


Structural Features of Oxyapatite

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Preparation and Characterization
2.2. Computational Details
3. Results and Discussion
3.1. SEM Investigation
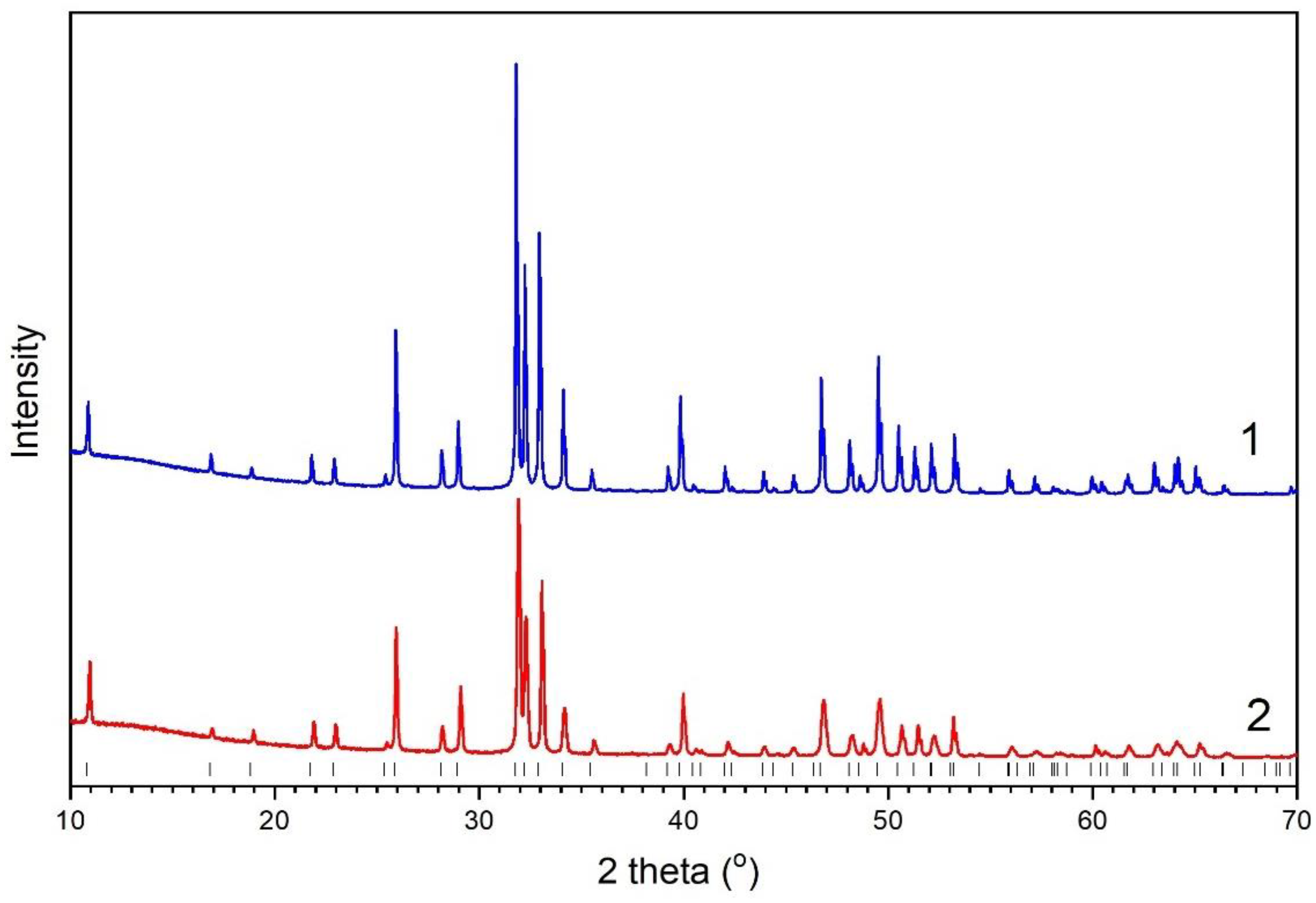
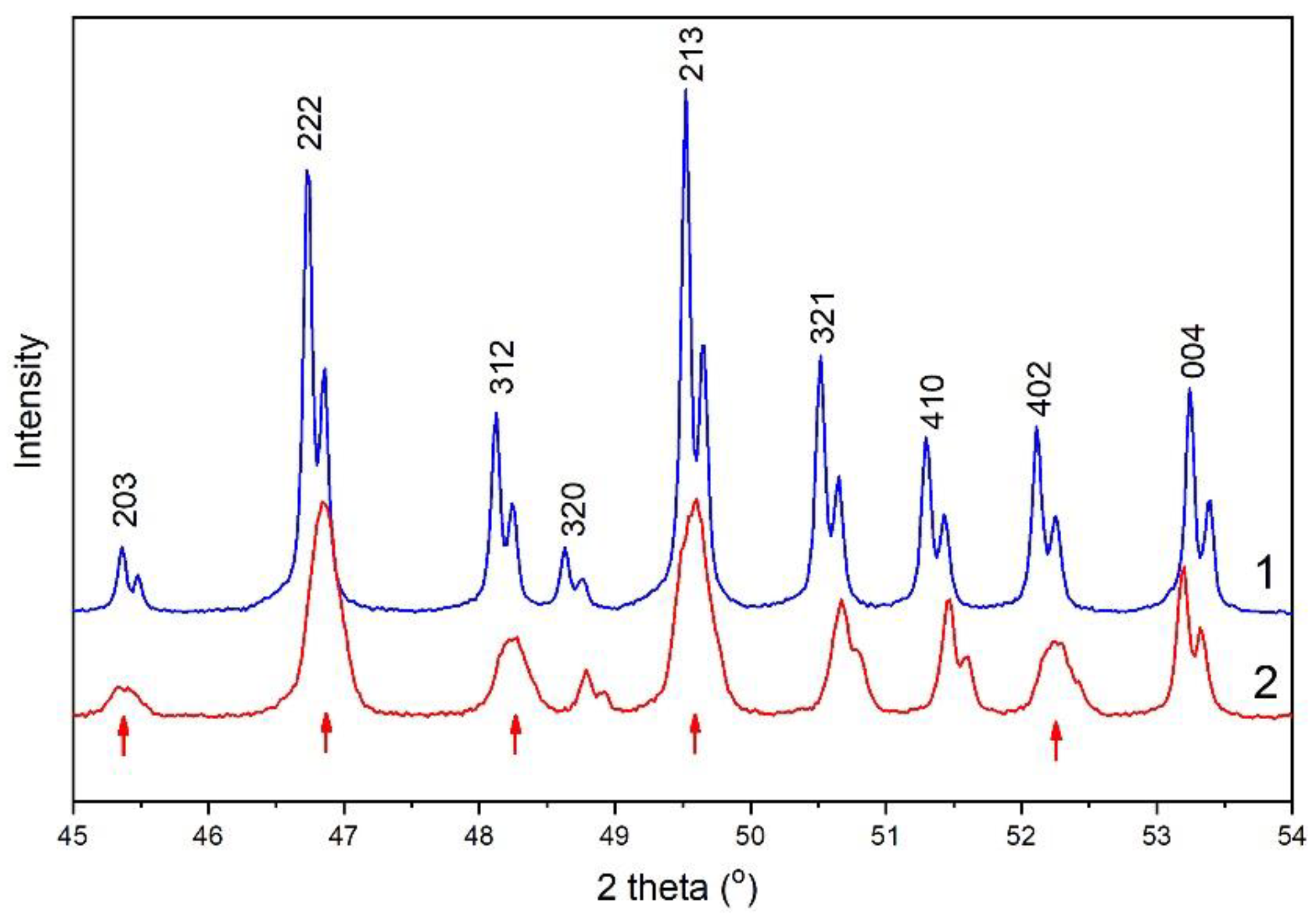
3.2. Powder Diffraction
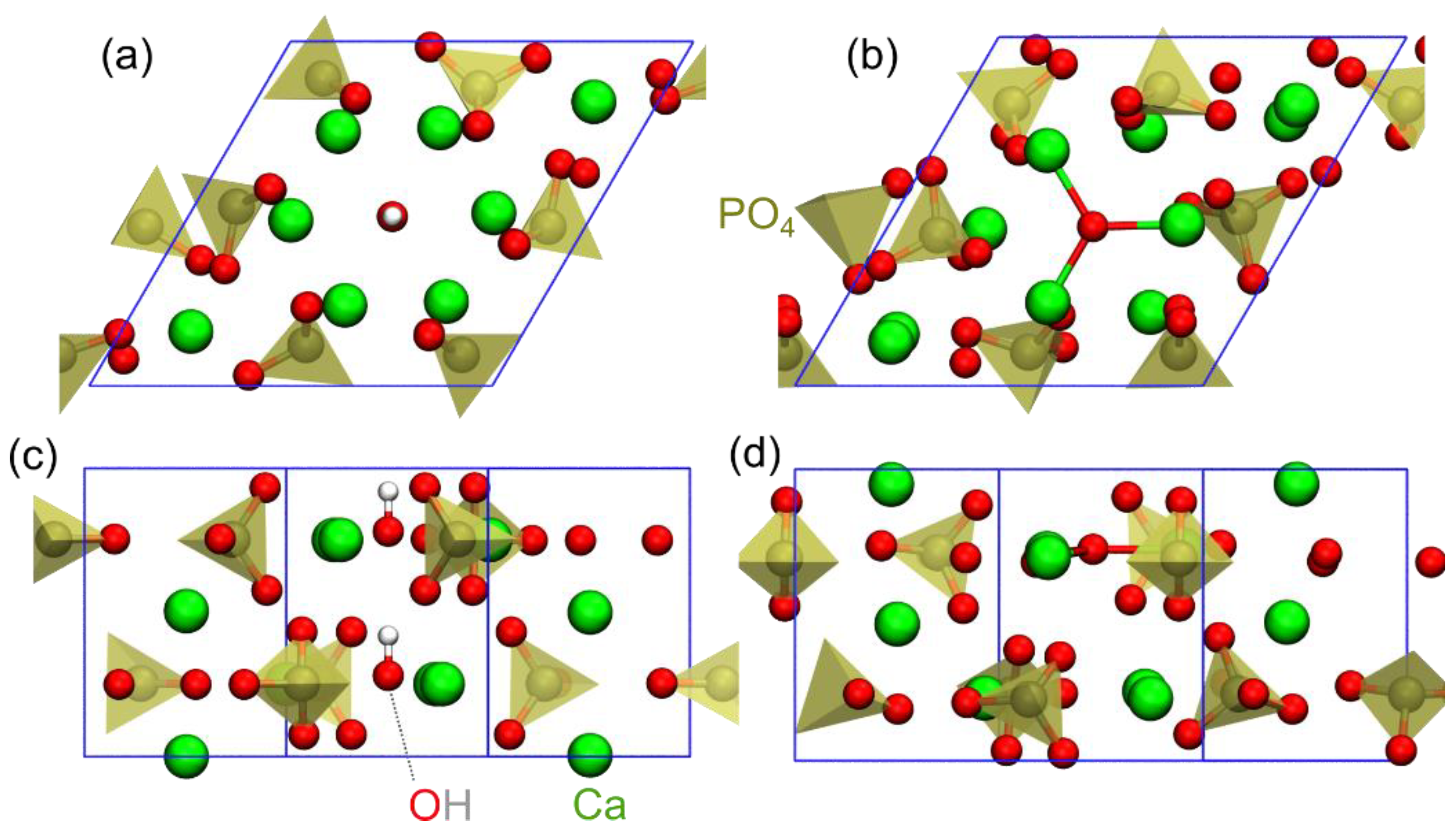
3.3. DFT Study of Crystal Structures
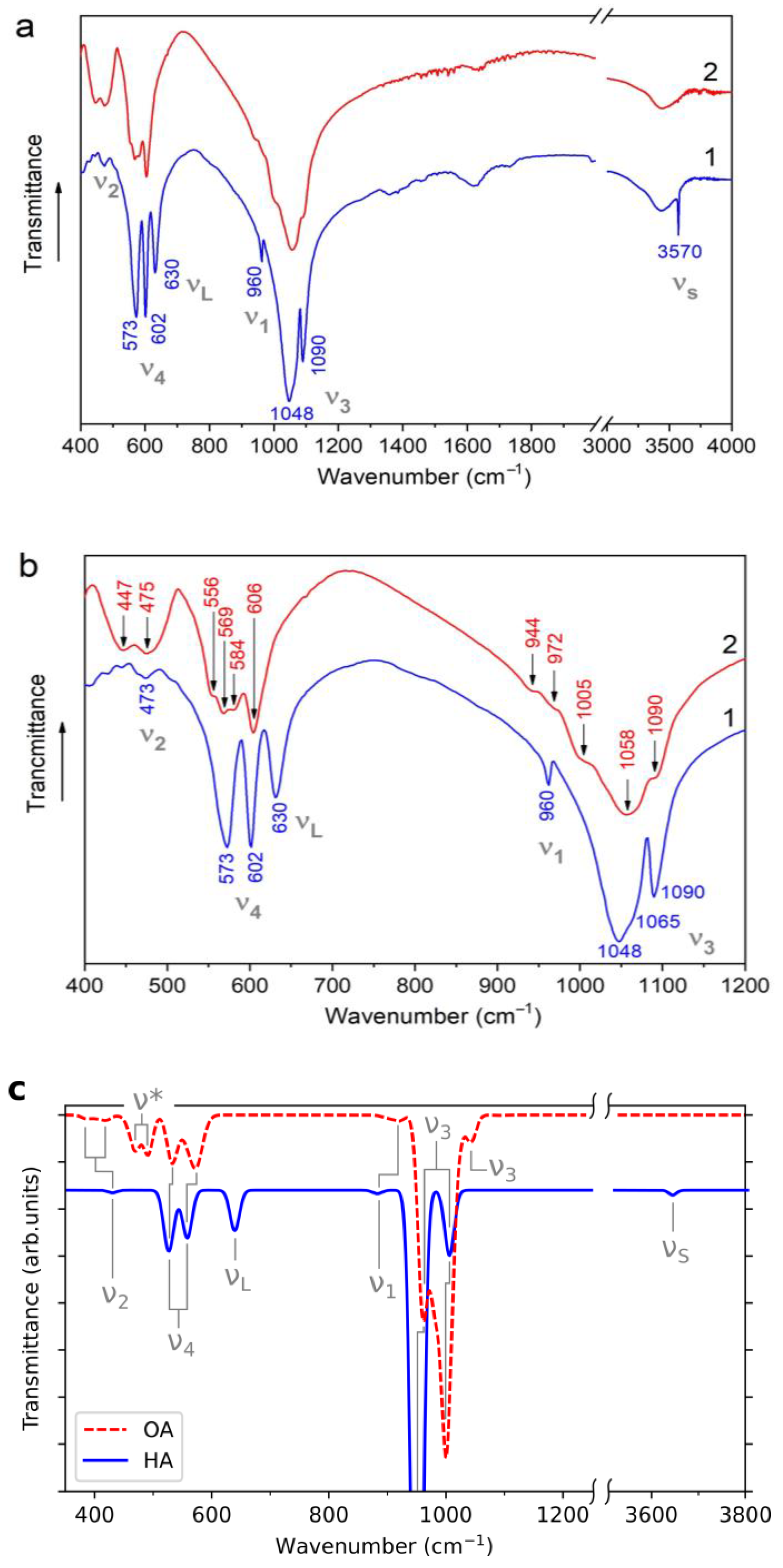
3.4. IR Spectroscopy
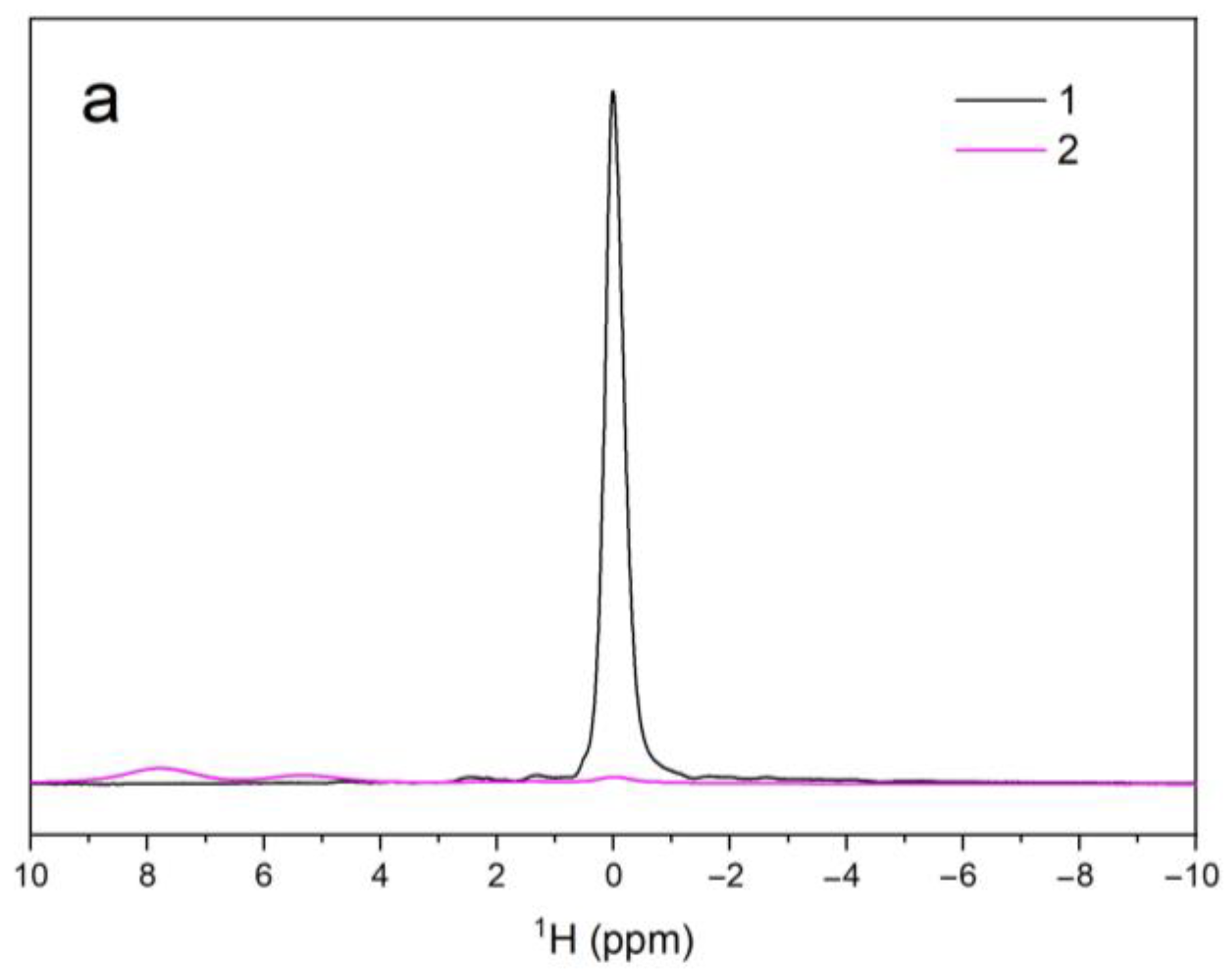
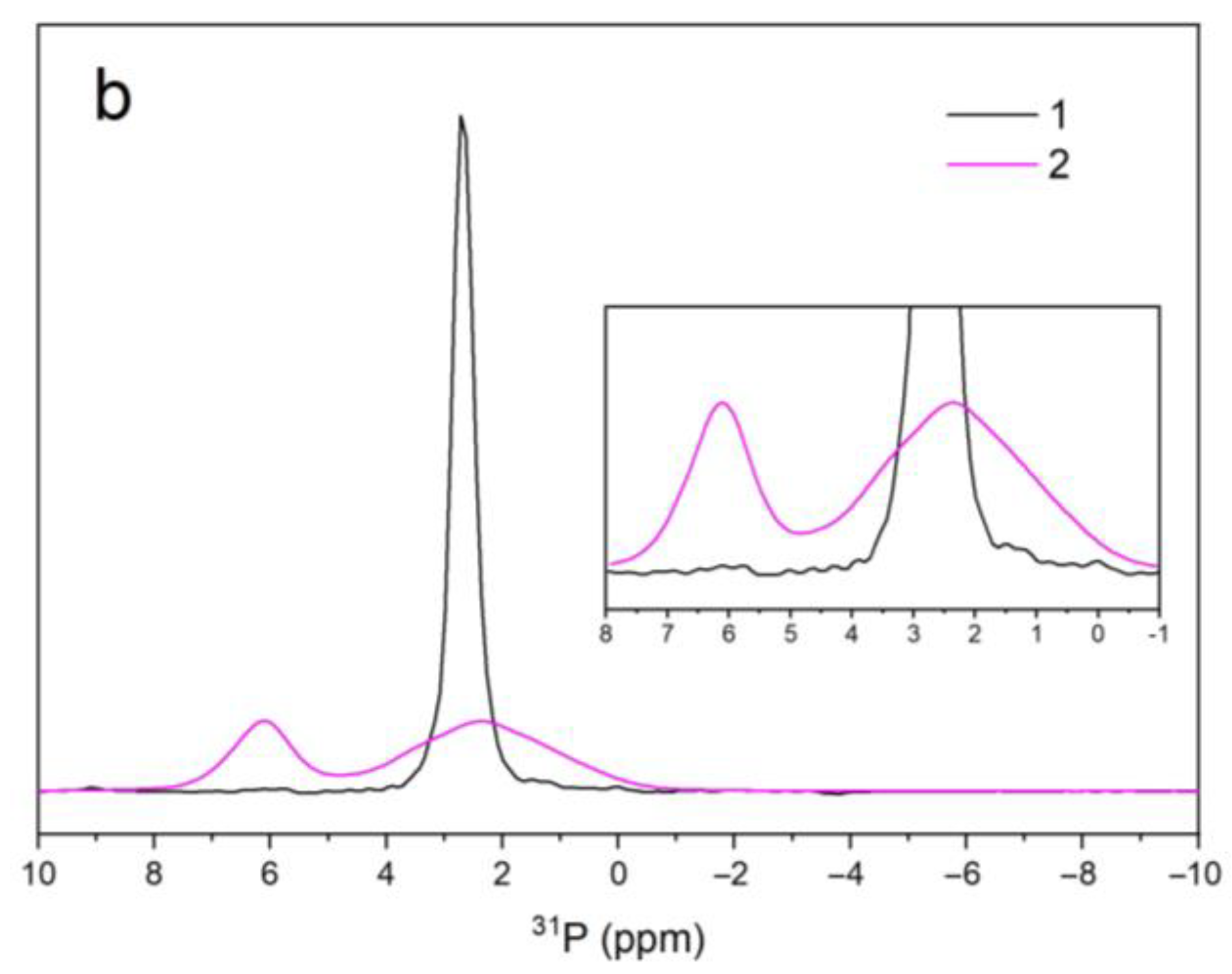
3.5. NMR Spectroscopy
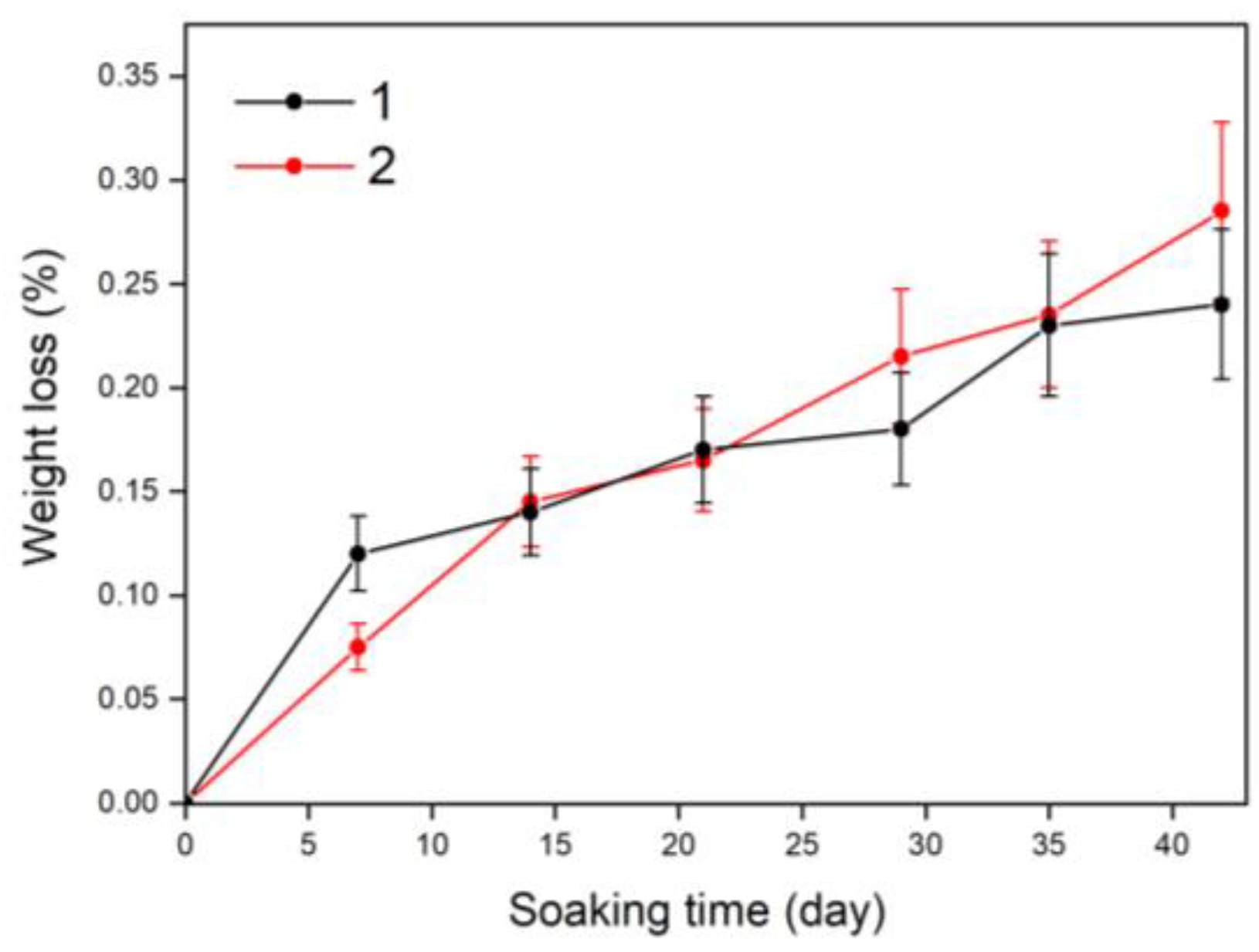
3.6. Solubility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Camaioni, A.; Cacciotti, I.; Campagnolo, L.; Bianco, A. 15—Silicon-substituted hydroxyapatite for biomedical applications. In Hydroxyapatite (Hap) for Biomedical Applications; Mucalo, M., Ed.; Woodhead Publishing: Cambridge, UK, 2015; pp. 343–373. [Google Scholar] [CrossRef]
- Turon, P.; del Valle, L.J.; Alemán, C.; Puiggalí, J. Biodegradable and Biocompatible Systems Based on Hydroxyapatite Nanoparticles. Appl. Sci. 2017, 7, 60. [Google Scholar] [CrossRef]
