
Atomic Model of Gold Adsorption onto the Pyrite Surface with DFT Study

Abstract
:1. Introduction
2. Calculational Methods and Models
2.1. Computational Methods
2.2. Effect of Surface Energy
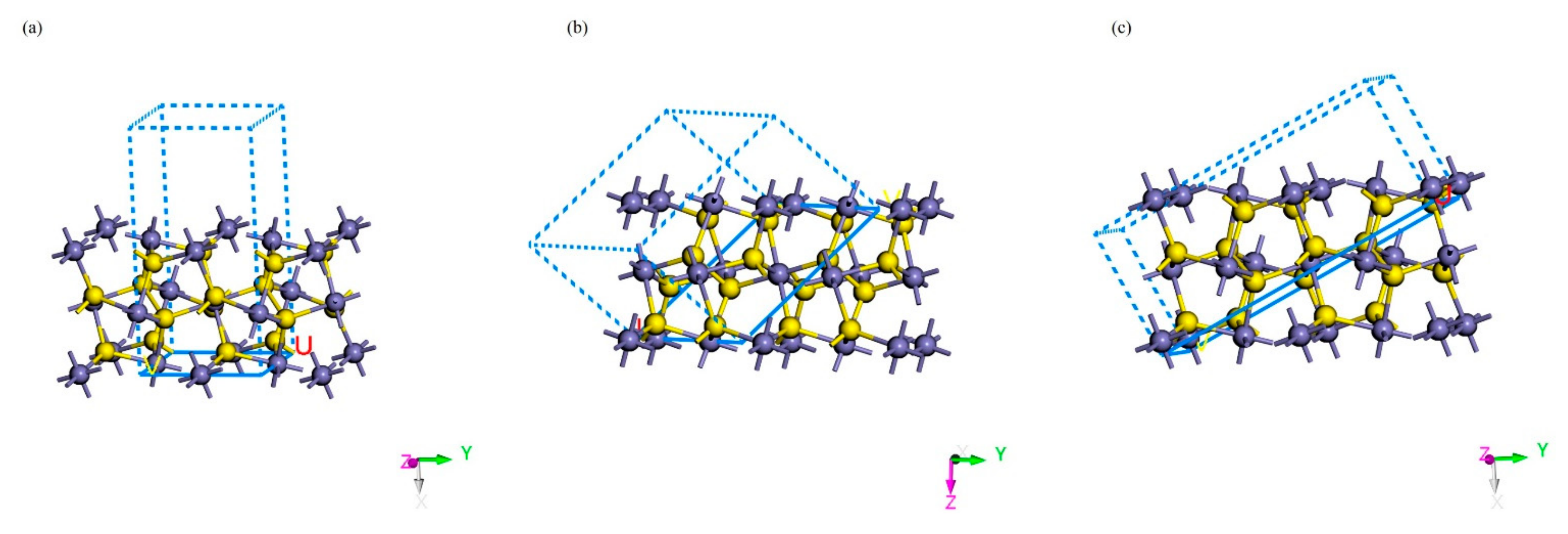
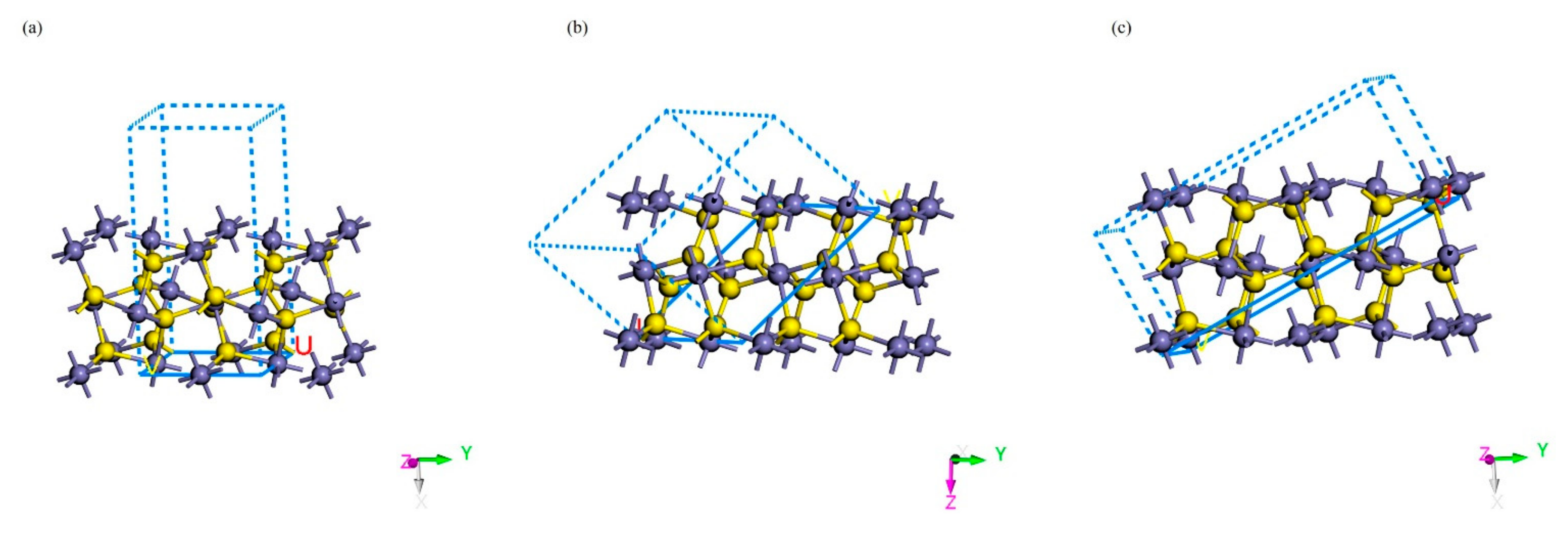
2.3. Adsorption Forms of Gold on the Surface of Pyrite
3. Results and Discussion
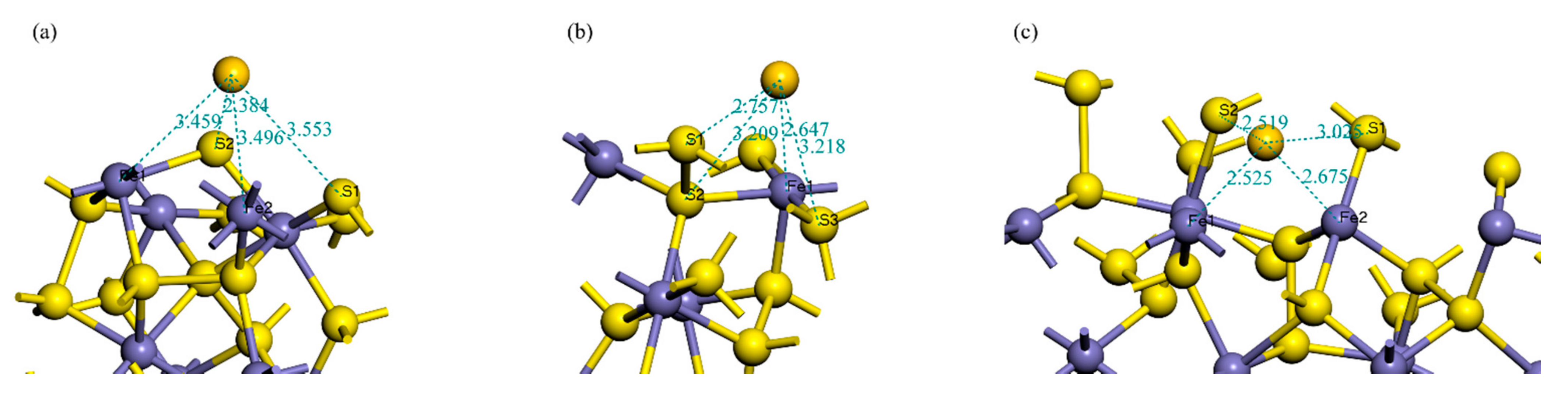
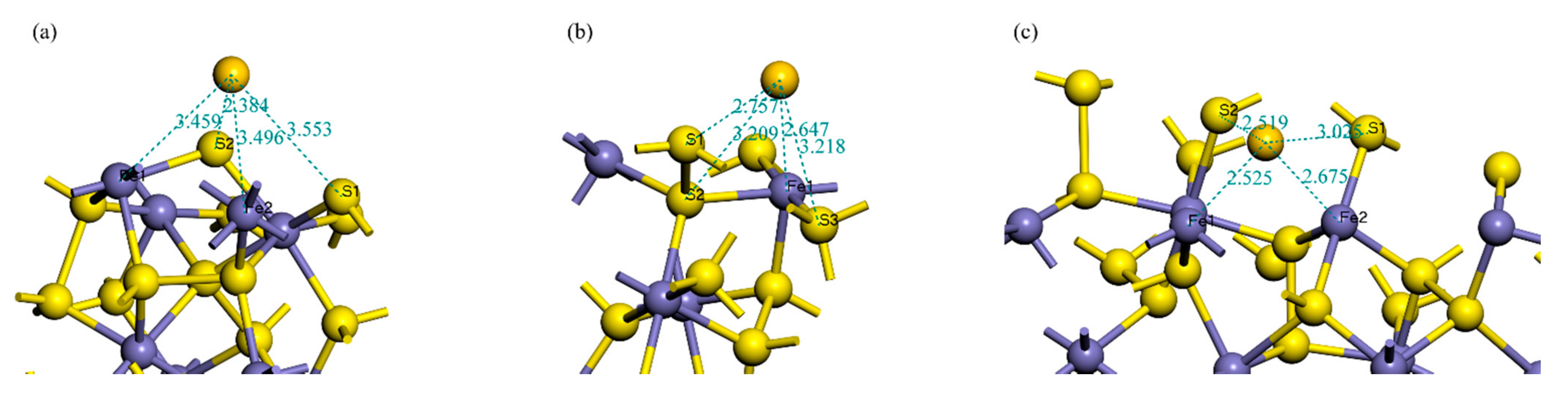
3.1. Adsorption Energy and Stable Adsorption Configuration
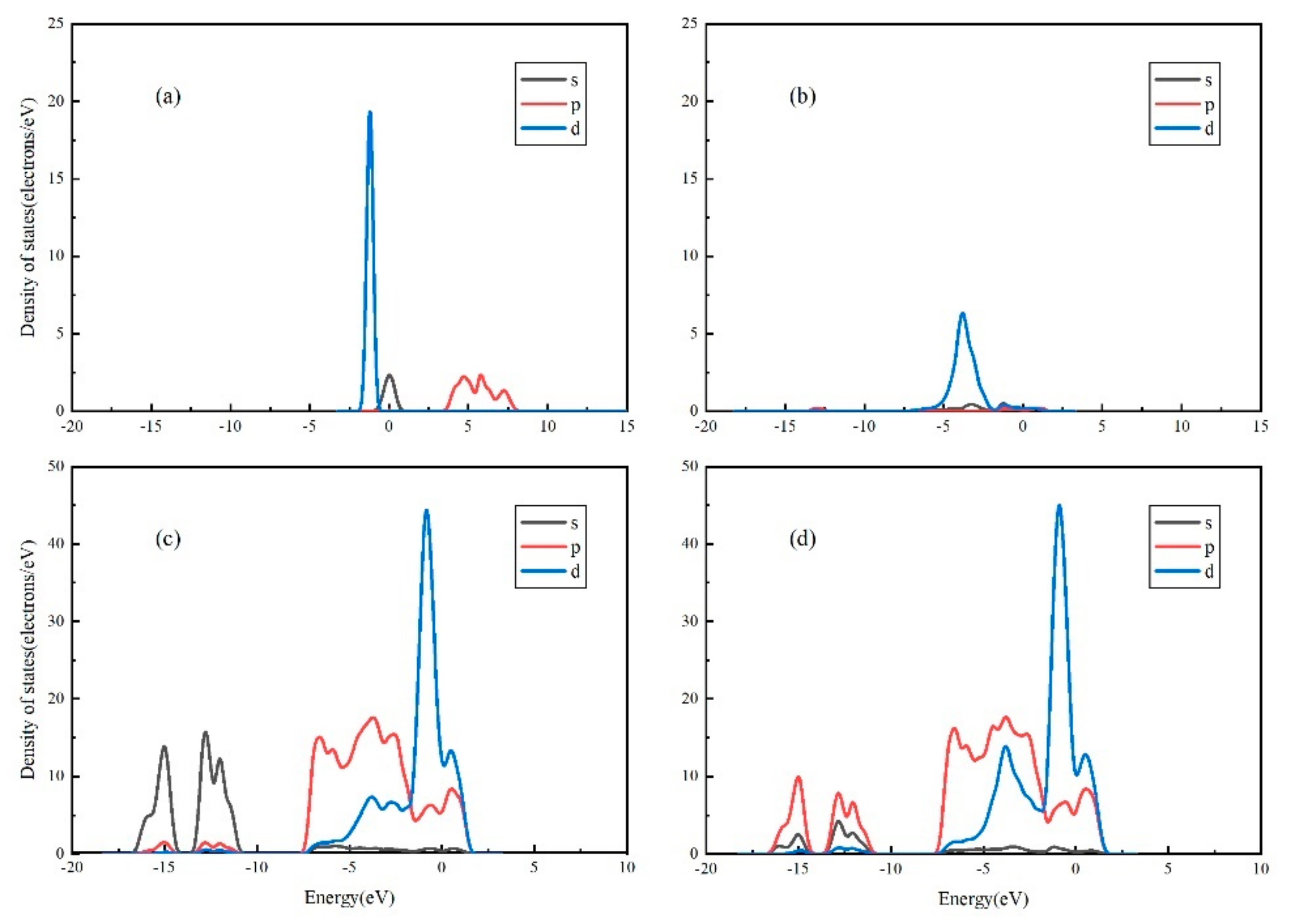
3.2. Surface Atoms Mulliken Analysis
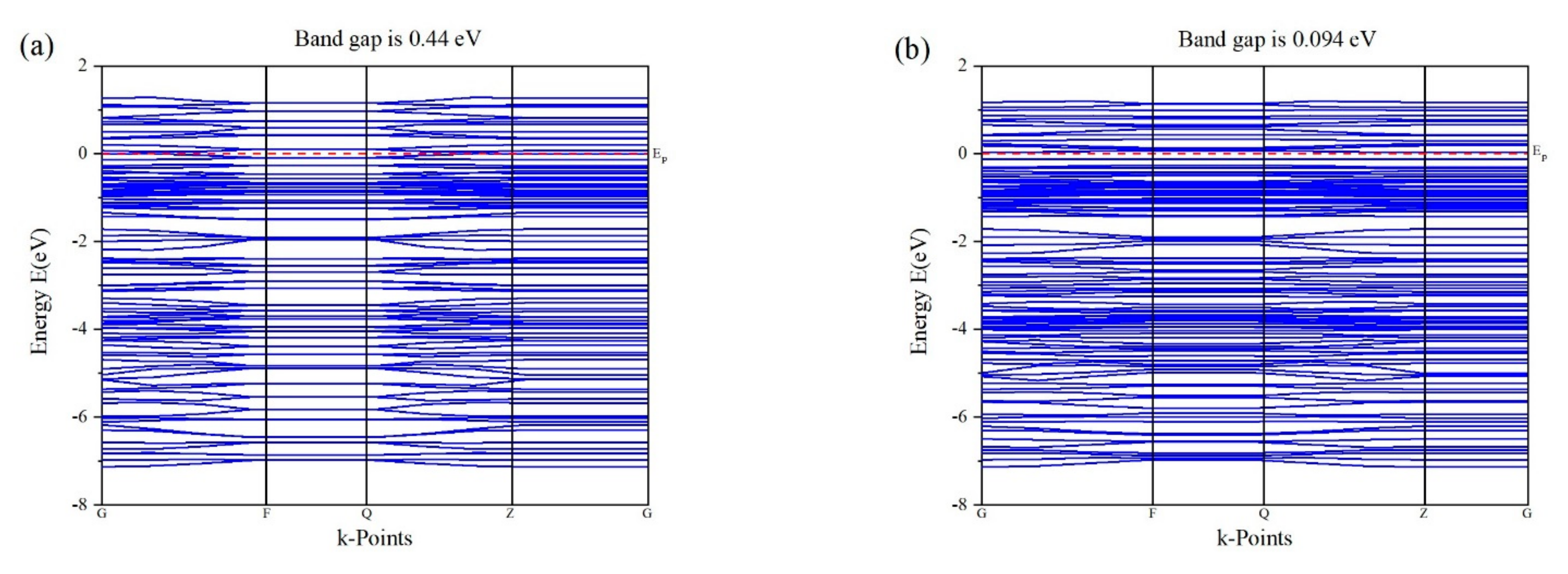
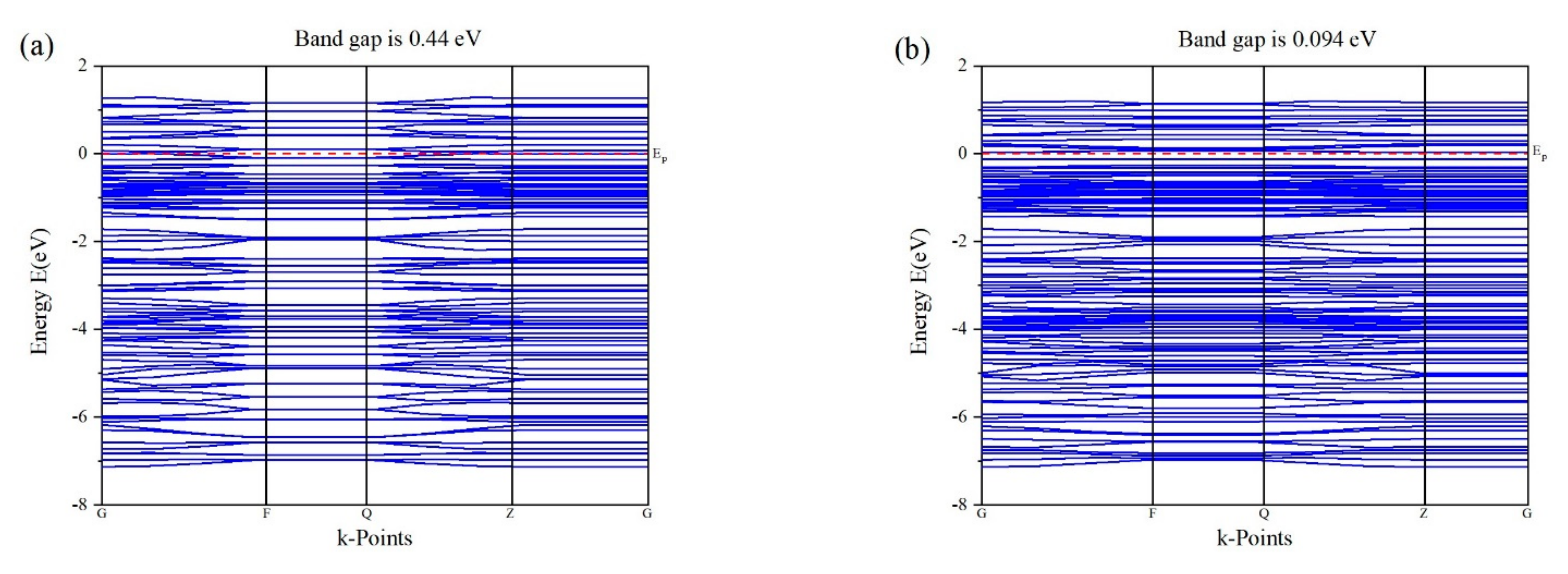
3.3. Band Structure of Au Pyrite (210) Surface Adsorption
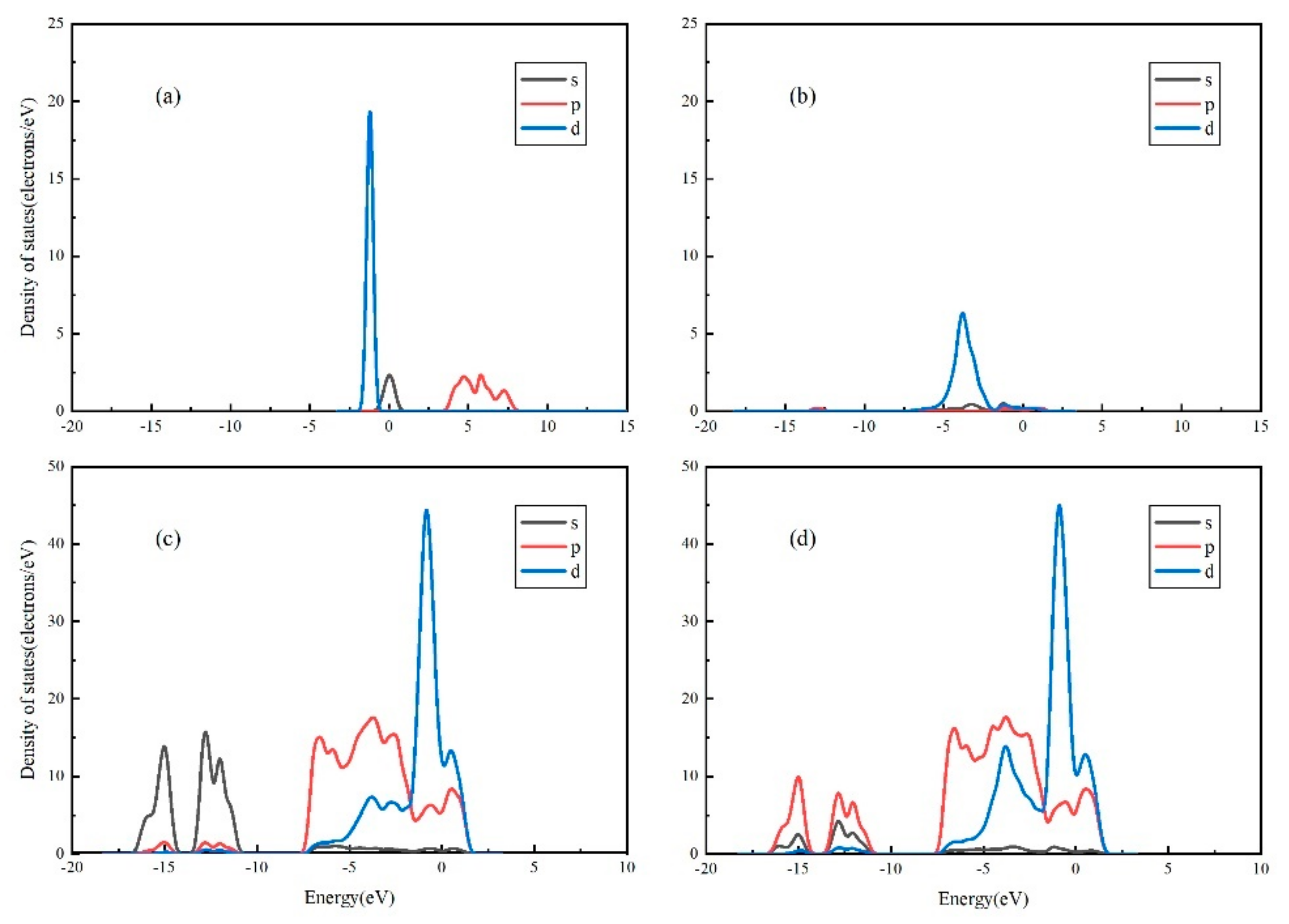
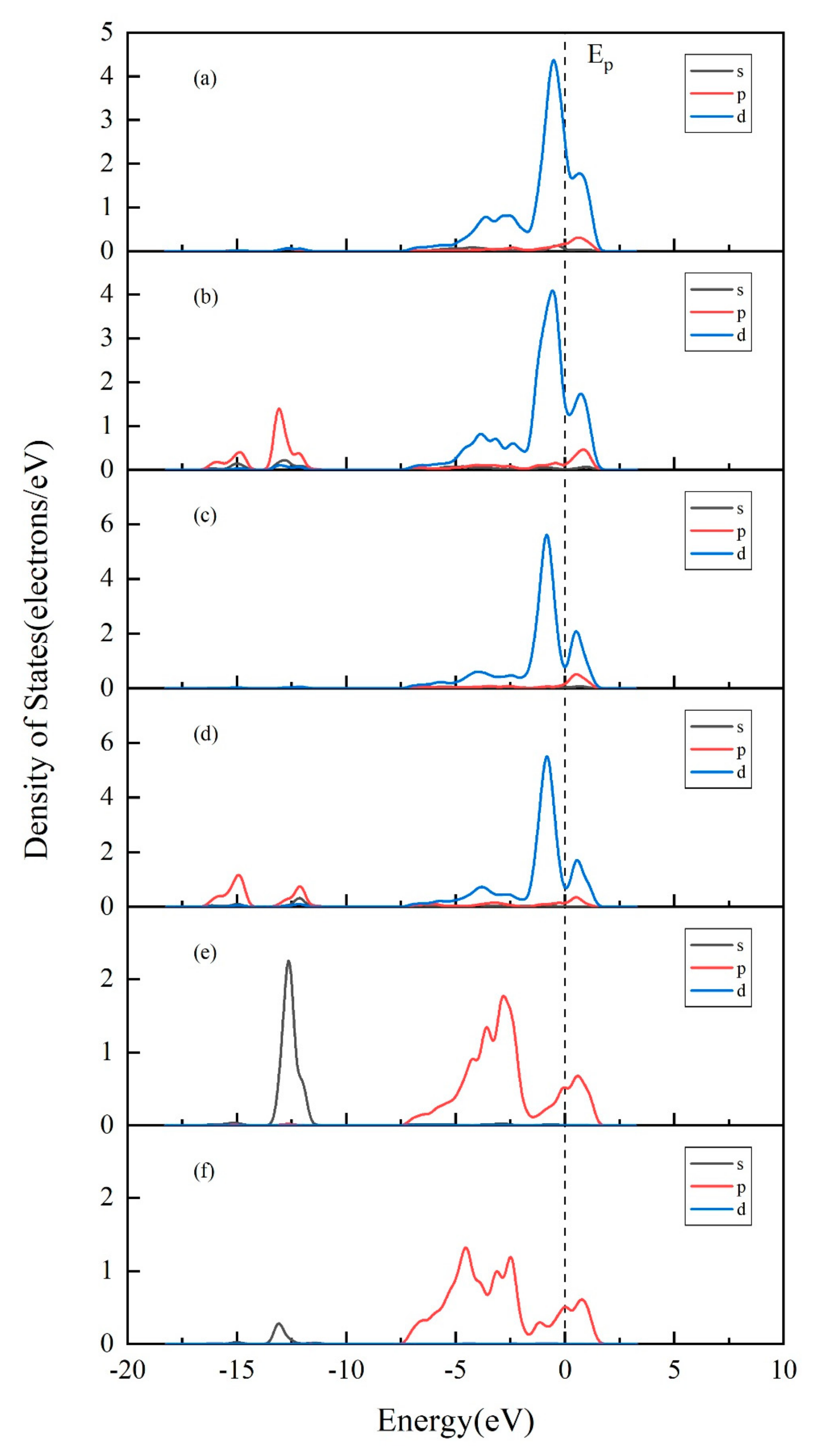
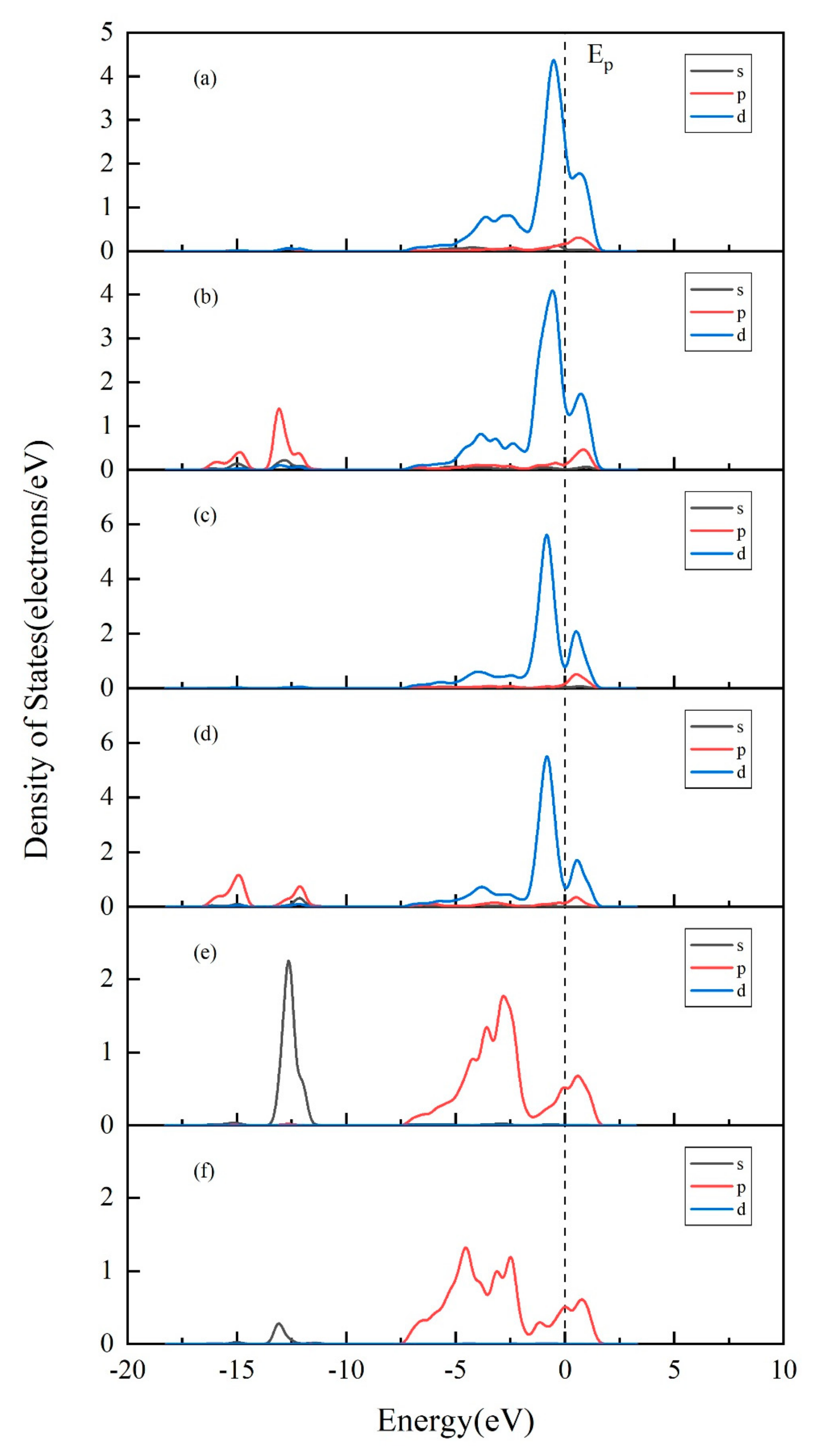
3.4. Projected Density of States (PDOS) Analysis of Au Pyrite (210) Surface Adsorption
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mycroft, J.R.; Bancroft, G.M.; McIntyre, N.S.; Lorimer, J.W. Spontaneous Deposition of Gold on Pyrite from Solutions Containing Au (III) and Au (I) Chlorides. Part I: A Surface Study. Geochim. Cosmochim. Acta 1995, 59, 3351–3365. [Google Scholar] [CrossRef]
