
Surface and Bulk Modifications of Fibrous Erionite in Mimicked Gamble’s Solution at Acidic pH

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Dissolution Experiments
2.3. SEM Investigation
2.4. HR-TEM Investigation
2.5. ICP-OES Investigation
2.6. X-ray Powder Diffraction
2.7. XPS Investigation
3. Results and Discussion
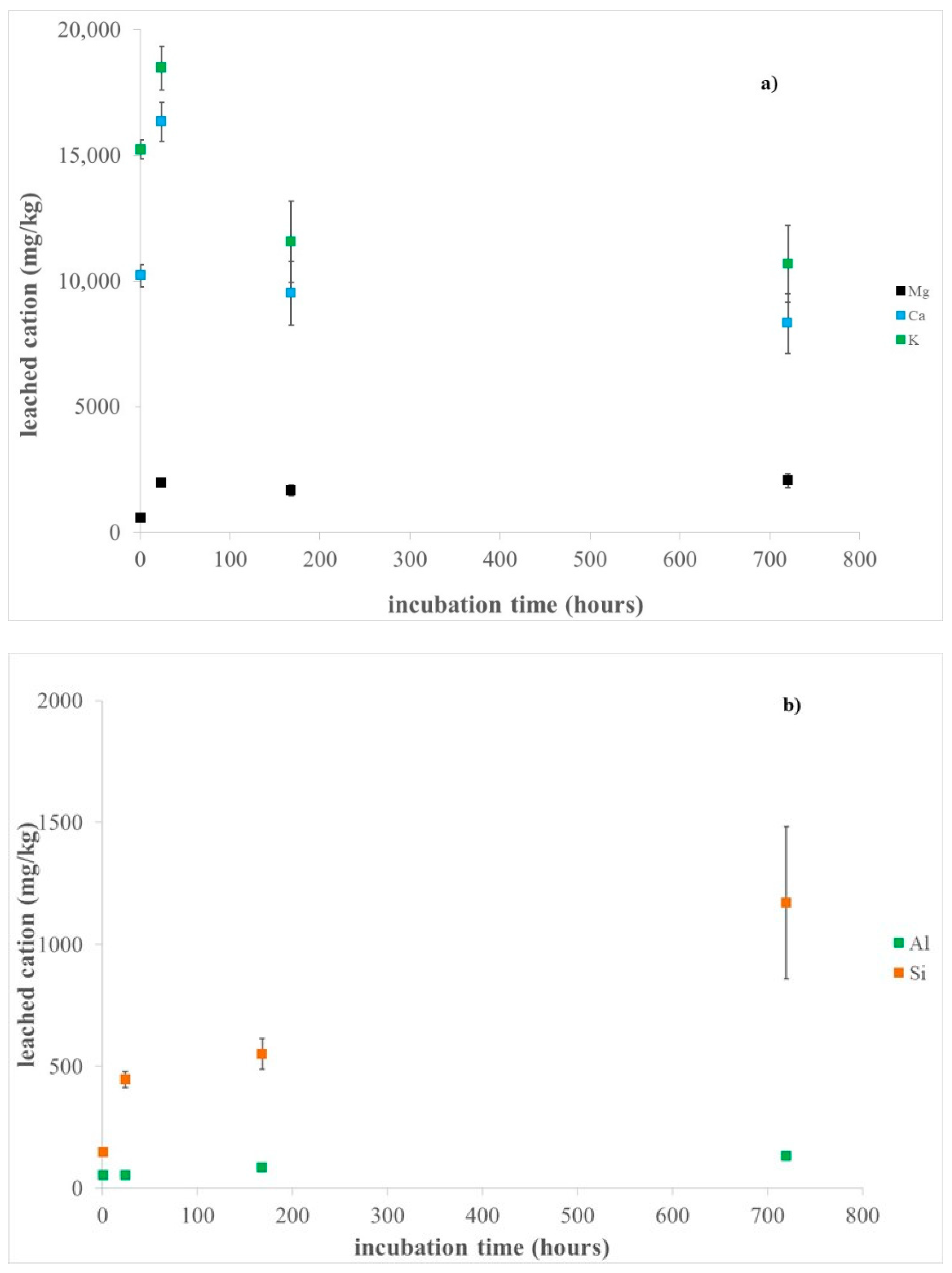
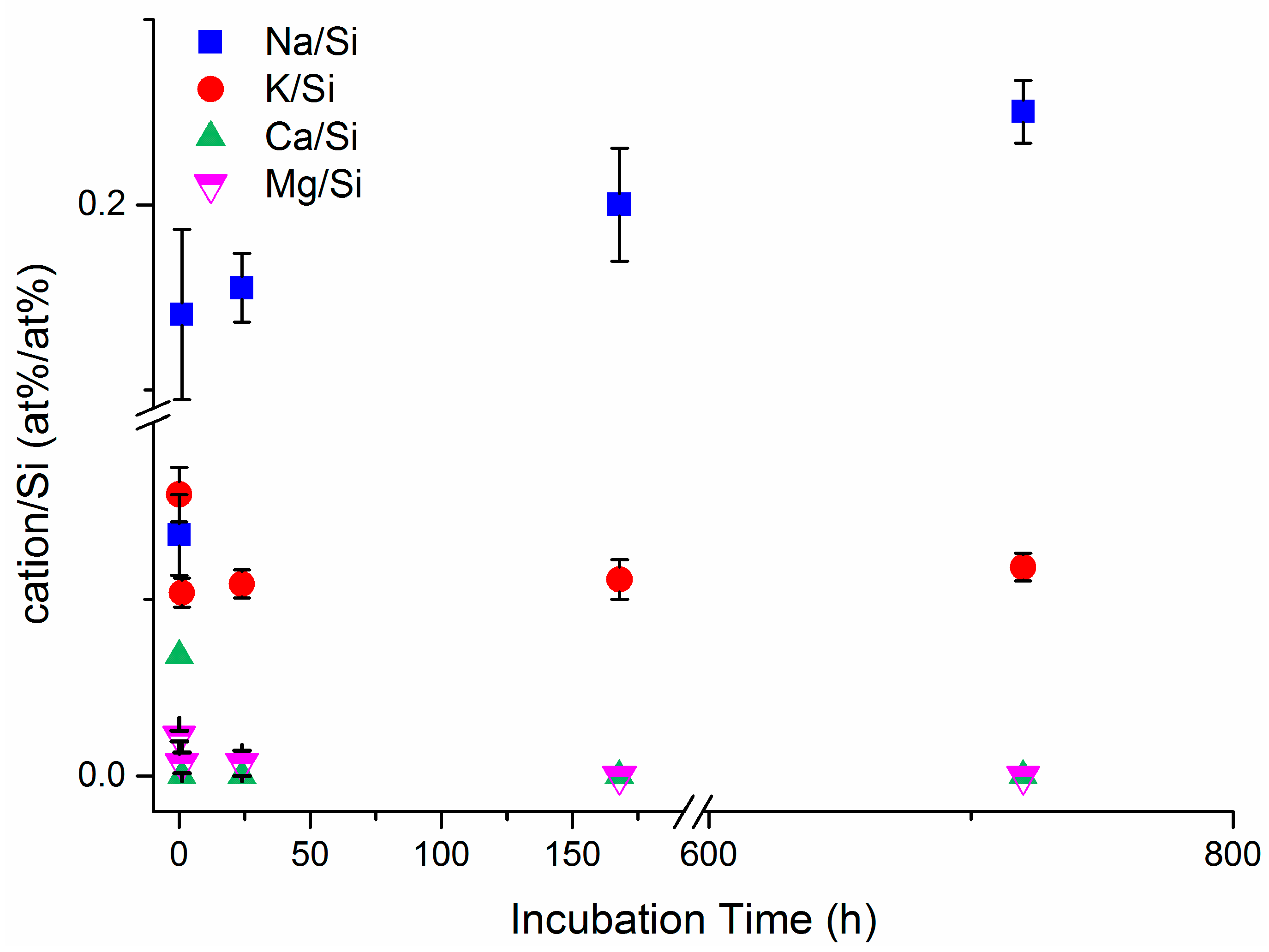
3.1. Erionite Dissolution and Chemical Characterization
3.2. Structural Modifications
3.3. TEM Observations: Nanomorphology of Pristine and E-1M Erionite Fibres
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sheppard, R.A.; Gude, A.I. Zeolites and Associated Authigenic Silicate Minerals in Tuffaceous Rocks of the Big Sandy Formation, Mohave Country, Arizona; U.S. Government Publishing Office: Washington, DC, USA, 1973; Volume 830, p. 36. [Google Scholar]
- Tschernich, R.W. Zeolites of the World; Geoscience Press: Phoenix, AZ, USA, 1992; p. 563. [Google Scholar]
- Bargar, K.E.; Keith, T.E.C. Calcium zeolites in rhyolitic drill cores from Yellowstone National Park. In Natural Zeolites ’93; Ming, D.W., Mumpton, F.S., Eds.; International Committee on Natural Zeolites: Brockport, NY, USA, 1995; pp. 69–86. [Google Scholar]
- Kawahara, A.; Curien, H. La structure cristalline de l′érionite. Bull. Soc. Fr. Minéral. Crist. 1969, 92, 250–256. [Google Scholar]
- Coombs, D.S.; Alberti, A.; Armbruster, T.; Artioli, G.; Colella, C.; Galli, E.; Grice, J.D.; Liebau, F.; Mandarino, J.A.; Minato, H.; et al. Recommended nomenclature for zeolite minerals; report of the Subcommittee on Zeolites of the International Mineralogical Association, Commission on New Minerals and Mineral Names. Can. Mineral. 1997, 35, 1571–1606. [Google Scholar]
- Passaglia, E.; Artioli, G.; Gualtieri, A. Crystal chemistry of the zeolites erionite and offretite. Am. Mineral. 1998, 83, 577–589. [Google Scholar] [CrossRef]
- Gottardi, G.; Galli, E. Natural Zeolites; Springer: Berlin/Heidelberg, Germany, 1985. [Google Scholar]
- Staples, L.W.; Gard, J.A. The fibrous zeolite erionite: Its occurrence, unit cell, and structure. Mineral. Mag. 1959, 32, 261–281. [Google Scholar] [CrossRef]
- Smith, J.V.; Rinaldi, F.; Dent Glasser, L.S. Crystal structures with a chabazite framework. II. Hydrated Ca-chabazite at room temperature. Acta Cryst. 1963, 16, 45–53. [Google Scholar] [CrossRef]
- Rüdinger, B.; Tillmanns, E.; Hentschel, G. Bellbergite—A new mineral with the structure type EAB. Mineral. Petrol. 1993, 48, 147–152. [Google Scholar] [CrossRef]
- Ballirano, P.; Merlino, S.; Bonaccorsi, E.; Maras, A. The crystal structure of liottite, a six-layer member of the cancrinite-group. Can. Mineral. 1996, 34, 1021–1030. [Google Scholar]