- Ghiasi, B.; Sefidbakht, Y.; Mozaffari-Jovin, S.; Gharehcheloo, B.; Mehrarya, M.; Khodadadi, A.; Rezaei, M.; Siadat, S.O.R.; Uskoković, V. Hydroxyapatite as a biomaterial—A gift that keeps on giving. Drug Dev. Ind. Pharm. 2020, 46, 1035–1062. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, H.A.; Pickering, K.L.; Mucalo, M.R. A Review on the use of hydroxyapatite-carbonaceous structure composites in bone replacement materials for strengthening purposes. Materials 2018, 11, 1813. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphate bioceramics. Ceram. Int. 2015, 41, 13913–13966. [Google Scholar] [CrossRef]
- Pasero, M.; Kampf, A.R.; Ferraris, C.; Pekov, I.V.; Rakovan, J.; White, T.J. Nomenclature of the apatite supergroup minerals. Eur. J. Miner. 2010, 22, 163–179. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates. J. Mater. Sci. 2007, 42, 1061–1095. [Google Scholar] [CrossRef]
- Matsunaga, K.; Kuwabara, A. First-principles study of vacancy formation in hydroxyapatite. Phys. Rev. B 2007, 75, 014102. [Google Scholar] [CrossRef]
- Sadetskaya, A.V.; Bobrysheva, N.P.; Osmolowsky, M.G.; Osmolovskaya, O.M.; Voznesenskiy, M.A. Correlative experimental and theoretical characterization of transition metal doped hydroxyapatite nanoparticles fabricated by hydrothermal method. Mater. Charact. 2021, 173, 110911. [Google Scholar] [CrossRef]
- Terra, J.; Dourado, E.R.; Eon, J.-G.; Ellis, D.E.; Gonzalez, G.; Rossi, A.M. The structure of strontium-doped hydroxyapatite: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2009, 11, 568–577. [Google Scholar] [CrossRef]
- Bystrov, V.; Paramonova, E.; Avakyan, L.; Coutinho, J.; Bulina, N. Simulation and Computer Study of Structures and Physical Properties of Hydroxyapatite with Various Defects. Nanomaterials 2021, 11, 2752. [Google Scholar] [CrossRef]
- Avakyan, L.A.; Paramonova, E.V.; Coutinho, J.; Öberg, S.; Bystrov, V.S.; Bugaev, L.A. Optoelectronics and defect levels in hydroxyapatite by first-principles. J. Chem. Phys. 2018, 148, 154706. [Google Scholar] [CrossRef] [PubMed]
