
Column-Test Data Analyses and Geochemical Modeling to Determine Uranium Reactive Transport Parameters at a Former Uranium Mill Site (Grand Junction, Colorado)


Abstract
:1. Objective
2. Introduction
2.1. Background
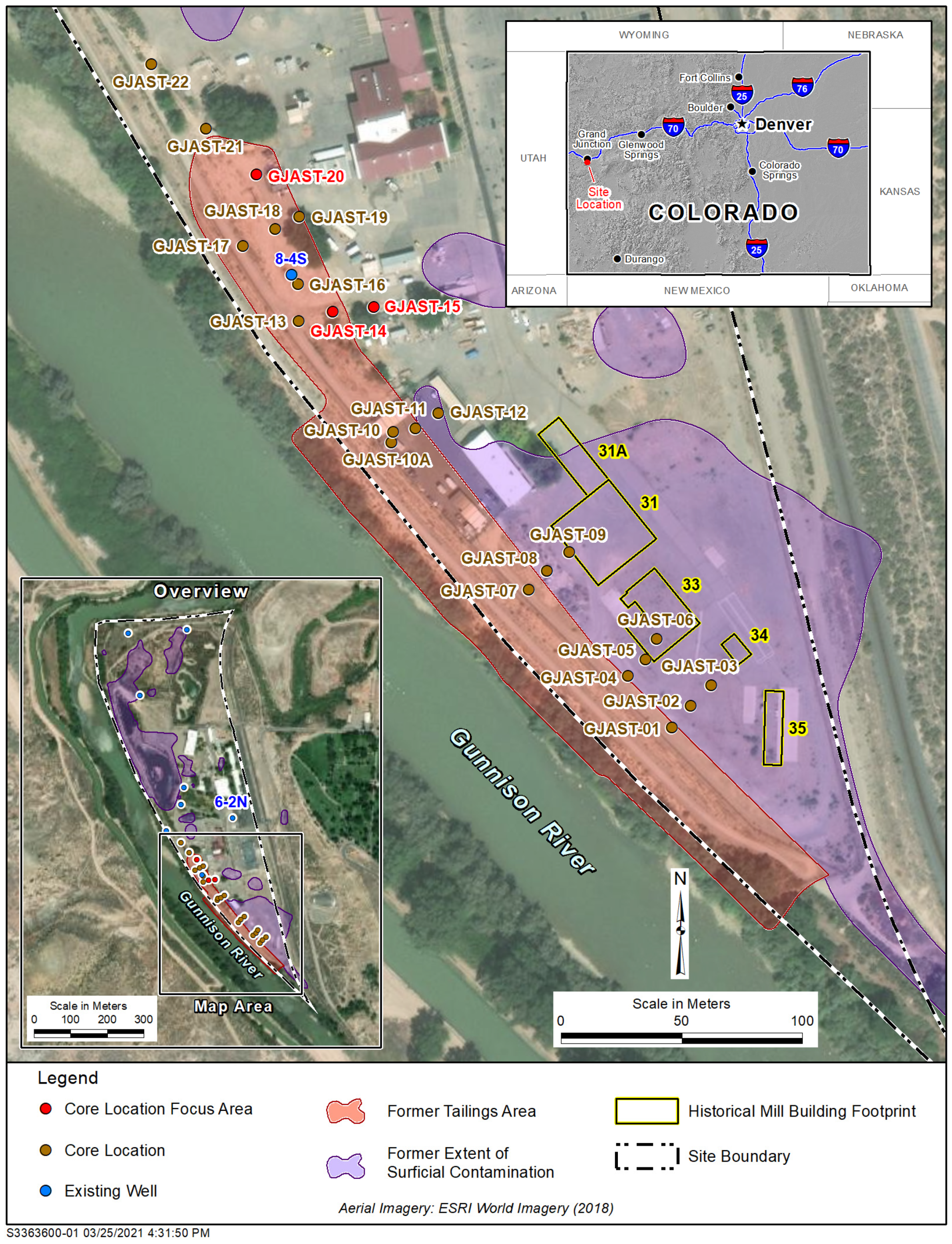
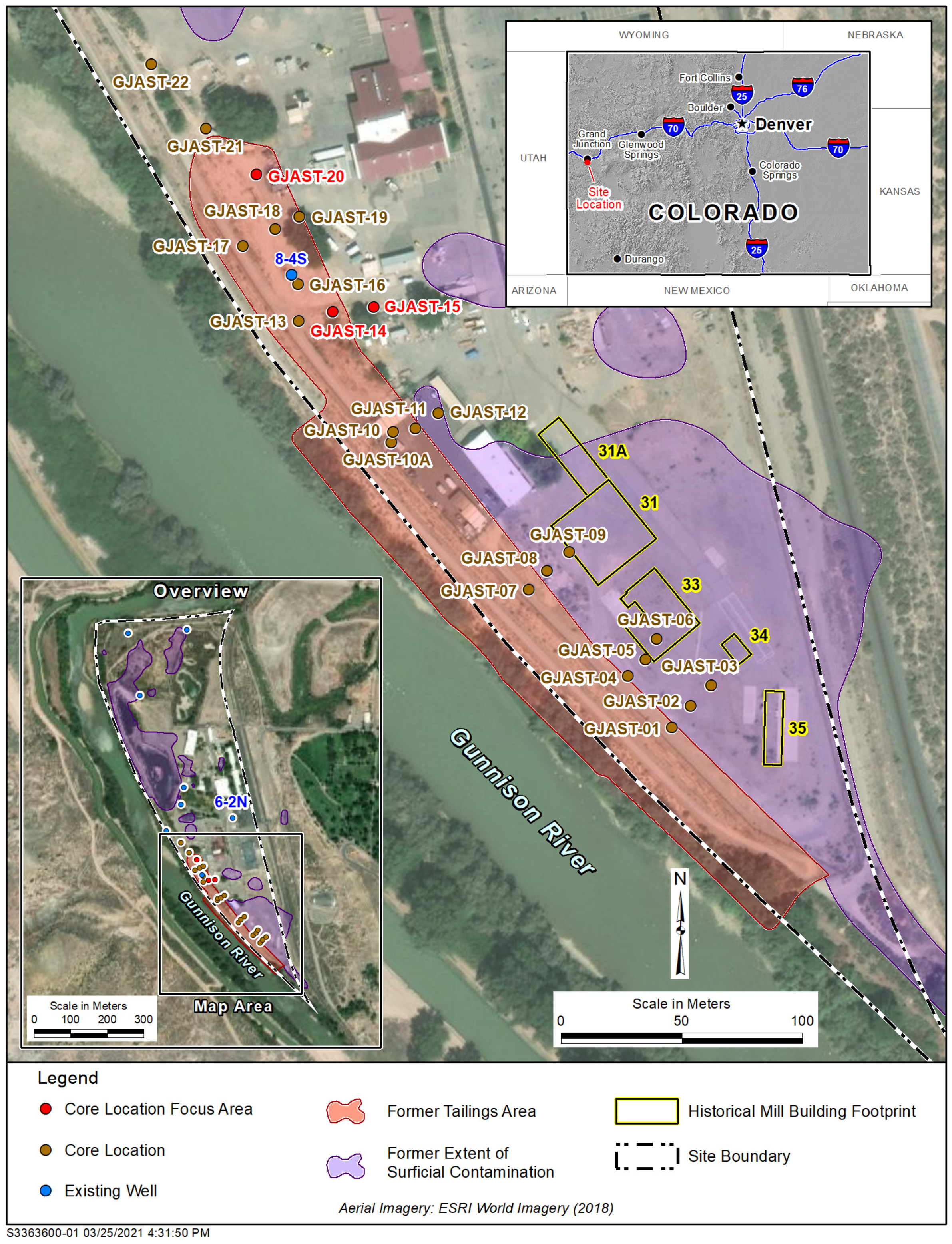
2.2. Column Testing Focus Locations
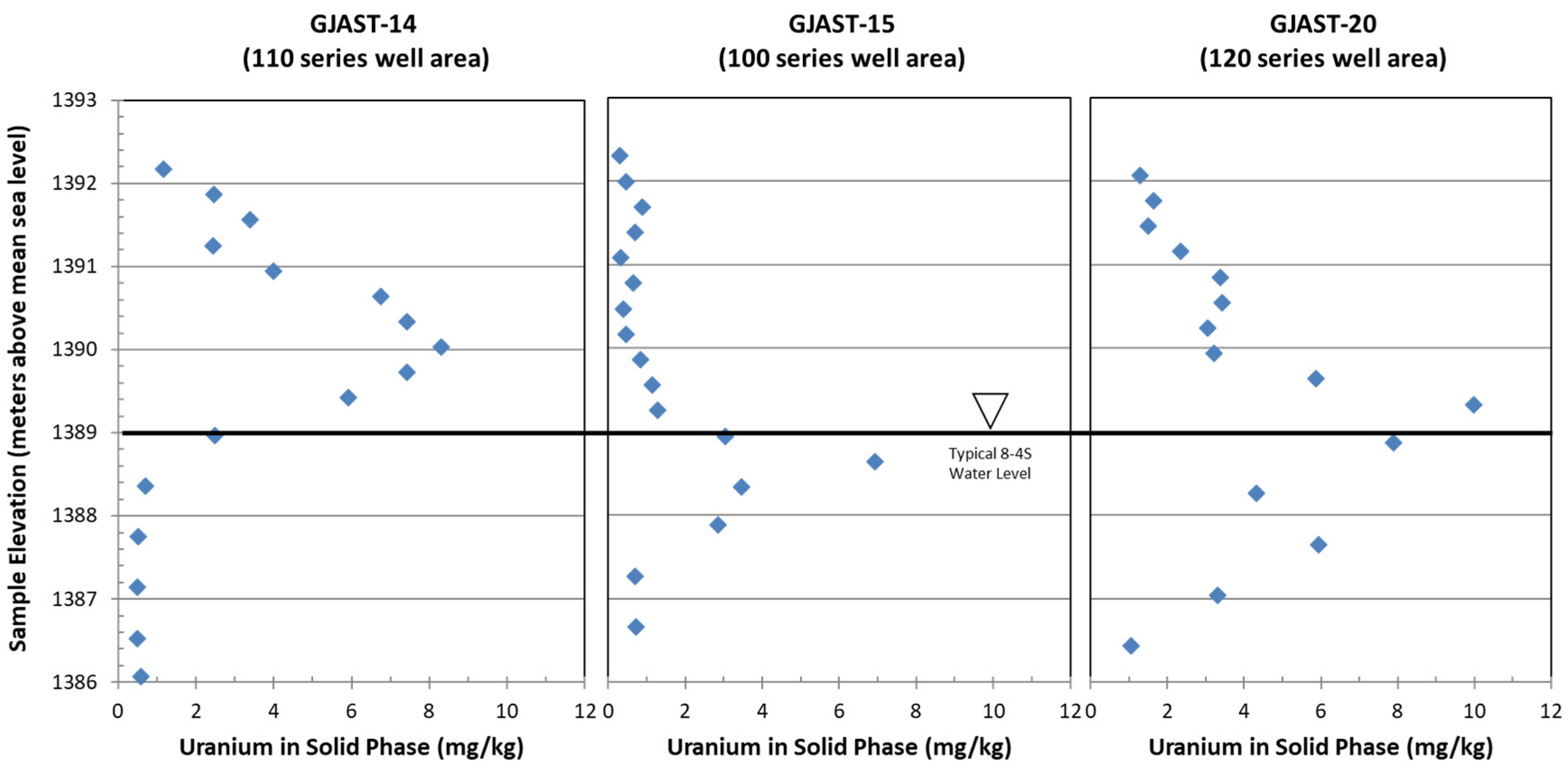
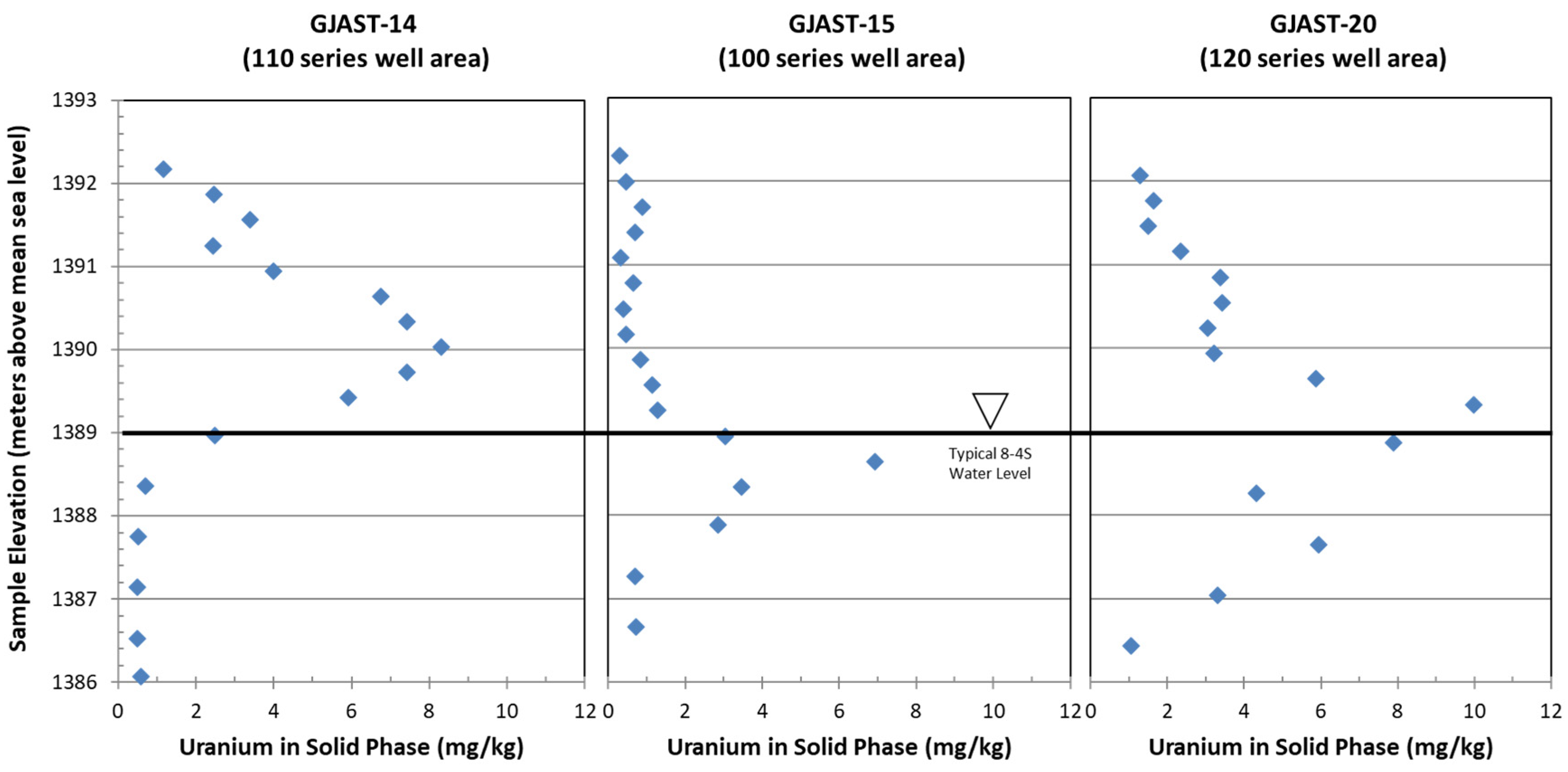
- Solid-phase uranium at GJAST-14 is associated with VZ sediments, possibly with uranium deposited by evapotranspiration of the underlying groundwater. Solid-phase uranium in the saturated zone is characterized by lower uranium concentrations (Figure 2);
- Solid-phase uranium at GJAST-15 is associated with higher organic content in the sediments just below the water table. Solid-phase uranium in the VZ is characterized by lower uranium concentrations (Figure 2);
- Solid-phase uranium at GJAST-20 is associated with the presence of gypsum, which is likely precipitated when low-pH waters derived from tailings pore fluids and/or hydrofluoric acid waste disposal are buffered by naturally occurring calcite [1,13]. The solid-phase uranium in the VZ and the saturated zone is characterized by elevated uranium concentrations compared to the background values (Figure 2).