- Ballirano, P.; Bloise, A.; Gualtieri, A.F.; Lezzerini, M.; Pacella, A.; Perchiazzi, N.; Dogan, M.; Dogan, A.U. The crystal structure of mineral fibres. In Mineral Fibres: Crystal Chemistry, Chemical-Physical Properties, Biological Interaction and Toxicity; Gualtieri, A.F., Ed.; EMU Notes in Mineralogy; European Mineralogical Union and Mineralogical Society of Great Britain & Ireland: London, UK, 2017; Volume 18, pp. 17–64. [Google Scholar]
- Bariş, Y.I.; Sahin, A.A.; Ozesmi, M.; Kerse, I.; Ozen, E.; Kolacan, B.; Altinörs, M.; Göktepeli, A. An outbreak of pleural mesothelioma and chronic fibrosing pleurisy in the village of Karain/Urgüp in Anatolia. Thorax 1978, 33, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumortier, P.; Coplü, L.; Broucke, I.; Emri, S.; Selcuk, T.; De Maertelaer, V.; De Vuyst, P.; Baris, I. Erionite bodies and fibres in bronchoalveolar lavage fluid (BALF) of residents from Tuzköy, Cappadocia, Turkey. Occup. Environ. Med. 2001, 58, 261–266. [Google Scholar] [CrossRef]
- Metintas, M.; Hillerdal, G.; Metintas, S.; Dumortier, P. Endemic malignant mesothelioma: Exposure to erionite is more important than genetic factors. Arch. Environ. Occup. Health 2010, 65, 86–93. [Google Scholar] [CrossRef]
- Carbone, M.; Baris, I.; Bertino, P.; Brass, B.; Corertpay, S.; Dogan, A.; Gaudino, G.; Jube, S.; Kanodia, S.; Partridge, C.; et al. Erionite exposure in North Dakota and Turkish villages with mesothelioma. Proc. Natl. Acad. Sci. USA 2011, 108, 13618–13623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirer, E.; Ghattas, C.F.; Radwan, M.O.; Elamin, E.M. Clinical and prognostic features of erionite-induced malignant mesothelioma. Yonsei Med. J. 2015, 56, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Ilgren, E.B.; Ortega Brena, M.; Castro Larragoitia, J.; Loustaunau Navarrete, G.; Fuentes Breña, A.; Krauss, E.; Fehér, G. A reconnaissance study of a potential emerging Mexican mesothelioma epidemic due to fibrous zeolite exposure. Indoor Built Environ. 2008, 17, 496–515. [Google Scholar] [CrossRef]
- Kliment, C.R.; Clemens, K.; Oury, T.D. North American erionite-associated mesothelioma with pleural plaques and pulmonary fibrosis: A case report. Int. J. Clin. Exp. Pathol. 2009, 2, 407–410. [Google Scholar]
- Ryan, P.H.; Dihle, M.; Griffin, S.; Partridge, C.; Hilbert, T.J.; Taylor, R.; Adjei, S.; Lockey, J.E. Erionite in road gravel associated with interstitial and pleural changes—An occupational hazard in western United States. J. Occup. Environ. Med. 2011, 53, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Kiefer, M. Erionite—An emerging North American hazard. NIOSH science blog, national institute for occupational safety and health, 2011. Toxicol. Appl. Pharmacol. 2014, 275, 257–264. [Google Scholar]