- Mikhlin, Y.L.; Romanchenko, A.S. Gold Deposition on Pyrite and the Common Sulfide Minerals: An STM/STS and SR-XPS Study of Surface Reactions and Au Nanoparticles. Geochim. Cosmochim. Acta 2007, 71, 5985–6001. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Pirajno, F.; Qi, J.-P. Origin of Gold Metallogeny and Sources of Ore-Forming Fluids, Jiaodong Province, Eastern China. Int. Geol. Rev. 2005, 47, 530–549. [Google Scholar] [CrossRef]
- Song, M.; Li, S.; Santosh, M.; Zhao, S.; Yu, S.; Yi, P.; Cui, S.; Lv, G.; Xu, J.; Song, Y.; et al. Types, Characteristics and Metallogenesis of Gold Deposits in the Jiaodong Peninsula, Eastern North China Craton. Ore Geol. Rev. 2015, 65, 612–625. [Google Scholar] [CrossRef]
- Groves, D.I.; Goldfarb, R.J.; Santosh, M. The Conjunction of Factors That Lead to Formation of Giant Gold Provinces and Deposits in Non-Arc Settings. Geosci. Front. 2016, 7, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Reich, M.; Kesler, S.E.; Utsunomiya, S.; Palenik, C.S.; Chryssoulis, S.L.; Ewing, R.C. Solubility of Gold in Arsenian Pyrite. Geochim. Cosmochim. Acta 2005, 69, 2781–2796. [Google Scholar] [CrossRef]
- Spence-Jones, C.P.; Jenkin, G.R.T.; Boyce, A.J.; Hill, N.J.; Sangster, C.J.S. Tellurium, Magmatic Fluids and Orogenic Gold: An Early Magmatic Fluid Pulse at Cononish Gold Deposit, Scotland. Ore Geol. Rev. 2018, 102, 894–905. [Google Scholar] [CrossRef]
- Li, Z.-K.; Li, J.-W.; Cooke, D.R.; Danyushevsky, L.; Zhang, L.; O’Brien, H.; Lahaye, Y.; Zhang, W.; Xu, H.-J. Textures, Trace Elements, and Pb Isotopes of Sulfides from the Haopinggou Vein Deposit, Southern North China Craton: Implications for Discrete Au and Ag–Pb–Zn Mineralization. Contrib. Mineral. Petrol. 2016, 171, 99. [Google Scholar] [CrossRef]