3. Materials and Methods
3.1. Column Testing: General Details
3.2. Sediment Collection and Processing
3.3. Column-Testing Configurations
3.4. Column-Filling Procedures
3.5. Column Effluent Sample Collection and Analyses
3.6. Geochemical Modeling Approach
4. Results and Discussion
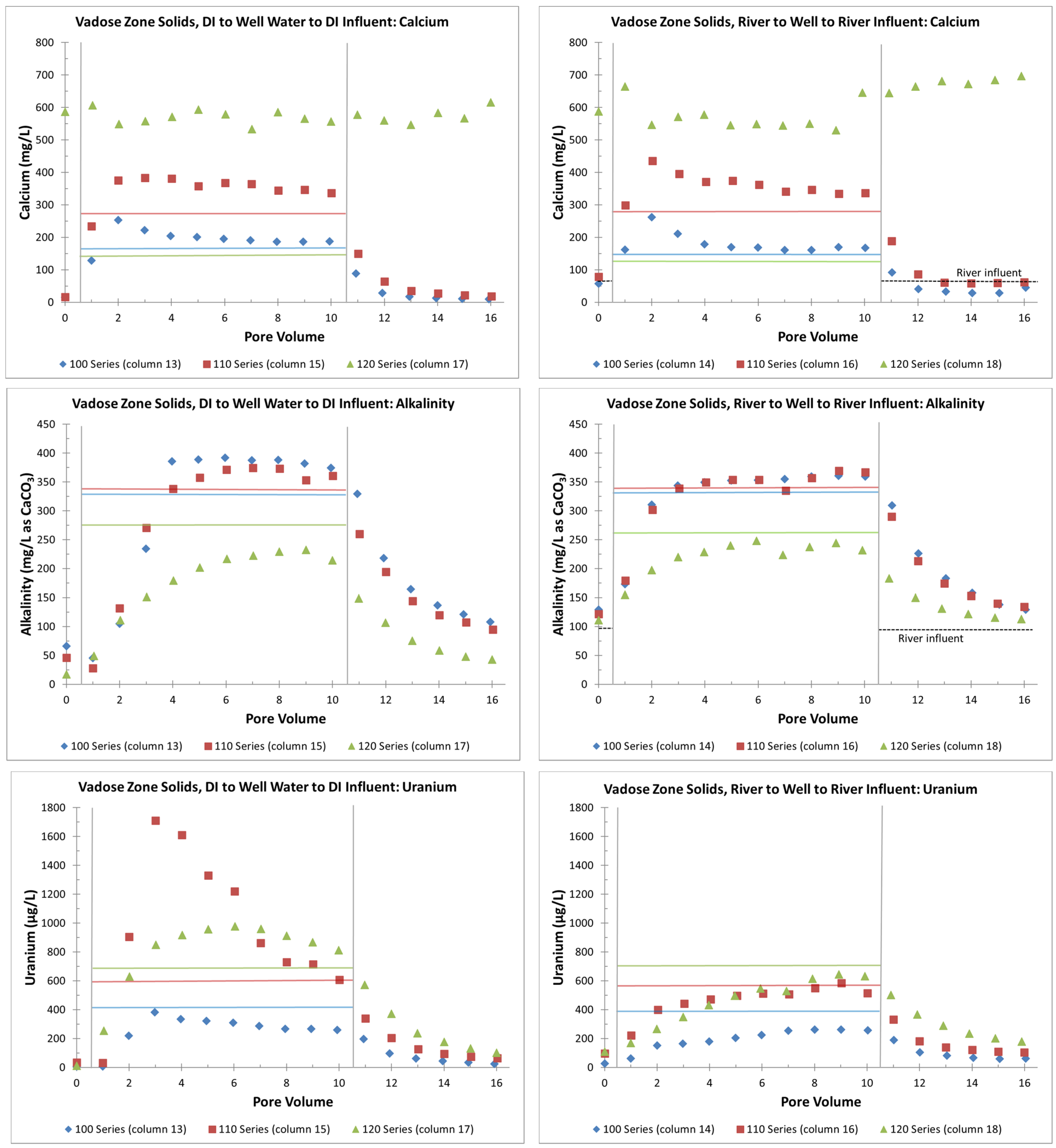
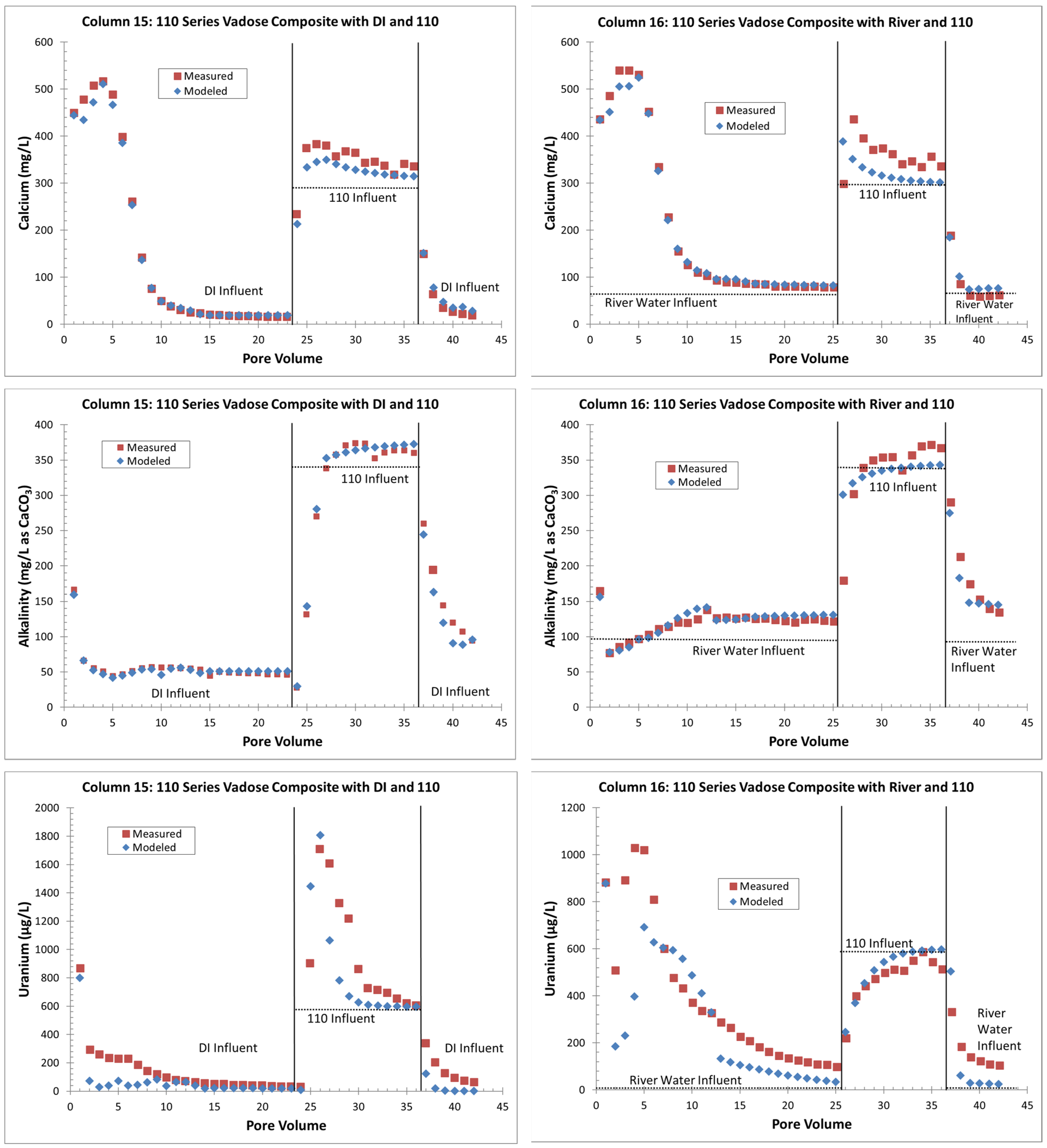
4.1. Vadose Zone (All Stop Flow)
4.1.1. Laboratory Results
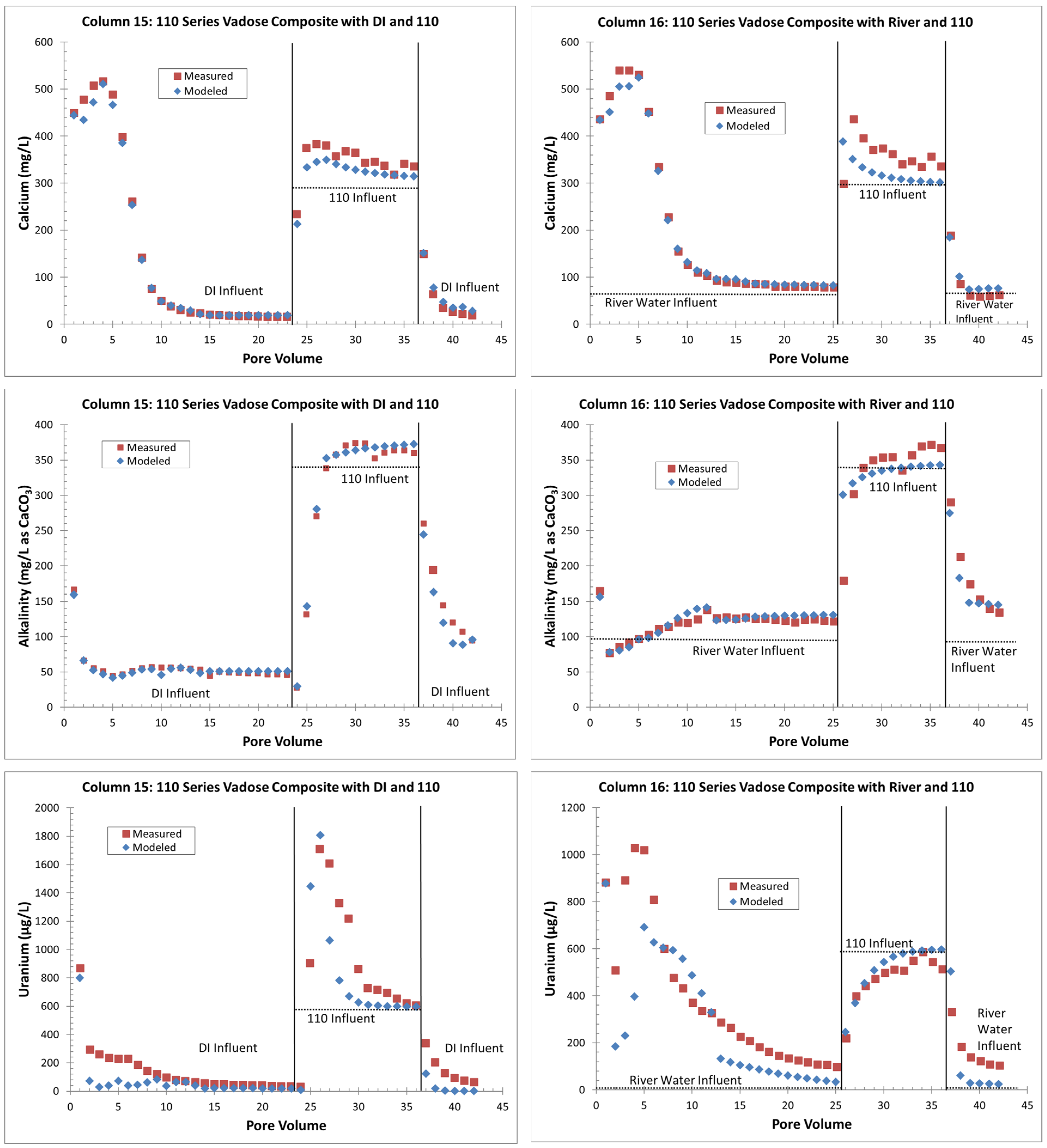
4.1.2. Geochemical Modeling
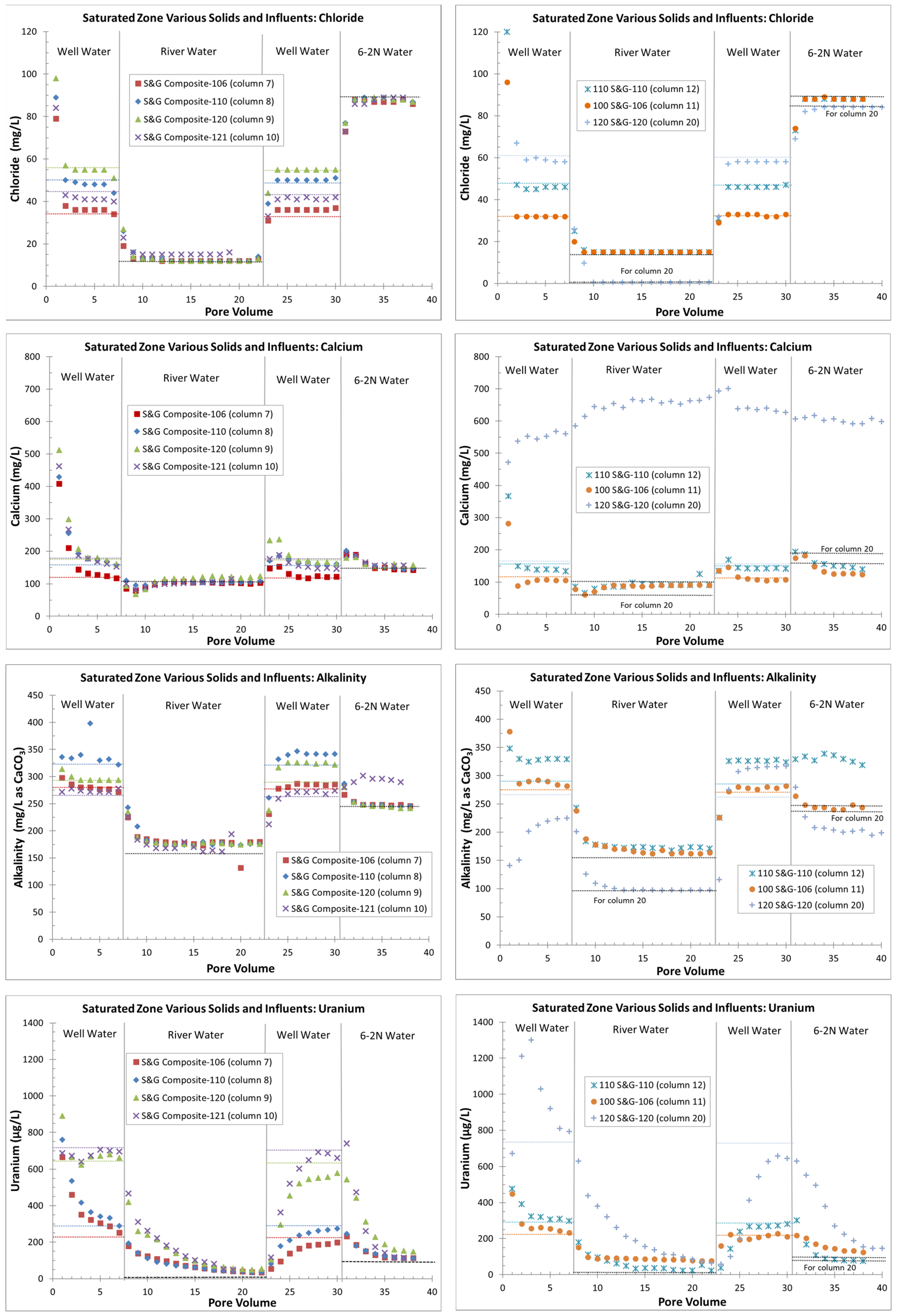
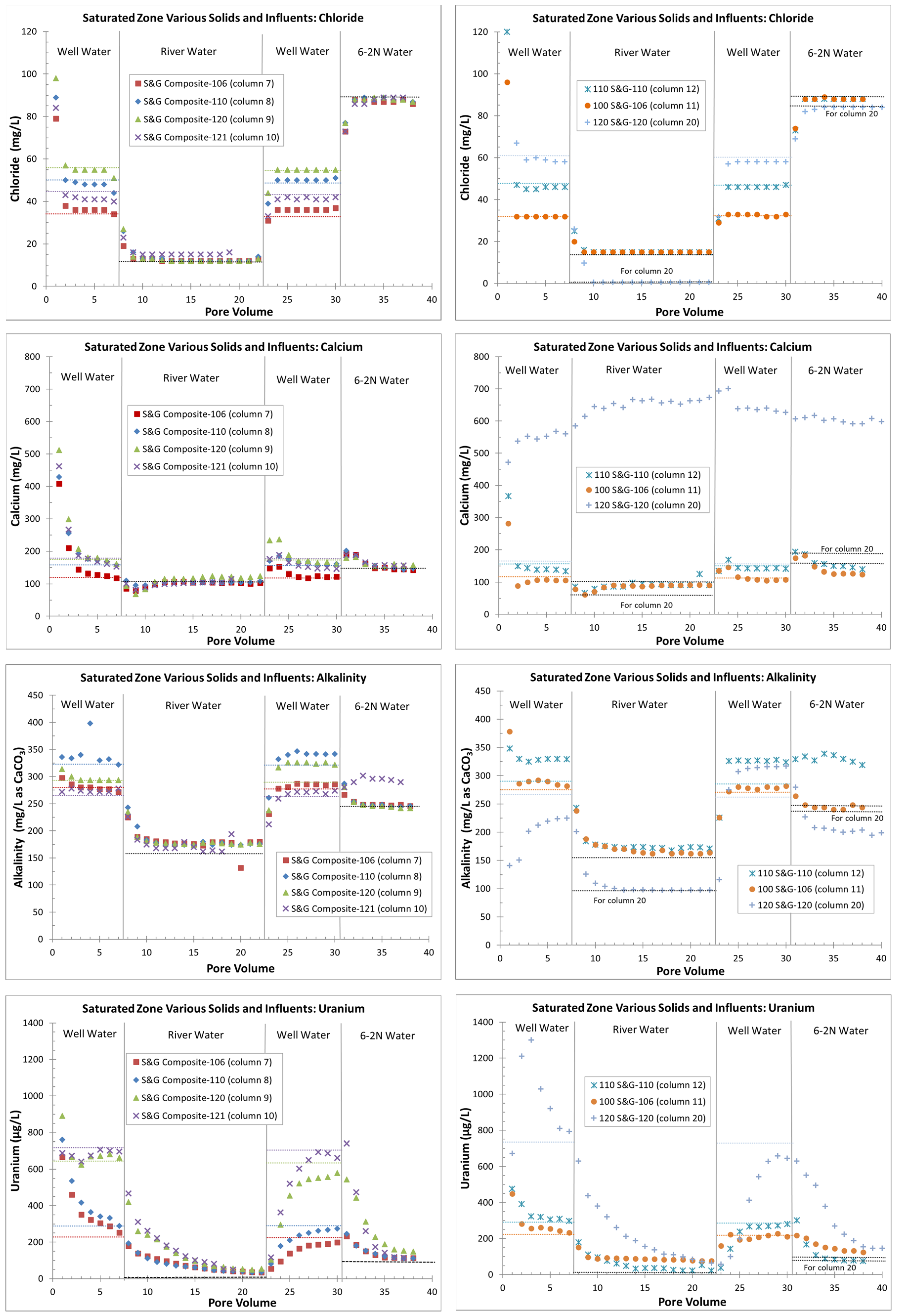
4.2. Saturated Zone (All Continuous Flows)
4.2.1. Laboratory Results
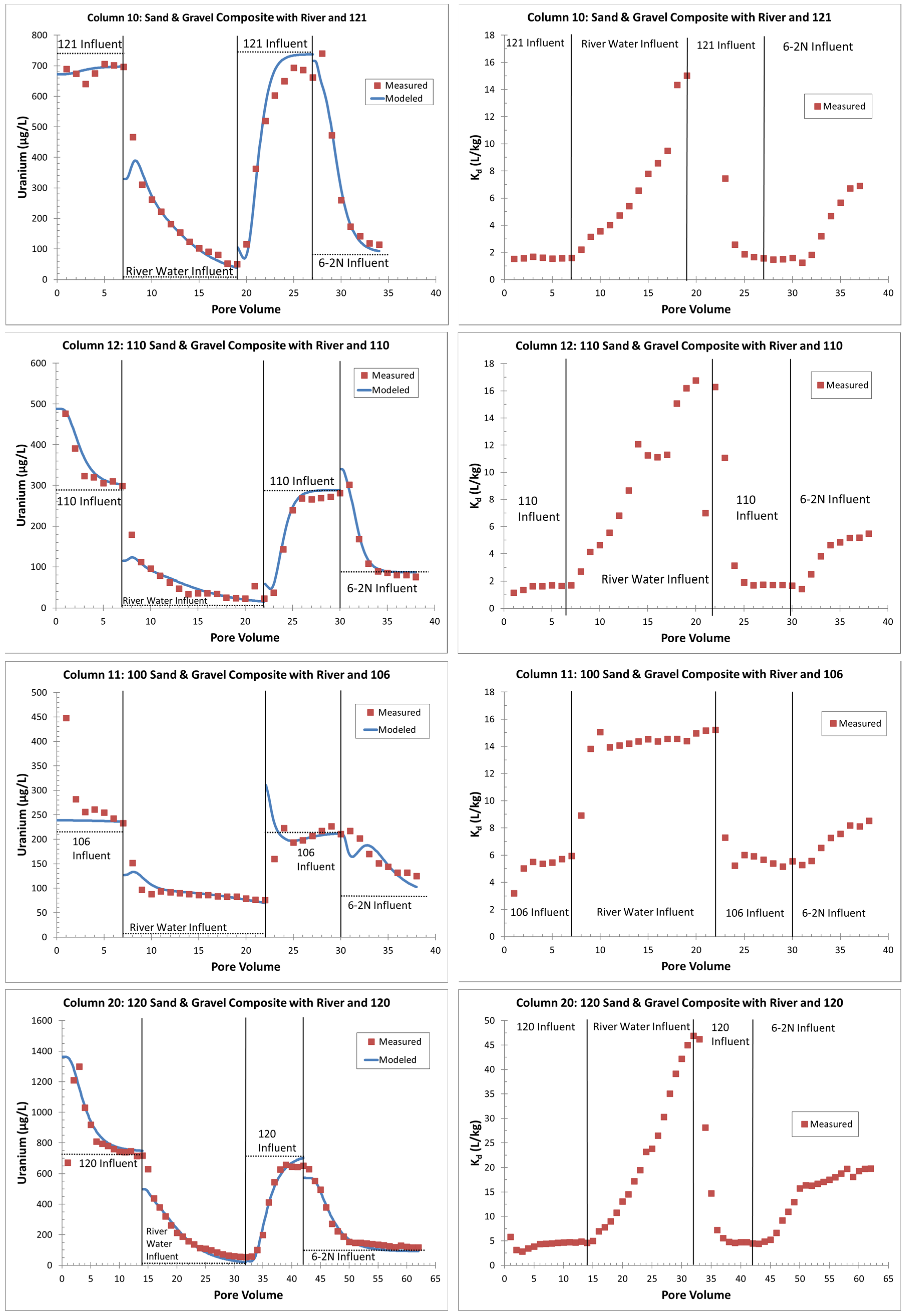
4.2.2. Geochemical Modeling and Sorption Coefficients
5. Conclusions and Practical Implications
5.1. Vadose-Zone Columns
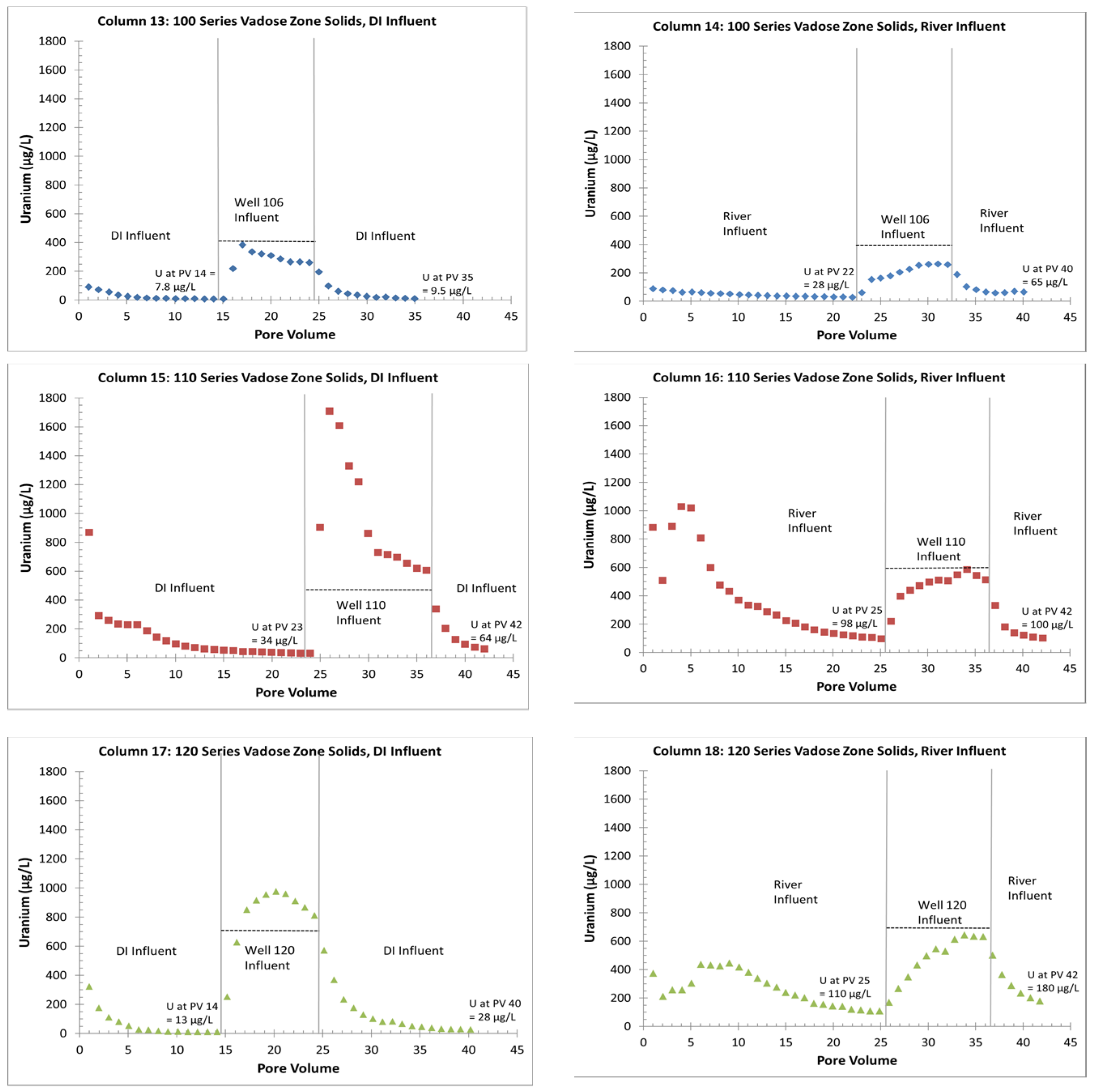
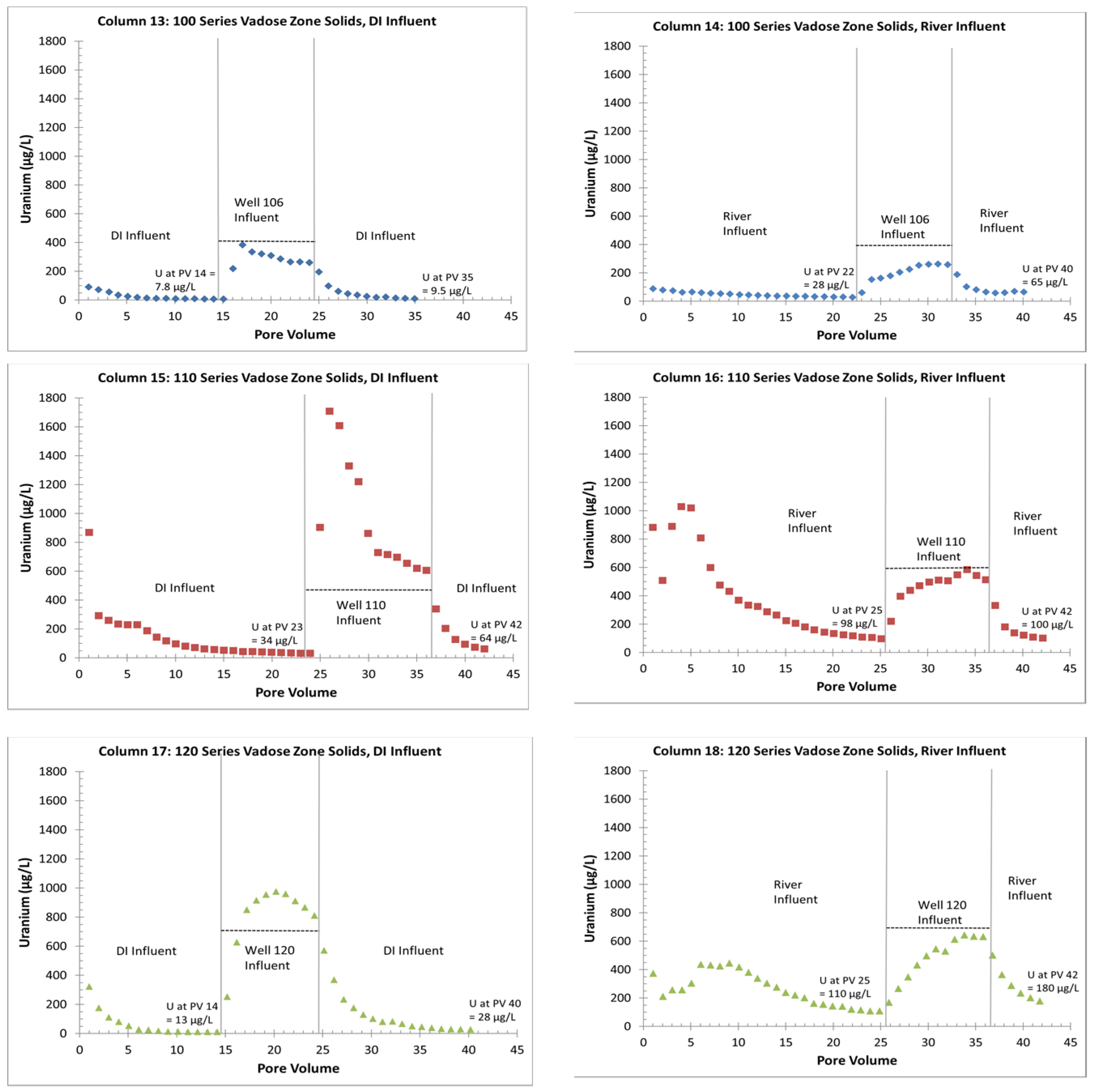
- The uranium release concentrations from the VZ would be lower from a large precipitation event (DI water column influent) than with a flooding event (river-water column influent). Similarly, flooding events would continue to be more efficient for removing uranium for a longer time (more PVs) than precipitation events. This is based on columns with the same solid-phase material releasing more uranium with the river-water influent than with the DI influent (Figure 4 and Figure 5);
- Without flooding events, more uranium is retained on the solid phase with just precipitation events. If a later rise in the water table creates contact between the underlying groundwater and the VZ, large uranium releases are possible (up to three times greater than the groundwater concentrations). The reverse is true with continuous flooding events that have removed solid-phase uranium from the VZ; during a water table rise, uranium is removed from the water phase to the solid phase by sorption reactions (Figure 4 and Figure 6);