- Van Gosen, B.S.; Blitz, T.A.; Plumlee, G.S.; Meeker, G.P.; Pierson, M.P. Geologic occurrences of erionite in the United States: An emerging national public health concern for respiratory disease. Environ. Geochem. Health 2013, 35, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Carbone, M. Environmental risk of mesothelioma in the United States: An emerging concern-epidemiological issues. J. Toxicol. Environ. Health B 2016, 19, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Ilgren, E.B.; Kazemian, H.; Hoskins, J.A. Kandovan the next ‘Capadoccia’? A potential public health issue for erionite related mesothelioma risk. Epidemiol. Biostat. Public Health 2015, 12, 1–12. [Google Scholar] [CrossRef]
- Coffin, D.L.; Cook, P.M.; Creason, J.P. Relative mesothelioma induction in rats by mineral fibers: Comparison with residual pulmonary mineral fiber number and epidemiology. Inhal. Toxicol. 1992, 4, 273–300. [Google Scholar] [CrossRef]
- Kokturk, N.; Firat, P.; Akay, H.; Kadilar, C.; Ozturk, C.; Zorlu, F.; Gungen, Y.; Emri, S. Prognostic significance of Bax and Fas ligand in erionite and asbestos induced Turkish malignant pleural mesothelioma. Lung Cancer 2005, 50, 189–198. [Google Scholar] [CrossRef]
- Bertino, P.; Marconi, A.; Palumbo, L.; Bruni, B.M.; Barbone, D.; Germano, S.; Dogan, A.U.; Tassi, G.F.; Porta, C.; Mutti, L.; et al. Erionite and asbestos differently cause transformation of human mesothelial cells. Int. J. Cancer 2007, 121, 2766–2774. [Google Scholar] [CrossRef]
- Zebedeo, C.N.; Davis, C.; Pena, C.; Ng, K.W.; Pfau, J.C. Erionite induces production of autoantibodies and IL-17 in C57BL/6 mice. Toxicol. Appl. Pharmacol. 2014, 275, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Agency for Research on Cancer (IARC). IARC Monographs on the evaluation of the carcinogenic risk of chemicals to humans. Silica Some Silic. 1997, 42, 225–239. [Google Scholar]
- International Agency for Research on Cancer (IARC). IARC Monographs on the evaluation of the carcinogenic risk to humans. Arsen. Met. Fibres Dusts 2011, 100, 311–316. [Google Scholar]
- Eborn, S.K.; Aust, A.E. Effect of iron acquisition on induction of DNA singlestrand breaks by erionite, a carcinogenic mineral fiber. Arch. Biochem. Biophys. 1995, 316, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Fach, E.; Waldman, W.J.; Williams, M.; Long, J.; Meister, R.K.; Dutta, P.K. Analysis of the biological and chemical reactivity of zeolite-based aluminosilicate fibers and particulates. Environ. Health Perspect. 2002, 110, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fach, E.; Kristovich, R.; Long, J.; Waldman, W.J.; Dutta, P.K.; Williams, M. The effect of iron on the biological activities of erionite and mordenite. Environ. Int. 2003, 29, 451–458. [Google Scholar] [CrossRef]
- Ballirano, P.; Pacella, A.; Cremisini, C.; Nardi, E.; Fantauzzi, M.; Atzei, D.; Rossi, A.; Cametti, G. Fe (II) segregation at a specific crystallographic site of fibrous erionite: A first step toward the understanding of the mechanisms inducing its carcinogenicity. Microp. Mesop. Mat. 2015, 211, 49–63. [Google Scholar] [CrossRef]