- Li, R.; Chen, H.; Xia, X.; Yang, Q.; Danyushevsky, L.V.; Lai, C. Using Integrated In-Situ Sulfide Trace Element Geochemistry and Sulfur Isotopes to Trace Ore-Forming Fluids: Example from the Mina Justa IOCG Deposit (Southern Perú). Ore Geol. Rev. 2018, 101, 165–179. [Google Scholar] [CrossRef]
- Tauson, V.L.; Kravtsova, R.G.; Smagunov, N.V.; Spiridonov, A.M.; Grebenshchikova, V.I.; Budyak, A.E. Structurally and Superficially Bound Gold in Pyrite from Deposits of Different Genetic Types. Russ. Geol. Geophys. 2014, 55, 273–289. [Google Scholar] [CrossRef]
- Wen, B.-J.; Fan, H.-R.; Hu, F.-F.; Liu, X.; Yang, K.-F.; Sun, Z.-F.; Sun, Z.-F. Fluid Evolution and Ore Genesis of the Giant Sanshandao Gold Deposit, Jiaodong Gold Province, China: Constrains from Geology, Fluid Inclusions and H–O–S–He–Ar Isotopic Compositions. J. Geochem. Explor. 2016, 171, 96–112. [Google Scholar] [CrossRef]
- Sung, Y.-H.; Brugger, J.; Ciobanu, C.L.; Pring, A.; Skinner, W.; Nugus, M. Invisible Gold in Arsenian Pyrite and Arsenopyrite from a Multistage Archaean Gold Deposit: Sunrise Dam, Eastern Goldfields Province, Western Australia. Miner. Depos. 2009, 44, 765–791. [Google Scholar] [CrossRef]
- Williams-Jones, A.E.; Bowell, R.J.; Migdisov, A.A. Gold in Solution. Elements 2009, 5, 281–287. [Google Scholar] [CrossRef]