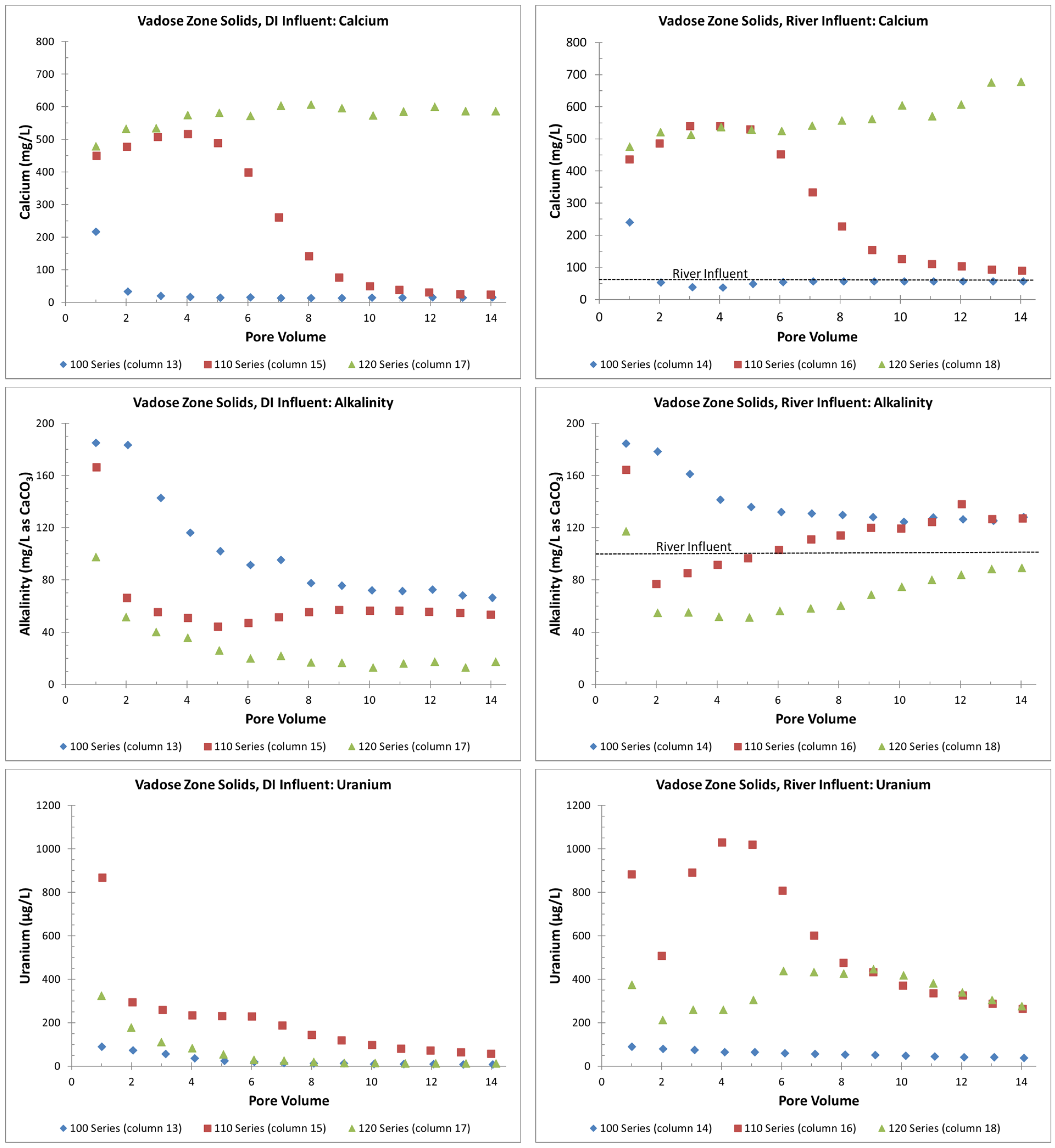
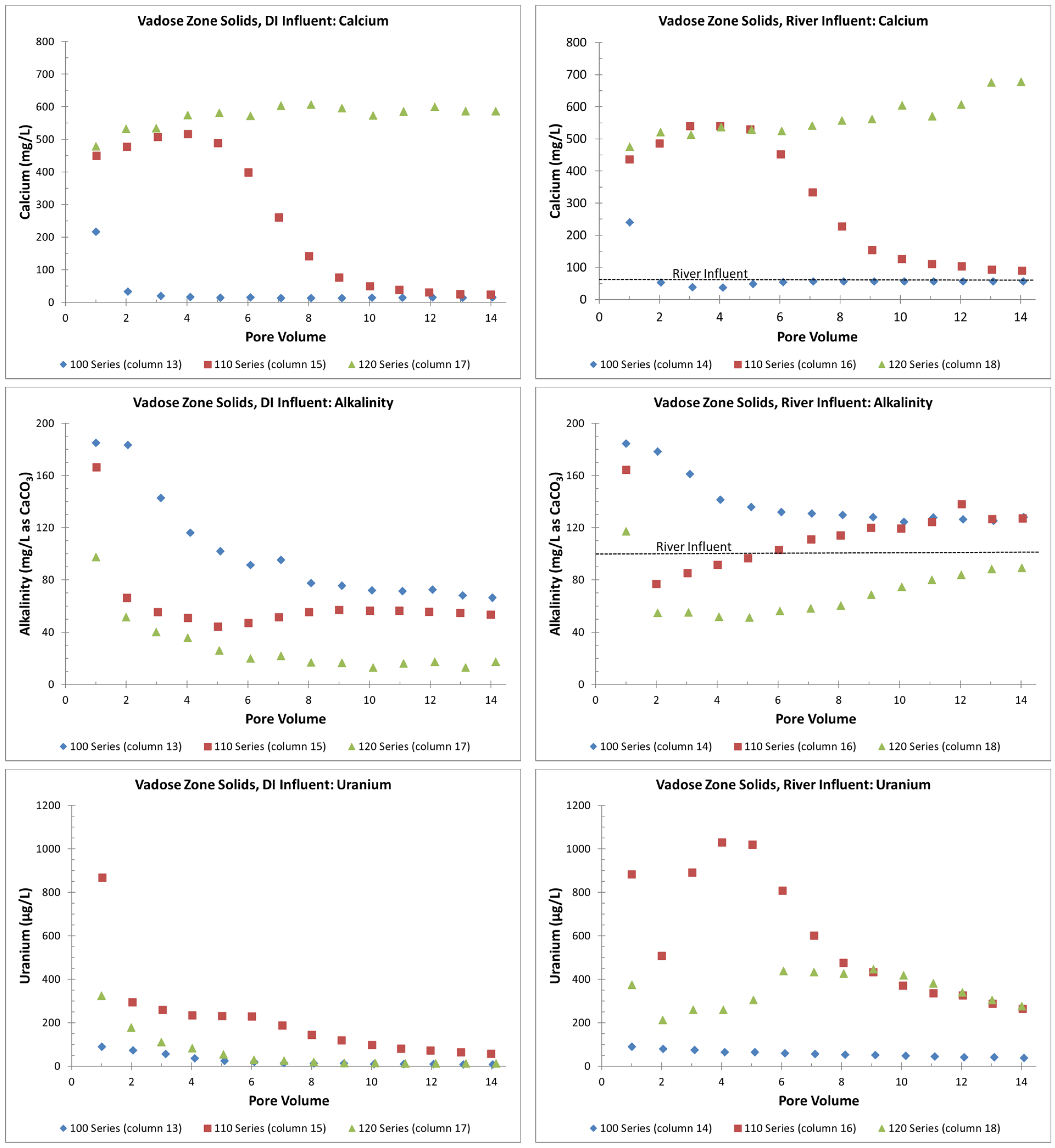
- The presence of gypsum can suppress alkalinity concentrations and lower initial uranium release rates during precipitation or flooding events (Figure 7). However, this involves a complex interplay between alkalinity concentrations and the possible release of uranium incorporated in the gypsum or other grain coatings. This is a topic of ongoing research;
- Overall, higher uranium in the solid phase corresponds to greater uranium release concentrations (Table 2 and Figure 5). Likewise, elevated uranium concentrations in the VZ can contribute uranium to the underlying groundwater at concentrations well above typical standards for an extended time period (greater than 25 PVs of continuous flow for a river water influent, Figure 4). Actual uranium removal from the VZ requires a consideration of the frequency and release rates from flushing events (such as high precipitation, flooding, or water table rise) compared to uranium emplacement events (such as the sorption of uranium from contaminated groundwater influx with a water table rise or from high evapotranspiration).
5.2. Saturated-Zone Columns
- Zones with higher uranium sorption (e.g., column 11, 100-series sediments with more organics) might delay flushing or other remedial efforts;