- Pacella, A.; Fantauzzi, M.; Atzei, D.; Cremisini, C.; Nardi, E.; Montereali, M.R.; Rossi, A.; Ballirano, P. Iron within the erionite cavity and its potential role in inducing its toxicity: Evidences of Fe (III) segregation as extra-framework cation. Microporous Mesoporous Mater. 2017, 237, 168–179. [Google Scholar] [CrossRef]
- Pacella, A.; Cremisini, C.; Nardi, E.; Montereali, M.R.; Pettiti, I.; Ballirano, P. The mechanism of iron binding processes in erionite fibres. Sci. Rep. 2017, 7, 1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1023. [Google Scholar] [CrossRef] [Green Version]
- Matassa, R.; Famigliari, G.; Relucenti, M.; Battaglione, E.; Downing, C.; Pacella, A.; Cametti, G.; Ballirano, P. A deep look into erionite fibres: An electron microscopy investigation of their self-assembly. Sci. Rep. 2015, 5, 16757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualtieri, A.F.; Bursi Gandolfi, N.; Pollastri, S.; Pollok, K.; Langenhorst, F. Where is iron in erionite? A multidisciplinary study of fibrous erionite-Na from Jersey (Nevada, USA). Sci. Rep. 2016, 6, 37981. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, S.; Gualtieri, A.F.; Vigliaturo, R.; Ignatyev, K.; Strafella, E.; Pugnaloni, A.; Croce, A. Stability of mineral fibres in contact with human cell cultures. An in situ XANES, XRD and XRF iron mapping study. Chemosphere 2016, 164, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, A.F.; Gandolfi, N.B.; Pollastri, S.; Burghammer, M.; Tibaldi, E.; Belpoggi, F.; Dražić, G. New insights into the toxicity of mineral fibres: A combined in situ synchrotron µ-XRD and HR-TEM study of chrysotile, crocidolite, and erionite fibres found in the tissues of Sprague-Dawley rats. Toxicol. Lett. 2017, 274, 20–30. [Google Scholar] [CrossRef]
- Cangiotti, M.; Salucci, S.; Battistelli, M.; Falcieri, E.; Mattioli, M.; Giordani, M.; Ottaviani, M.F. EPR, TEM and cell viability study of asbestiform zeolite fibers in cell media. Colloids Surf. B Biointerfaces 2018, 161, 147–155. [Google Scholar] [CrossRef]
- Ballirano, P.; Cametti, G. Crystal chemical and structural modifications of erionite fibers leached with simulated lung fluids. Am. Mineral. 2015, 100, 1003–1012. [Google Scholar] [CrossRef]
- Giordani, M.; Cametti, G.; Di Lorenzo, F.; Churakov, S.V. Real-time observation of fibrous zeolites reactivity in contact with Simulated Lung Fluids (SLFs) obtained by Atomic Force Microscope (AFM). Minerals 2019, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, A.F.; Pollastri, S.; Gandolfi, N.B.; Gualtieri, M.L. In vitro acellular dissolution of mineral fibres: A comparative study. Sci. Rep. 2018, 8, 7071. [Google Scholar] [CrossRef]