- Su, W.; Xia, B.; Zhang, H.; Zhang, X.; Hu, R. Visible Gold in Arsenian Pyrite at the Shuiyindong Carlin-Type Gold Deposit, Guizhou, China: Implications for the Environment and Processes of Ore Formation. Ore Geol. Rev. 2008, 33, 667–679. [Google Scholar] [CrossRef]
- Widler, A.M.; Seward, T.M. The Adsorption of Gold(I) Hydrosulphide Complexes by Iron Sulphide Surfaces. Geochim. Cosmochim. Acta 2002, 66, 383–402. [Google Scholar] [CrossRef]
- Pokrovski, G.S.; Tagirov, B.R.; Schott, J.; Hazemann, J.-L.; Proux, O. A New View on Gold Speciation in Sulfur-Bearing Hydrothermal Fluids from in Situ X-Ray Absorption Spectroscopy and Quantum-Chemical Modeling. Geochim. Cosmochim. Acta 2009, 73, 5406–5427. [Google Scholar] [CrossRef] [Green Version]
- Vlassopoulos, D.; Wood, S.A. Gold Speciation in Natural Waters: I. Solubility and Hydrolysis Reactions of Gold in Aqueous Solution. Geochim. Cosmochim. Acta 1990, 54, 3–12. [Google Scholar] [CrossRef]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population Analysis of Plane-Wave Electronic Structure Calculations of Bulk Materials. Phys. Rev. B 1996, 54, 16317–16320. [Google Scholar] [CrossRef] [Green Version]
- Segall, M.D.; Pickard, C.J.; Shah, R.; Payne, M.C. Population Analysis in Plane Wave Electronic Structure Calculations. Mol. Phys. 1996, 89, 571–577. [Google Scholar] [CrossRef]