- The use of lower-alkalinity river water instead of higher-alkalinity, lower-uranium groundwater may inhibit flushing (there is less uranium desorption, based on higher Kd values (stronger sorption), with the use of river water);
- An apparent increase in Kd values at lower effluent uranium concentrations, typical of the tail end of flushing (seen with river water and background groundwater), might create difficulties in reaching cleanup goals;
- Areas with higher amounts of uranium on the solid phase (columns 20 with 120-series area sediments) might require slower flushing rates (lower flow velocities) to avoid kinetic limitations in uranium removal when uranium reactions are not strictly equilibrium-sorption controlled.
5.3. Overall
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DOE (U.S. Department of Energy). Plume Persistence Final Project Report; LMS/ESL/S15233; ESL-RPT-2018-02; Office of Legacy Management: Grand Junction, CO, USA, 2018. Available online: https://www.energy.gov/lm/services/applied-studies-and-technology-ast/ast-reports (accessed on 13 January 2022).
- Dangelmayr, M.A.; Reimus, P.W.; Wasserman, N.L.; Punsal, J.J.; Johnson, R.H.; Clay, J.T.; Stone, J.J. Laboratory column experiments and transport modeling to evaluate retardation of uranium in an aquifer downgradient of a uranium in-situ recovery site. Appl. Geochem. 2017, 80, 1–13. [Google Scholar] [CrossRef]
- DOE (U.S. Department of Energy). Final Site Observational Work Plan for the UMTRA Project Site at Riverton, Wyoming; U0013801; Grand Junction Office: Grand Junction, CO, USA, 1998. Available online: https://lmpublicsearch.lm.doe.gov/SitePages/default.aspx?sitename=Riverton (accessed on 13 January 2022).