- Martra, G.; Tomatis, M.; Fenoglio, I.; Coluccia, S.; Fubini, B. Ascorbic acid modifies the surface of asbestos: Possible implications in the molecular mechanisms of toxicity. Chem. Res. Toxicol. 2003, 16, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Turci, F.; Favero-Longo, S.E.; Tomatis, M.; Martra, G.; Castelli, D.; Piervittori, R.; Fubini, B. A biomimetic approach to the chemical inactivation of chrysotile fibres by lichen metabolites. Chem. Eur. J. 2007, 13, 4081–4093. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, G.B.; Pacella, A.; Corazzari, I.; Tomatis, M.; Turci, F. Surface reactivity of amphibole asbestos: A comparison between crocidolite and tremolite. Sci. Rep. 2017, 7, 14696. [Google Scholar] [CrossRef] [PubMed]
- Turci, F.; Tomatis, M.; Pacella, A. Surface and bulk properties of mineral fibres relevant to toxicity. In Mineral Fibres: Crystal Chemistry, Chemical–Physical Properties, Biological Interaction and Toxicity; Gualtieri, A.F., Ed.; European Mineralogical Union: London, UK, 2017; pp. 171–214. [Google Scholar]
- Pacella, A.; Andreozzi, G.B.; Corazzari, I.; Tomatis, M.; Turci, F. Surface reactivity of amphibole asbestos: A comparison between two tremolite samples with different surface area. Period. Mineral. 2018, 87, 195–205. [Google Scholar]
- Pacella, A.; Tomatis, M.; Viti, C.; Bloise, A.; Arrizza, L.; Ballirano, P.; Turci, F. Thermal inertization of amphibole asbestos modulates Fe topochemistry and surface reactivity. J. Hazard. Mater. 2020, 398, 123119. [Google Scholar] [CrossRef] [PubMed]
- Ballirano, P.; Pacella, A. Erionite-Na upon heating: Dehydration dynamics and exchangeable cations mobility. Sci. Rep. 2016, 6, 22786. [Google Scholar] [CrossRef] [Green Version]
- Rozalen, M.; Ramos, M.E.; Huertas, F.J.; Fiore, S.; Gervilla, F. Dissolution kinetics and biodurability of tremolite particles in mimicked lung fluids: Effect of citrate and oxalate. J. Asian Earth Sci. 2013, 77, 318–326. [Google Scholar] [CrossRef]
- Passaglia, E. The crystal chemistry of chabazites. Am. Mineral. 1970, 55, 1278–1301. [Google Scholar]
- Cametti, G.; Pacella, A.; Mura, F.; Rossi, M.; Ballirano, P. New morphological, chemical, and structural data of woolly erionite-Na from Durkee, Oregon, U.S.A. Am. Mineral. 2013, 98, 2155–2163. [Google Scholar] [CrossRef]
- Pacella, A.; Ballirano, P.; Fantauzzi, M.; Rossi, A.; Nardi, E.; Capitani, G.C.; Arrizza, L.; Montereali, M.R. Surface and bulk modifications of amphibole asbestos in mimicked Gamble’s solution at acidic pH. Sci. Rep. Submitted 2021, 11, 1–11. [Google Scholar]
- Bruker, AXS. Topas V.4.2: General Profile and Structure Analysis Software for Powder Diffraction Data; Bruker AXS: Karlsruhe, Germany, 2009. [Google Scholar]
- Sabine, T.M.; Hunter, B.A.; Sabine, W.R.; Ball, C.J. Analytical expressions for the transmission factor and peak shift in absorbing cylindrical specimens. J. Appl. Crystallogr. 1998, 31, 47–51. [Google Scholar] [CrossRef]
