Pergamon Press: Oxford, UK, 1969. [Google Scholar]
- Stucki, J.W.; Su, K.; Pentráková, L.; Pentrák, M. Methods for handling redox-sensitive smectite dispersions. Clay Miner. 2014, 49, 359–377. [Google Scholar] [CrossRef]
- Gorski, C.A.; Klüpfel, L.; Voegelin, A.; Sander, M.; Hofstetter, T.B. Redox Properties of Structural Fe in Clay Minerals. 2. Electrochemical and Spectroscopic Characterization of Electron Transfer Irreversibility in Ferruginous Smectite, SWa-1. Environ. Sci. Technol. 2012, 46, 9369–9377. [Google Scholar] [CrossRef]
- Amonette, J.E.; Templeton, J.C. Improvements to the quantitative assay of nonrefractory minerals for Fe(II) and total Fe using 1,10-phenanthroline. Clays Clay Miner. 1998, 46, 51–62. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. Dissolution. In The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses; Book Section 12; Wiley: Weinheim, Germany, 2003; pp. 297–344. [Google Scholar]
- Hyacinthe, C.; Bonneville, S.; Van Cappellen, P. Reactive iron(III) in sediments: Chemical versus microbial extractions. Geochim. Cosmochim. Acta 2006, 70, 4166–4180. [Google Scholar] [CrossRef]
- Tsarev, S.; Waite, T.D.; Collins, R.N. Uranium Reduction by Fe(II) in the Presence of Montmorillonite and Nontronite. Environ. Sci. Technol. 2016, 50, 8223–8230. [Google Scholar] [CrossRef]
- Anastacio, A.S.; Harris, B.; Yoo, H.I.; Fabris, J.D.; Stucki, J.W. Limitations of the ferrozine method for quantitative assay of mineral systems for ferrous and total iron. Geochim. Cosmochim. Acta 2008, 72, 5001–5008. [Google Scholar] [CrossRef]
- Rancourt, D.G.; Christie, I.A.D.; Lamarche, G.; Swainson, I.; Flandrois, S. Magnetisms of synthetic and natural annite mica–ground state and nature of excitation in an exchange-wise 2-dimensional easy-plane ferromagnet with disorder. J. Magn. Magn. Mater. 1994, 138, 31–44. [Google Scholar] [CrossRef]
- Rancourt, D.G.; Ping, J.Y. Voigt-based method for arbritrary-shape static hyperfine parameter distributions on Mossbauer-spectroscopy. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1991, 58, 85–97. [Google Scholar] [CrossRef]
- Thompson, A.; Byrne, J.M.; Dreher, C.L.; Grigg, A.R.C.; Joshi, P.; Latta, D.E.; Neumann, A.; Notini, L.; ONeill, K.E.B.; Rothwell, K.A. Current Practices for Analyzing Soils and Sediments via Mössbauer Spectroscopy. J. Plant Nutr. Soil Sci. 2025, in press. [Google Scholar]
- Soltermann, D.; Marques Fernandes, M.; Baeyens, B.; Dähn, R.; Joshi, P.A.; Scheinost, A.C.; Gorski, C.A. Fe(II) Uptake on Natural Montmorillonites. I. Macroscopic and Spectroscopic Characterization. Environ. Sci. Technol. 2014, 48, 8688–8697. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Murphy, C.A.; Waite, T.D.; Collins, R.N. Fe(II) Interactions with Smectites: Temporal Changes in Redox Reactivity and the Formation of Green Rust. Environ. Sci. Technol. 2017, 51, 12573–12582. [Google Scholar] [CrossRef]
- Dousova, B.; Fuitova, L.; Kolousek, D.; Lhotka, M.; Grygar, T.M.; Spurna, P. Stability of iron in clays under different leaching conditions. Clays Clay Miner. 2014, 62, 145–152. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1996. [Google Scholar]