- Blanchard, M.; Alfredsson, M.; Brodholt, J.; Wright, K.; Catlow, C.R.A. Arsenic Incorporation into FeS2 Pyrite and Its Influence on Dissolution: A DFT Study. Geochim. Cosmochim. Acta 2007, 71, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, Y.; Liu, J.; Gao, T.; Guo, Y.; Liu, J.; Shi, Y. First-Principles Investigation of the Effect of Crystal Structure on Sulfur Isotope Fractionation in Sulfide Polymorphs. Eur. J. Mineral. 2018, 30, 1047–1061. [Google Scholar] [CrossRef]
- Hung, A.; Muscat, J.; Yarovsky, I.; Russo, S.P. Density-Functional Theory Studies of Pyrite FeS2(1 0 0) and (1 1 0) Surfaces. Surf. Sci. 2002, 513, 511–524. [Google Scholar] [CrossRef]
- Hung, A.; Muscat, J.; Yarovsky, I.; Russo, S.P. Density-Functional Theory Studies of Pyrite FeS2 (1 1 1) and (2 1 0) Surfaces. Surf. Sci. 2002, 520, 111–119. [Google Scholar] [CrossRef]
- Meng, L.; Huang, F.; Wang, X.; Gao, W.; Zhang, B.; Song, D.; Li, G.; Zhang, B. An Experimental Study of the Morphological Evolution of Pyrite under Hydrothermal Conditions and Its Implications. J. Geochem. Explor. 2020, 219, 106636. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Für Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative Minimization Techniques for Ab Initio Total-Energy Calculations: Molecular Dynamics and Conjugate Gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef] [Green Version]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, A.D. Self-Consistent Field Theory of the Electron Spin Distribution in π-Electron Radicals. Mol. Phys. 1960, 3, 233–252. [Google Scholar] [CrossRef]