- Ballirano, P.; Maras, A. In-situ X-ray transmission powder diffraction study of the kinetics of the light induced alteration of realgar (α-As4S4). Eur. J. Mineral. 2006, 18, 589–599. [Google Scholar] [CrossRef]
- Ballirano, P. Effects of the choice of different ionization level for scattering curves and correction for small preferred orientation in Rietveld refinement: The MgAl2O4 test case. J. Appl. Crystallogr. 2003, 36, 1056–1061. [Google Scholar] [CrossRef] [Green Version]
- Yakubovich, O.V.; Massa, W.; Gavrilenko, P.G.; Pekov, I.V. Crystal structure of chabazite K. Crystallogr. Rep. 2005, 50, 544–553. [Google Scholar] [CrossRef]
- Le Page, Y.; Donnay, G. Refinement of the crystal structure of low-quartz. Acta Crystallogr. 1976, B32, 2456–2459. [Google Scholar] [CrossRef]
- Fantauzzi, M.; Pacella, A.; Atzei, D.; Gianfagna, A.; Andreozzi, G.; Rossi, A. Combined use of X-ray photoelectron and Mossbauer spectroscopic techniques in the analytical characterization of iron oxidation state in amphibole asbestos. Anal. Bioanal. Chem. 2010, 396, 2889–2898. [Google Scholar] [CrossRef]
- Pacella, A.; Cremisini, C.; Nardi, E.; Montereali, M.R.; Pettiti, I.; Giordani, M.; Mattioli, M.; Ballirano, P. Different erionite species bind iron into the atructure: A potential explanation for fibrous erionite toxicity. Minerals 2018, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Gainey, S.R.; Hausrath, E.M.; Hurowitz, J.A.; Milliken, R.E. Nontronite dissolution rates and implications for Mars. Geoch. Cosmochim. Acta 2014, 126, 102–211. [Google Scholar] [CrossRef]
- Schofield, R.E.; Hausrath, E.M.; Gainey, S.R. Zeolite weathering in laboratory and natural settings, and implications for Mars. In Proceedings of the 46th Lunar and Planetary Science Conference, The Woodlands, TX, USA, 16–20 March 2015. [Google Scholar]
- Hartman, R.L.; Fogler, H.S. Understanding the dissolution of zeolites. Langmiur 2007, 23, 5477–5484. [Google Scholar] [CrossRef]
- Della Ventura, G.R.; Vigliaturo, R.; Gieré, R.; Pollastri, S.; Gualtieri, A.F.; Iezzi, G. Infra Red spectroscopy of the regulated asbestos amphiboles. Minerals 2018, 8, 413. [Google Scholar] [CrossRef] [Green Version]
- Bergamini, C.; Fato, R.; Biagini, G.; Pugnaloni, A.; Giantomassi, F.; Foresti, E.; Lesci, G.I.; Roveri, N. Mitochondria changes induced by natural and synthetic asbestos fibers: Studies on isolated mitochindria. Cell. Mol. Biol. 2004, 50, 691–700. [Google Scholar]
- Pacella, A.; Andreozzi, G.B.; Fournier, J.; Stievano, L.; Giantomassi, F.; Lucarini, G.; Rippo, M.R.; Pugnaloni, A. Iron topochemistry and surface reactivity of amphibole asbestos: Relations with in vitro toxicity. Anal. Bioanal. Chem. 2012, 402, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B. Al-O and Si-O tetrahedral distances in aluminosilicate framework structures. Acta Crystallogr. 1968, 24, 355–358. [Google Scholar] [CrossRef]