- Schultz, C.; Grundl, T. pH Dependence of ferrous sorption onto two smectite clays. Chemosphere 2004, 57, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Sposito, G. The Chemistry of Soils; Oxford University Press: Oxford, UK, 2016. [Google Scholar] [CrossRef]
- Stucki, J.W. The quantitative assay of minerals for Fe2+ and Fe3+ using 1,10-phenanthroline: 2. A photochemical method. Soil Sci. Soc. Am. J. 1981, 45, 638–641. [Google Scholar] [CrossRef]
- Murad, E.; Cashion, J. Mössbauer Spectroscopy of Environmental Materials and Their Industrial Utilization; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004. [Google Scholar] [CrossRef]
- Notini, L.; Latta, D.E.; Neumann, A.; Pearce, C.I.; Sassi, M.; N’Diaye, A.T.; Rosso, K.M.; Scherer, M.M. The Role of Defects in Fe(II)-Goethite Electron Transfer. Environ. Sci. Technol. 2018, 52, 2751–2759. [Google Scholar] [CrossRef]
- Notini, L.; Latta, D.E.; Neumann, A.; Pearce, C.I.; Sassi, M.; N’Diaye, A.T.; Rosso, K.M.; Scherer, M.M. A Closer Look at Fe(II) Passivation of Goethite. ACS Earth Space Chem. 2019, 3, 2717–2725. [Google Scholar] [CrossRef]
- Thompson, A.; Rancourt, D.G.; Chadwick, O.A.; Chorover, J. Iron solid-phase differentiation along a redox gradient in basaltic soils. Geochim. Cosmochim. Acta 2011, 75, 119–133. [Google Scholar] [CrossRef]
- Pasakarnis, T.; McCormick, M.L.; Parkin, G.F.; Thompson, A.; Scherer, M.M. Feaq(II)-Fe-oxide(III) oxide electron transfer and Fe exchange: Effect of organic carbon. Environ. Chem. 2015, 12, 52–63. [Google Scholar] [CrossRef]
- Handler, R.M.; Frierdich, A.J.; Johnson, C.M.; Rosso, K.M.; Beard, B.L.; Wang, C.; Latta, D.E.; Neumann, A.; Pasakarnis, T.; Premaratne, W.A.P.J.; et al. Fe(II)-Catalyzed Recrystallization of Goethite Revisited. Environ. Sci. Technol. 2014, 48, 11302–11311. [Google Scholar] [CrossRef] [PubMed]
- Latta, D.E.; Gorski, C.A.; Scherer, M.M. Influence of Fe2+-catalysed iron oxide recrystallization on metal cycling. Biochem. Soc. Trans. 2012, 40, 1191–1197. [Google Scholar] [CrossRef]
- Gorski, C.A.; Scherer, M.M. Fe2+ sorption at the Fe oxide-water interface: A revised conceptual framework. In Aquatic Redox Chemistry; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2011; Volume 1071, pp. 315–343. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Joshi, P.; Fantle, M.S.; Larese-Casanova, P.; Gorski, C.A. Susceptibility of Goethite to Fe2+-Catalyzed Recrystallization over Time. Environ. Sci. Technol. 2017, 51, 11681–11691. [Google Scholar] [CrossRef]
- Manceau, A.; Drits, V.A.; Lanson, B.; Chateigner, D.; Wu, J.; Huo, D.; Gates, W.P.; Stucki, J.W. Oxidation-reduction mechanism of iron in dioctahedral smectites: II. Crystal chemistry of reduced Garfield nontronite. Am. Mineral. 2000, 85, 153–172. [Google Scholar] [CrossRef]
- Drits, V.A.; Manceau, A. A model for the mechanism of Fe3+ to Fe2+ reduction in dioctahedral smectites. Clays Clay Miner. 2000, 48, 185–195. [Google Scholar] [CrossRef]
- Rozenson, I.; Hellerkallai, L. Reduction and oxidation of Fe3+ in dioctahedral smectites. 1. Reduction with hydrazine and dithionite. Clays Clay Miner. 1976, 24, 271–282. [Google Scholar] [CrossRef]
- Alexandrov, V.; Neumann, A.; Scherer, M.M.; Rosso, K.M. Electron Exchange and Conduction in Nontronite from First-Principles. J. Phys. Chem. C 2013, 117, 2032–2040. [Google Scholar] [CrossRef]