- Wu, R.; Zheng, Y.F.; Zhang, X.G.; Sun, Y.F.; Xu, J.B.; Jian, J.K. Hydrothermal Synthesis and Crystal Structure of Pyrite. J. Cryst. Growth 2004, 266, 523–527. [Google Scholar] [CrossRef]
- Zhu, W.; Wu, P. Surface Energetics of Hydroxyapatite: A DFT Study. Chem. Phys. Lett. 2004, 396, 38–42. [Google Scholar] [CrossRef]
- Maddox, L.M.; Bancroft, G.M.; Scaini, M.J.; Lorimer, J.W. Invisible Gold; Comparison of Au Deposition on Pyrite and Arsenopyrite. Am. Mineral. 1998, 83, 1240–1245. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exchange–Correlation Functions/eV | Lattice Parameter/Å | Parameter Deviation Δc/% | Total Energy/eV |
|---|---|---|---|
| LDA-CA-PZ | 5.2566 | 2.9270 | −5696.3077 |
| GGA-PBE | 5.3771 | 0.7017 | −5692.2264 |
| GGA-RPBE | 5.4183 | 0.0591 | −5696.2807 |
| GGA-PW91 | 5.3813 | 0.6242 | −5702.4578 |
| GGA-WC | 5.3209 | 1.7396 | −5683.4191 |
| GGA-PBESOL | 5.3086 | 1.9667 | −5671.2324 |
| Experimental results [38] | 5.4151 |
| Calculation Setting | Lattice Parameter/Å | Parameter Deviation Δc/% | Total Energy/eV | |
|---|---|---|---|---|
| Cut off Energy/eV | 310.0000 | 5.4602 | 0.8329 | −5696.3292 |
| 330.0000 | 5.4214 | 0.1163 | −5696.2412 | |
| 350.0000 | 5.4162 | 0.0203 | −5696.2679 | |
| 370.0000 | 5.4084 | 0.1237 | −5696.2967 | |
| 390.0000 | 5.4080 | 0.1311 | −5696.3132 | |
| k Point | 3 × 3 × 3 | 5.4170 | 0.0351 | −5696.3030 |
| 3 × 4 × 4 | 5.4167 | 0.0295 | −5696.3053 | |
| 3 × 3 × 4 | 5.4168 | 0.0314 | −5696.3023 | |