- Alberti, A.; Gottardi, G.; Lai, T. The determination of (Si,Al) distribution in zeolites. In Guidelines for Mastering the Properties of Molecular Sieves; Barthomeuf, D., Derouane, E.G., Hölderich, W., Eds.; NATO ASI Series; Plenum Press: New York, NY, USA, 1990; Volume 221, pp. 145–155. [Google Scholar]
- Giordani, M.; Mattioli, M.; Ballirano, P.; Pacella, P.; Cenni, M.; Boscardin, M.; Valentini, L. Geological occurrence, mineralogical characterization and risk assessment of potentially carcinogenic erionite in Italy. J. Toxicol. Environ. Health B 2017, 20, 81–103. [Google Scholar] [CrossRef]
- Ballirano, P.; Andreozzi, G.B.; Dogan, M.; Dogan, A.U. Crystal structure and iron topochemistry of erionite-K from Rome, Oregon, U.S.A. Am. Mineral. 2009, 94, 1262–1270. [Google Scholar] [CrossRef]
- Pacella, A.; Ballirano, P.; Cametti, G. Quantitative chemical analysis of erionite fibres using a micro-analytical SEM-EDX method. Eur. J. Mineral. 2016, 28, 257–264. [Google Scholar] [CrossRef]
- Ballirano, P.; Cametti, G. Dehydration dynamics and thermal stability of erionite-K: Experimental evidence of the “internal ionic exchange” mechanism. Micropor. Mesopor. Mat. 2012, 163, 160–168. [Google Scholar] [CrossRef]
- Ballirano, P.; Pacella, A.; Bloise, A.; Giordani, M.; Mattioli, M. Thermal stability of woolly erionite-K and considerations about the heat induced behaviour of the erionite group. Minerals 2018, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Quiroz-Estrada, K.; Pacella, A.; Ballirano, P.; Hernández-Espinosa, M.A.; Felipe, C.; Esparza-Schulz, M. Crystal chemical and structural characterization of natural and cation-exchanged mexican erionite. Minerals 2020, 10, 772. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rietveld Data | Pristine a | Pristine | E-1h | E-24h | E-1W | E-1M | Avg. |
|---|---|---|---|---|---|---|---|
| Rwp | 1.690 | 1.441 | 1.792 | 1.531 | 1.514 | 1.601 | - |
| Rp | 1.258 | 1.089 | 1.340 | 1.150 | 1.137 | 1.180 | - |
| DWd | 0.364 | 0.385 | 0.238 | 0.326 | 0.293 | 0.281 | - |
| GoF | 3.179 | 2.716 | 4.067 | 3.256 | 3.336 | 3.497 | - |
| RBragg | 0.487 | 0.423 | 0.575 | 0.485 | 0.498 | 0.529 | - |
| Erionite (wt.%) | 96.51 (10) | 94.92 (17) | 94.9 (2) | 94.69 (18) | 94.61 (17) | 94.55 (17) | 94.73 (16) |
| Chabazite (wt.%) | 3.28 (10) | 4.87 (17) | 4.9 (2) | 5.11 (18) | 5.19 (17) | 5.27 (17) | 5.08 (17) |
| Quartz (wt.%) | 0.21 (1) | 0.21 (1) | 0.18 (1) | 0.20 (1) | 0.20 (1) | 0.18 (1) | 0.19 (1) |
| a (Å) | 13.23097 (9) | 13.23009 (8) | 13.22715 (7) | 13.22669 (7) | 13.22808 (7) | 13.22744 (7) | - |
| c (Å) | 15.06475 (11) | 15.06460 (8) | 15.07581 (9) | 15.07603 (9) | 15.07650 (8) | 15.07469 (8) | - |
| c/a | 1.13860 | 1.13866 | 1.13976 (1) | 1.13982 (1) | 1.13973 (1) | 1.13965 (1) | - |
| Vol. (Å3) | 2283.89 (3) | 2283.57 (3) | 2284.25 (3) | 2284.13 (3) | 2284.68 (3) | 2284.18 (3) | 2284.2 (4) |