- Avakyan, L.; Paramonova, E.; Bystrov, V.; Coutinho, J.; Gomes, S.; Renaudin, G. Iron in Hydroxyapatite: Interstitial or Substitution Sites? Nanomaterials 2021, 11, 2978. [Google Scholar] [CrossRef] [PubMed]
- Leeuw, N.H.; Bowe, J.R.; Rabone, J.A.L. A computational investigation of stoichiometric and calcium-deficient oxy- and hydroxy-apatites. Faraday Discuss. 2007, 134, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C. Structure and Chemistry of Apatite and Other Calcium Orthophosphates; Studies in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 1994; pp. 1–404. ISBN 0-444-81582-1. [Google Scholar]
- Champion, E. Sintering of calcium phosphate bioceramics. Acta Biomater. 2013, 9, 5855–5875. [Google Scholar] [CrossRef] [PubMed]
- Bulina, N.V.; Makarova, S.V.; Baev, S.G.; Matvienko, A.A.; Gerasimov, K.B.; Logutenko, O.A.; Bystrov, V.S. A Study of Thermal Stability of Hydroxyapatite. Minerals 2021, 11, 1310. [Google Scholar] [CrossRef]
- Tõnsuaadu, K.; Gross, K.A.; Plūduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]
- Tanaka, H.; Chikazawa, M.; Kandori, K.; Ishikawa, T. Influence of thermal treatment on the structure of calcium hydroxyapatite. Phys. Chem. Chem. Phys. 2000, 2, 2647–2650. [Google Scholar] [CrossRef]
- Kanazawa, T. Inorganic Phosphate Materials. In Materials Science Monographs; Elsevier: Amsterdam, The Netherlands, 1989; Volume 52, ISSN 0166-6010. [Google Scholar]
- Trombe, J.C.; Montel, G. Some features of the incorporation of oxygen in different oxidation states in the apatitic lattice—I On the existence of calcium and strontium oxyapatites. J. Inorg. Nucl. Chem. 1978, 40, 15–21. [Google Scholar] [CrossRef]
- Sakka, S.; Bouaziz, J.; Ben, F. Mechanical Properties of Biomaterials Based on Calcium Phosphates and Bioinert Oxides for Applications in Biomedicine. In Advances in Biomaterials Science and Biomedical Applications; Pignatello, R., Ed.; InTech: Rijeka, Croatia, 2013; pp. 23–50. [Google Scholar] [CrossRef]
- Saad, B.H.F. 2—Structure, microstructure, and properties of bioceramics. In Bioceramics: For Materials Science and Engineering; Woodhead Publishing: Sawston, UK; Elsevier: Amsterdam, The Netherlands, 2019; pp. 39–76. [Google Scholar] [CrossRef]