- DOE (U.S. Department of Energy). Monticello Mill Tailings Site, Operable Unit III, Interim Remedial Action Progress Report, July 1999–July 2000; GJO-200C163-TAR; Grand Junction Office: Grand Junction, CO, USA, 2000.
- DOE (U.S. Department of Energy). Final Report of the Decontamination and Decommissioning of the Exterior Land Areas at the Grand Junction Projects Office Facility; DOE/ID/12584-220; GJPO-GJ-13; Grand Junction Office: Grand Junction, CO, USA, 1995.
- DOE (U.S. Department of Energy). Final Remedial Investigation/Feasibility Study for the U.S. Department of Energy Grand Junction (Colorado) Projects Office Facility; DOE/ID/12584-16; UNC-GJ-GRAP-1; Grand Junction Office: Grand Junction, CO, USA, 1989.
- DOE (U.S. Department of Energy). Monticello Mill Tailings Site, Operable Unit III, Remedial Investigation; GJO-97-6-TAR; Grand Junction Office: Grand Junction, CO, USA, 1998.
- DOE (U.S. Department of Energy). 2015 Advanced Site Investigation and Monitoring Report Riverton, Wyoming, Processing Site; LMS/RVT/S14148; Office of Legacy Management: Grand Junction, CO, USA, 2016. Available online: https://lmpublicsearch.lm.doe.gov/SitePages/default.aspx?sitename=Riverton (accessed on 13 January 2022).
- DOE (U.S. Department of Energy). Monticello Mill Tailings Site, Operable Unit III, Geochemical Conceptual Site Model Update; LMS/MNT/S26486; Office of Legacy Management: Grand Junction, CO, USA, 2020. Available online: https://lmpublicsearch.lm.doe.gov/SitePages/default.aspx?sitename=Monticello (accessed on 13 January 2022).
- Dam, W.L.; Campbell, S.; Johnson, R.H.; Looney, B.B.; Denham, M.E.; Eddy-Dilek, C.A.; Babits, S.J. Refining the site conceptual model at a former uranium mill site in Riverton, Wyoming, USA. Environ. Earth Sci. 2015, 74, 7255–7265. [Google Scholar] [CrossRef] [Green Version]