| Experimental results [38] | 5.4151 |
| Surface | Atomic Layers | Thickness of the Vacuum Layer/Å | Surface Energy/J∙m2 |
|---|---|---|---|
| (1 0 0) | 18 | 10 | 1.0508 |
| (1 1 1) | 15 | 10 | 1.5337 |
| (2 1 0) | 15 | 10 | 1.8255 |
| Surface | Atom | s | p | d | Total | Charge/e | |
|---|---|---|---|---|---|---|---|
| (100) | S1 | Before | 1.85 | 4.23 | 0.00 | 6.08 | −0.08 |
| After | 1.84 | 4.10 | 0.00 | 5.94 | 0.06 | ||
| S2 | Before | 1.81 | 4.16 | 0.00 | 5.98 | 0.02 | |
| After | 1.81 | 4.13 | 0.00 | 5.93 | 0.07 | ||
| S3 | Before | 1.81 | 4.16 | 0.00 | 5.98 | 0.02 | |
| After | 1.80 | 4.10 | 0.00 | 5.90 | 0.10 | ||
| Fe1 | Before | 0.36 | 0.48 | 7.15 | 7.99 | 0.01 | |
| After | 0.37 | 0.79 | 7.09 | 8.24 | −0.24 | ||
| Au | Before | 1.00 | 0.00 | 10.00 | 11.00 | 0.00 | |
| After | 1.19 | 0.09 | 9.81 | 11.08 | −0.08 | ||
| (111) | S1 | Before | 1.89 | 4.31 | 0.00 | 6.20 | −0.20 |
| After | 1.04 | 4.30 | 0.00 | 5.34 | 0.66 | ||
| S2 | Before | 1.89 | 4.31 | 0.00 | 6.20 | −0.20 | |
| After | 1.01 | 4.33 | 0.00 | 5.34 | 0.66 | ||
| Fe1 | Before | 0.38 | 0.48 | 6.98 | 7.84 | 0.16 | |
| After | 0.49 | 1.85 | 7.08 | 9.42 | −1.42 | ||
| Fe2 | Before | 0.38 | 0.48 | 6.98 | 7.84 | 0.16 | |
| After | 0.50 | 1.93 | 7.08 | 9.51 | −1.51 | ||
| Au | Before | 1.00 | 0.00 | 10.00 | 11.00 | 0.00 | |
| After | 0.98 | 0.18 | 9.81 | 10.97 | 0.03 | ||
| (210) | S1 | Before | 1.86 | 4.26 | 0.00 | 6.12 | −0.12 |
| After | 1.40 | 4.19 | 0.00 | 5.59 | 0.41 | ||