- Bredig, M.A.; Frank, H.H.; Füldner, H. Beiträge zur kenntnis der kalk-phosphorsaure-verbindungen II. Z. Elektrochem. Angew. Phys. Chem. 1933, 39, 959–969. [Google Scholar] [CrossRef]
- Pastero, L.; Bruno, M.; Aquilano, D. About the Genetic Mechanisms of Apatites: A Survey on the Methodological Approaches. Minerals 2017, 7, 139. [Google Scholar] [CrossRef]
- Wilson, R.M.; Elliott, J.C.; Dowker, S.E.P.; Rodriguez-Lorenzo, L.M. Rietveld refinements and spectroscopic studies of the structure of Ca-deficient apatite. Biomaterials 2005, 26, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Dong, Z.L. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. B Struct. Sci. 2003, B59, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Henning, P.A.; Landa-CaÂnovas, A.R.; Larssona, A.K.; Lidin, S. Elucidation of the crystal structure of oxyapatite by high-resolution electron microscopy. Acta Crystallogr. B Struct. Sci. 1999, B55, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Henning, P.A.; Adolfsson, E.; Grins, J.; Fritch, A. Triclinic oxy-hydroxyapatite. J. Mater. Sci. 2001, 36, 663–668. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophospates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal Structure of Hydroxyapatite. Nature 1964, 204, 1050–1052. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Schlipf, M.; Gygi, F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput. Phys. Commun. 2015, 196, 36–44. [Google Scholar] [CrossRef]
- Hamann, D.R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. 2013, B88, 085117. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, B.N.; Baradaran, S.; Zalnezhad, E.; Basirun, W.J. Applications of electron microscopy in mechanochemistry. In Microscopy: Advances in Scientific Research and Education; Mendez-Vilas, A., Ed.; Formatex Microscopy Series; Formatex Research Center S.L.: Badajoz, Spain, 2014; Volume 2, pp. 791–802. Available online: https://www.researchgate.net/publication/267066990 (accessed on 3 December 2022).