- DOE (U.S. Department of Energy). Fact Sheet for the Grand Junction, CO, Site; Office of Legacy Management: Grand Junction, CO, USA, 2020. Available online: https://www.energy.gov/lm/articles/grand-junction-colorado-site-fact-sheet (accessed on 13 January 2022).
- DOE (U.S. Department of Energy). Long-Term Surveillance and Maintenance Plan for the Grand Junction, Colorado, Site; LMS/GJT/S02013-1.0; Office of Legacy Management: Grand Junction, CO, USA, 2021.
- Johnson, R.H.; Hall, S.M.; Tigar, A.D. Using Fission-Track Radiography Coupled with Scanning Electron Microscopy for Efficient Identification of Solid-Phase Uranium Mineralogy at a Former Uranium Pilot Mill (Grand Junction, Colorado). Geosciences 2021, 11, 294. [Google Scholar] [CrossRef]
- DOE (U.S. Department of Energy). Sediment Sampling and Analysis Report for the Grand Junction, Colorado, Office Facility; GJO–2002-288-TAR; Grand Junction Office: Grand Junction, CO, USA, 2002.
- Paradis, C.J.; Johnson, R.H.; Tigar, A.D.; Sauer, K.B.; Marina, O.C.; Reimus, P.W. Field experiments of surface water to groundwater recharge to characterize the mobility of uranium and vanadium at a former mill tailing site. J. Contam. Hydrol. 2020, 229, 103581. [Google Scholar] [CrossRef] [PubMed]
- DOE (U.S. Department of Energy). Environmental Sciences Laboratory Procedures Manual; LMS/PRO/S04343-5.0; Office of Legacy Management: Grand Junction, CO, USA, 2021.
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. US Geol. Surv. Tech. Methods 2013, 6, 497. Available online: https://pubs.usgs.gov/tm/06/a43/ (accessed on 13 January 2022).