- Shi, B.; Liu, K.; Wu, L.; Li, W.; Smeaton, C.M.; Beard, B.L.; Johnson, C.M.; Roden, E.E.; Van Cappellen, P. Iron Isotope Fractionations Reveal a Finite Bioavailable Fe Pool for Structural Fe(III) Reduction in Nontronite. Environ. Sci. Technol. 2016, 50, 8661–8669. [Google Scholar] [CrossRef]
- Gorski, C.A.; Aeschbacher, M.; Soltermann, D.; Voegelin, A.; Baeyens, B.; Marques Fernandes, M.; Hofstetter, T.B.; Sander, M. Redox Properties of Structural Fe in Clay Minerals. 1. Electrochemical Quantification of Electron-Donating and -Accepting Capacities of Smectites. Environ. Sci. Technol. 2012, 46, 9360–9368. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, D.G. Mossbauer spectroscopy in clay science. Hyperfine Interact. 1998, 117, 3–38. [Google Scholar] [CrossRef]
- Dyar, M.D.; Agresti, D.G.; Schaefer, M.W.; Grant, C.A.; Sklute, E.C. Mossbauer spectroscopy of earth and planetary materials. Annu. Rev. Earth Planet. Sci. 2006, 34, 83–125. [Google Scholar] [CrossRef]
- Lagaly, G. Colloid Clay Science. In Developments in Clay Science; Book Section 5; Bergaya, F., Theng, B.K.G., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 141–245. [Google Scholar]
- Adamou, P.; Entwistle, J.; Graham, D.W.; Neumann, A. Mineral-Based Advanced Oxidation Processes for Enhancing the Removal of Antibiotic Resistance Genes from Domestic Wastewater. ACS ES&T Water 2025, 5, 2310–2321. [Google Scholar] [CrossRef]
- Yang, S.; Liu, D.; Zheng, W.; Fan, Q.; Wang, H.; Zhao, L. Microbial reduction and alteration of Fe(III)-containing smectites in the presence of biochar-derived dissolved organic matter. Appl. Geochem. 2023, 152, 105661. [Google Scholar] [CrossRef]
- Latta, D.E.; Boyanov, M.I.; Kemner, K.M.; O’Loughlin, E.J.; Scherer, M.M. Abiotic reduction of uranium by Fe(II) in soil. Appl. Geochem. 2012, 27, 1512–1524. [Google Scholar] [CrossRef]
- Zhou, N.; Kupper, R.J.; Catalano, J.G.; Thompson, A.; Chan, C.S. Biological Oxidation of Fe(II)-Bearing Smectite by Microaerophilic Iron Oxidizer Sideroxydans lithotrophicus Using Dual Mto and Cyc2 Iron Oxidation Pathways. Environ. Sci. Technol. 2022, 56, 17443–17453. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, A.; Notini, L.; Premaratne, W.A.P.J.; Latta, D.E.; Scherer, M.M. Reduction-Driven Mobilization of Structural Fe in Clay Minerals with High Fe Content. Minerals 2025, 15, 713. https://doi.org/10.3390/min15070713
Neumann A, Notini L, Premaratne WAPJ, Latta DE, Scherer MM. Reduction-Driven Mobilization of Structural Fe in Clay Minerals with High Fe Content. Minerals. 2025; 15(7):713. https://doi.org/10.3390/min15070713
Chicago/Turabian StyleNeumann, Anke, Luiza Notini, W. A. P. Jeewantha Premaratne, Drew E. Latta, and Michelle M. Scherer. 2025. "Reduction-Driven Mobilization of Structural Fe in Clay Minerals with High Fe Content" Minerals 15, no. 7: 713. https://doi.org/10.3390/min15070713
APA StyleNeumann, A., Notini, L., Premaratne, W. A. P. J., Latta, D. E., & Scherer, M. M. (2025). Reduction-Driven Mobilization of Structural Fe in Clay Minerals with High Fe Content. Minerals, 15(7), 713. https://doi.org/10.3390/min15070713