- Likhachev, I.; Balabaev, N.; Bystrov, V.; Paramonova, E.; Avakyan, L.; Bulina, N. Molecular Dynamics Simulation of the Thermal Behavior of Hydroxyapatite. Nanomaterials 2022, 12, 4244. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 88th ed.; CRC Press: Boca Raton, FL, USA, 2007; p. 2640. ISBN 0-8493-0488-1. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Putz, M. Density Functionals of Chemical Bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 6th ed.; John Wiley & Sons: New York, NY, USA, 2009; p. 400. ISBN 978-0-470-40587-1. [Google Scholar]
- Mukherjee, K.; Palchowdhury, S.; Maroncelli, M. OH Stretching and Libration Bands of Solitary Water in Ionic Liquids and Dipolar Solvents Share a Single Dependence on Solvent Polarity. J. Phys. Chem. B 2022, 126, 4584–4598. [Google Scholar] [CrossRef]
- Calzolari, A.; Pavan, B.; Curtarolo, S.; Nardelli, M.B.; Fornari, M. Vibrational spectral fingerprinting for chemical recognition of biominerals. ChemPhysChem 2020, 21, 770–778. [Google Scholar] [CrossRef]
- Jager, C.; Welzel, T.; Meyer-Zaika, W.; Epple, M. A solid-state NMR investigation of the structure ofnanocrystalline hydroxyapatite. Magn. Reson. Chem. 2006, 44, 573–580. [Google Scholar] [CrossRef]
- Hartmann, P.; JaKger, C.; Barth, S.; Vogel, J.; Meyer, K. Solid State NMR, X-ray Diffraction, and Infrared Characterization of Local Structure in Heat-Treated Oxyhydroxyapatite Microcrystals: An Analog of the Thermal Decomposition of Hydroxyapatite during Plasma-Spray Procedure. J. Solid State Chem. 2001, 160, 460–468. [Google Scholar] [CrossRef]
- Bulina, N.V.; Chaikina, M.V.; Andreev, A.S.; Lapina, O.B.; Ishchenko, A.V.; Prosanov, I.Y.; Gerasimov, K.B.; Solovyov, L.A. Mechanochemical Synthesis of SiO44–-Substituted Hydroxyapatite, Part II—Reaction Mechanism, Structure, and Substitution Limit. Eur. J. Inorg. Chem. 2014, 2014, 4810–4825. [Google Scholar] [CrossRef]
- McPherson, R.; Gane, N.; Bastow, T.J. Structural characterization of plasma-sprayed hydroxylapatite coatings. J. Mater. Sci. Mater. Med. 1995, 6, 327–334. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal System | Space Group | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | Reference |
|---|---|---|---|---|---|---|---|---|
| Hexagonal | P63/m | 9.40 | 9.40 | 6.89 | 90 | 90 | 120 | [15] |
| P | 9.432 | 9.432 | 6.881 | 90.3 | 90 | 119.9 | [25,26,27,28] | |
| Triclinic | P | 9.4002 | 9.3970 | 6.8997 | 90.063 | 89.748 | 119.997 | [29] |
| Sample Name | Annealing Conditions | Space Group | a (Å) | c (Å) | V (Å3) | Crystallite Size (nm) | Rwp/GOF |
|---|---|---|---|---|---|---|---|
| A | Air | P63/m | 9.42378(6) | 6.88104(6) | 529.218(9) | 220(4) | 5.69/2.06 |
| B | Vacuum | P63/m | 9.4060(3) | 6.8950(2) | 528.30(4) | 106(8) | 11.9/2.87 |
| P | 9.4089(3) | 6.8965(3) | 528.67(4) | 100(5) | 10.4/3.13 |
| a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | V (Å3) | Rwp/GOF |
|---|---|---|---|---|---|---|---|
| 9.4115(4) | 9.4077(4) | 6.8953(2) | 90.01(1) | 89.83(4) | 120.04(1) | 528.50(4) | 6.3/1.9 |
| Material | a (Å) | c (Å) | B (GPa) | ΔfH (eV/cell) |
|---|---|---|---|---|
| HA | 9.481 | 6.859 | 88 | −128.49 |
| OA | 9.442 | 6.888 | 79 | −123.16 |
| HA w/o OH | 9.549 | 6.747 | 69 | −117.68 |
| Type of Vibration | Wavenumber (cm−1) | |||
|---|---|---|---|---|
| Sample A | Sample B | Model HA | Model OA | |
| O–P–O, ν2 | 473 | Out of range | 420 | 380, 420 |
| Ca–O, ν* | – | 447, 475 | – | 470, 490 |
| O–P–O, ν4 | 573, 602 | 556, 569, 584, 606 | 520, 560 | 530, 580 |
| OH, νL | 630 | – | 640 | – |
| P–O, ν1 | 960 | 944, 972 | 880 | 900, 920 |
| P–O, ν3 | 1048, 1065, 1090 | 1005, 1058, 1090 | 950, 1000 | 970, 1000, 1020 |
| O–H, νS | 3570 | – | 3640 | – |
| Sample Name | 1H | 31P | ||||
|---|---|---|---|---|---|---|
| V–OH–V | OH–V–OH | OH–OH | PO43− | PO43− | ||
| A | δiso (ppm) | – | – | 0.01 | 2.27 | – |
| Width (Hz) | – | – | 189 | 91 | – | |
| Area (arb. un.) | – | – | 1.7 × 106 | 8.2 × 107 | – | |
| B | δiso (ppm) | 7.80 | 5.29 | 0.02 | 2.33 | 6.11 |
| Width (Hz) | 714 | 630 | 309 | 574 | 247 | |
| Area (arb. un.) | 8.7 × 104 | 3.7 × 104 | 2.2 × 104 | 3.6 × 107 | 1.6 × 107 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulina, N.V.; Avakyan, L.A.; Makarova, S.V.; Orehov, I.B.; Bystrov, V.S. Structural Features of Oxyapatite. Minerals 2023, 13, 102. https://doi.org/10.3390/min13010102
Bulina NV, Avakyan LA, Makarova SV, Orehov IB, Bystrov VS. Structural Features of Oxyapatite. Minerals. 2023; 13(1):102. https://doi.org/10.3390/min13010102
Chicago/Turabian StyleBulina, Natalia V., Leon A. Avakyan, Svetlana V. Makarova, Igor B. Orehov, and Vladimir S. Bystrov. 2023. "Structural Features of Oxyapatite" Minerals 13, no. 1: 102. https://doi.org/10.3390/min13010102
APA StyleBulina, N. V., Avakyan, L. A., Makarova, S. V., Orehov, I. B., & Bystrov, V. S. (2023). Structural Features of Oxyapatite. Minerals, 13(1), 102. https://doi.org/10.3390/min13010102