| S2 | Before | 1.86 | 4.40 | 0.00 | 6.26 | −0.26 | |
| After | 1.41 | 4.31 | 0.00 | 5.72 | 0.28 | ||
| Fe1 | Before | 0.39 | 0.34 | 7.06 | 7.79 | 0.21 | |
| After | 0.49 | 1.09 | 7.05 | 8.63 | −0.63 | ||
| Fe2 | Before | 0.35 | 0.36 | 7.09 | 7.80 | 0.20 | |
| After | 0.46 | 1.14 | 7.08 | 8.68 | −0.68 | ||
| Au | Before | 1.00 | 0.00 | 10.00 | 11.00 | 0.00 | |
| After | 1.25 | 0.15 | 9.70 | 11.10 | −0.11 |
| Surface | Bond | Population | Bond Length/Å |
|---|---|---|---|
| (100) | Fe–Au | 0.36 | 2.6472 |
| S1–Au | 0.03 | 2.7567 | |
| (111) | S2–Au | 0.41 | 2.3845 |
| (210) | S2–Au | 0.18 | 2.5191 |
| Fe1–Au | 0.56 | 2.5255 | |
| Fe2–Au | 0.22 | 2.6751 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Li, Y.; Cheng, Q.; Zhao, Y. Atomic Model of Gold Adsorption onto the Pyrite Surface with DFT Study. Minerals 2022, 12, 387. https://doi.org/10.3390/min12030387
Liu C, Li Y, Cheng Q, Zhao Y. Atomic Model of Gold Adsorption onto the Pyrite Surface with DFT Study. Minerals. 2022; 12(3):387. https://doi.org/10.3390/min12030387
Chicago/Turabian StyleLiu, Chunlin, Yongbing Li, Qi Cheng, and Yang Zhao. 2022. "Atomic Model of Gold Adsorption onto the Pyrite Surface with DFT Study" Minerals 12, no. 3: 387. https://doi.org/10.3390/min12030387
APA StyleLiu, C., Li, Y., Cheng, Q., & Zhao, Y. (2022). Atomic Model of Gold Adsorption onto the Pyrite Surface with DFT Study. Minerals, 12(3), 387. https://doi.org/10.3390/min12030387