- Davis, J.D.; Meece, D.E.; Kohler, M.; Curtis, G.P. Approaches to surface complexation modeling of uranium (VI) adsorption on aquifer sediments. Geochim. Cosmochim. Acta 2004, 68, 3621–3641. [Google Scholar] [CrossRef]
- Johnson, R.H.; Truax, R.A.; Lankford, D.A.; Stone, J.J. Sorption Testing and Generalized Composite Surface Complexation Models for Determining Uranium Sorption Parameters at a Proposed Uranium in Situ Recovery Site. Mine Water Environ. 2016, 35, 435–446. [Google Scholar] [CrossRef]
- Guillaumont, R.; Fanghanel, T.; Fuger, J.; Grenthe, I.; Neck, V.; Palmer, D.; Rand, M.H. Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium. In Chemical Thermodynamics; OECD Nuclear Energy Agency, Elsevier Science: Amsterdam, The Netherlands, 2003; Volume 5. [Google Scholar]
- Dong, W.; Brooks, S.C. Determination of the Formation Constants of Ternary Complexes of Uranyl and Carbonate with Alkaline Earth Metals (Mg2+, Ca2+, Sr2+, and Ba2+) Using Anion Exchange Method. Environ. Sci. Technol. 2006, 40, 4689–4695. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y. Application of surface complexation modeling on adsorption of uranium at water-solid interface: A review. Environ. Pollut. 2021, 278, 116861. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J. Calibration and Uncertainty Analysis for Complex Environmental Models, PEST: Complete Theory and What It Means for Modeling the Real World; Watermark Numerical Computing: Brisbane, Australia, 2015; Available online: https://pesthomepage.org/pest-book (accessed on 13 January 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Column | Solid Phase | Influent Water Series | Stop Flow | Continuous Flow | Description |
|---|---|---|---|---|---|
| 1 | S&G Composite | 106/GR/106/6-2N | X | Stop-flow column to compare with continuous-flow column 7 | |
| 2 | 100 Series S&G | 106/GR/106/6-2N | X | Stop-flow column to compare with continuous-flow column 11 | |
| 3 | S&G Composite | 110/GR/110/6-2N | X | Stop-flow column to compare with continuous-flow column 8 | |
| 4 | 110 Series S&G | 110/GR/110/6-2N | X | Stop-flow column to compare with continuous-flow column 12 | |
| 5 | S&G Composite | 120/GR/120/6-2N | X | Stop-flow column to compare with continuous-flow column 9 | |
| 6 | S&G Composite | 121/GR/121/6-2N | X | Stop-flow column to compare with continuous-flow column 10 | |
| 7 | S&G Composite | 106/GR/106/6-2N | X | Composite solid phase with well 106 influent water | |
| 8 | S&G Composite | 110/GR/110/6-2N | X | Composite solid phase with well 110 influent water | |
| 9 | S&G Composite | 120/GR/120/6-2N | X | Composite solid phase with well 120 influent water | |
| 10 | S&G Composite | 121/GR/121/6-2N | X | Composite solid phase with well 121 influent water | |
| 11 | 100-Series S&G | 106/GR/106/6-2N | X | 100-series S&G solid phase with well 106 influent water | |
| 12 | 110-Series S&G | 110/GR/110/6-2N | X | 110-series S&G solid phase with well 110 influent water | |
| 13 | 100-Series VZ | DI/106/DI | X | 100-series VZ with DI influent | |
| 14 | 100-Series VZ | GR/106/GR | X | 100-series VZ with river influent | |
| 15 | 110-Series VZ | DI/110/DI | X | 110-series VZ with DI influent | |
| 16 | 110-Series VZ | GR/110/GR | X | 110-series VZ with river influent | |
| 17 | 120-Series VZ | DI/120/DI | X | 120-series VZ with DI influent | |
| 18 | 120-Series VZ | GR/120/GR | X | 120-series VZ with river influent | |
| 19 | 120-Series S&G | 120/GR/120/6-2N | X | Stop-flow column to compare with continuous-flow column 20 | |
| 20 | 120-Series S&G | 120/GR/120/6-2N | X | 120 S&G solid phase with well 120 influent water |
| Description | Column Number | Uranium, 5% Nitric Acid Extraction, Precolumn mg/kg | Remaining Uranium, Calculated from Effluent, at 13 PVs mg/kg | Remaining Uranium, Calculated from Effluent, Postcolumn mg/kg | Remaining Uranium, 5% Nitric Acid Extraction, Postcolumn mg/kg |
|---|---|---|---|---|---|
| 100-series VZ solids, DI influent | 13 | 0.94 | 0.49 | 1.0 | 0.96 |
| 100-series VZ solids, river influent | 14 | 0.94 | 0.40 | 1.1 | 1.0 |
| 110-series VZ solids, DI influent | 15 | 4.2 | 3.1 | 1.7 | 1.5 |
| 110-series VZ solids, river influent | 16 | 4.2 | 1.8 | 2.1 | 1.8 |
| 120-series VZ solids, DI influent | 17 | 3.8 | 3.3 | 1.8 | 2.3 |
| 120-series VZ solids, river influent | 18 | 3.8 | 2.4 | 2.8 | 2.5 |
| Description | Column Number | Uranium, Influent Water µg/L | Uranium, 5% Nitric Extraction, Precolumn mg/kg | Remaining Uranium, Calculated from Effluent, Postcolumn mg/kg | Remaining Uranium, 5% Nitric Extraction, Postcolumn mg/kg |
|---|---|---|---|---|---|
| S&G Composite—106 | 7 | 220 | 1.0 | 0.69 | 0.82 |
| S&G Composite—110 | 8 | 290 | 1.0 | 0.69 | 0.69 |
| S&G Composite—120 | 9 | 660 | 1.0 | 0.70 | 0.80 |
| S&G Composite—121 | 10 | 750 | 1.0 | 0.79 | 0.83 |
| 110 Series S&G—110 | 12 | 290 | 0.59 | 0.42 | 0.46 |
| 100 Series S&G—106 | 11 | 210 | 1.5 | 1.1 | 1.0 |
| 120 Series S&G—120 | 20 | 720 | 3.9 | 2.5 | 2.3 |
| Gunnison River water for columns 7–9 | n/a | 7.2 | n/a | n/a | n/a |
| Gunnison River water for columns 10–12 | n/a | 7.4 | n/a | n/a | n/a |
| Gunnison River water for column 20 | n/a | 3.1 | n/a | n/a | n/a |
| 6-2N groundwater for columns 7–9 | n/a | 86 | n/a | n/a | n/a |
| 6-2N groundwater for columns 10–12 | n/a | 87 | n/a | n/a | n/a |
| 6-2N groundwater for column 20 | n/a | 92 | n/a | n/a | n/a |
| Description | Column Number | PHREEQC Strong Sorption Site Density (GC_s) moles/kg water | PHREEQC Super-Strong Sorption Site Density (GC_ss) moles/kg water | Measured Kd at Last PV before River Influent L/kg |
|---|---|---|---|---|
| S&G Composite—106 | 7 | 3.0 × 10−3 | 2.0 × 10−4 | 3.4 |
| S&G Composite—110 | 8 | 3.5 × 10−3 | 9.5 × 10−5 | 3.0 |
| S&G Composite—120 | 9 | 1.5 × 10−3 | 2.0 × 10−4 | 1.5 |
| S&G Composite—121 | 10 | 1.1 × 10−3 | 1.0 × 10−4 | 1.6 |
| 110 Series S&G—110 | 12 | 1.6 × 10−3 | 1.0 × 10−4 | 1.7 |
| 100 Series S&G—106 | 11 | 1.0 × 10−3 | 2.9 × 10−4 | 5.9 |
| 120 Series S&G—120 | 20 | 1.6 × 10−3 | 1.5 × 10−4 | 4.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, R.H.; Tigar, A.D.; Richardson, C.D. Column-Test Data Analyses and Geochemical Modeling to Determine Uranium Reactive Transport Parameters at a Former Uranium Mill Site (Grand Junction, Colorado). Minerals 2022, 12, 438. https://doi.org/10.3390/min12040438
Johnson RH, Tigar AD, Richardson CD. Column-Test Data Analyses and Geochemical Modeling to Determine Uranium Reactive Transport Parameters at a Former Uranium Mill Site (Grand Junction, Colorado). Minerals. 2022; 12(4):438. https://doi.org/10.3390/min12040438
Chicago/Turabian StyleJohnson, Raymond H., Aaron D. Tigar, and C. Doc Richardson. 2022. "Column-Test Data Analyses and Geochemical Modeling to Determine Uranium Reactive Transport Parameters at a Former Uranium Mill Site (Grand Junction, Colorado)" Minerals 12, no. 4: 438. https://doi.org/10.3390/min12040438
APA StyleJohnson, R. H., Tigar, A. D., & Richardson, C. D. (2022). Column-Test Data Analyses and Geochemical Modeling to Determine Uranium Reactive Transport Parameters at a Former Uranium Mill Site (Grand Junction, Colorado). Minerals, 12(4), 438. https://doi.org/10.3390/min12040438