| Structural Data | Pristine a | Pristine | E-1h | E-24h | E-1W | E-1M | Avg. |
|---|---|---|---|---|---|---|---|
| Ca1 s.s. (e-) | 15.8 (7) | 23.2 (4) | 7.6 (7) | 6.4 (7) | 5.9 (7) | 6.3 (7) | - |
| Ca2 s.s. (e-) | 27.3 (5) | 29.9 (3) | 34.2 (3) | 33.8 (3) | 34.9 (3) | 33.8 (3) | - |
| Ca3 s.s. (e-) | 12.6 (7) | 6.8 (4) | 2.2 (4) | 1.5 (4) | 0.0 (4) | 0.2 (4) | - |
| K1 s.s. (e-) | 38 (0) | 38 (0) | 38 (0) | 38 (0) | 38 (0) | 38 (0) | - |
| K2 s.s. (e-) | 13.1 (6) | 14.3 (5) | 9.9 (5) | 9.6 (5) | 10.5 (5) | 9.5 (5) | - |
| EF cat. s.s. (e-) | 107 (2) | 112.2 (15) | 92.0 (19) | 89.3 (19) | 89.3 (19) | 87.8 (19) | - |
| EF cat. s.s. (e-) EDS | 92.3 | 92.7 | 89.5 | - | - | 90.9 | - |
| OW7 s.s. (e-) | 22.2 (18) | 22.8 (16) | 34.9 (7) | 35.2 (7) | 37.2 (7) | 37.8 (7) | - |
| OW8 s.s. (e-) | 43.2 (6) | 41.7 (5) | 33.4 (5) | 33.1 (5) | 33.9 (5) | 34.3 (5) | - |
| OW9 s.s. (e-) | 52.1 (14) | 46.3 (12) | 43.7 (14) | 43.6 (13) | 45.1 (13) | 44.3 (12) | - |
| OW10 s.s. (e-) | 37.4 (18) | 37.7 (13) | 45.2 (12) | 43.4 (12) | 45.0 (12) | 44.2 (12) | - |
| OW11 s.s. (e-) | 46.2 (20) | 46.4 (17) | 47.4 (11) | 48.3 (11) | 46.8 (10) | 45.8 (11) | - |
| OW12 s.s. (e-) | 59.7 (12) | 55.6 (12) | 63.2 (18) | 65.4 (16) | 63.3 (16) | 66.2 (14) | - |
| H2O s.s. (e-) | 261 (9) | 251 (7) | 268 (7) | 269 (6) | 271 (6) | 272 (6) | - |
| EF cat. + H2O s.s. (e-) | 368 (11) | 363 (9) | 360 (9) | 358 (8) | 361 (8) | 360 (8) | 360.3 (16) |
| Biso Si (Å2) | 0.628 (19) | 0.661 (17) | 0.449 (16) | 0.458 (16) | 0.435 (14) | 0.449 (15) | - |
| Biso O1,2,4 (Å2) | 0.73 (5) | 1.04 (4) | 0.91 (4) | 0.95 (4) | 0.95 (4) | 0.94 (4) | - |
| Biso O3,5,6 (Å2) | 2.15 (7) | 2.39 (7) | 2.18 (7) | 2.15 (7) | 2.11 (6) | 2.09 (7) | - |
| Biso Ca1,2 (Å2) | 13.0 (6) | 15.2 (2) | 15.6 (2) | 15.8 (2) | 15.9 (2) | 16.4 (2) | - |
| Biso K1,2 (Å2) | 2.37 (10) | 1.90 (8) | 2.32 (8) | 2.26 (8) | 2.27 (7) | 2.10 (7) | - |
| Biso OW7-12 (Å2) | 17.0 (3) | 15.2 (2) | 15.6 (2) | 15.8 (2) | 15.9 (2) | 16.4 (2) | - |
| < T1–O > (Å) | 1.6265 | 1.6316 | 1.6342 | 1.6345 | 1.6339 | 1.6334 | 1.6335 (12) |
| < T2–O > (Å) | 1.6539 | 1.6435 | 1.6403 | 1.6388 | 1.6423 | 1.6415 | 1.6413 (18) |
| Al apfu @ T1 | 3.52 | 4.29 | 4.70 | 4.74 | 4.65 | 4.58 | 4.59 (18) |
| Al apfu @ T2 | 3.87 | 3.07 | 2.82 | 2.70 | 2.97 | 2.91 | 2.89 (14) |
| Altot apfu | 7.39 | 7.36 | 7.51 | 7.44 | 7.62 | 7.49 | 7.49 (10) |
| R = Si/(Si + Al) | 0.795 | 0.796 | 0.791 | 0.793 | 0.788 | 0.792 | 0.792 (3) |
| Oxides (wt.%) | Pristine a | Pristine | E-1h | E-1M | Chabazite |
|---|---|---|---|---|---|
| SiO2 | 59.80 (32) | 59.47 (77) | 59.40 (81) | 59.90 (63) | 53.95 (43) |
| Al2O3 | 13.18 (35) | 13.41 (63) | 13.62 (25) | 13.08 (17) | 15.06 (25) |
| Na2O | 1.56 (31) | 1.73 (41) | 3.31 (74) | 4.04 (71) | 3.49 (47) |
| K2O | 4.89 (35) | 4.69 (43) | 3.41 (27) | 3.62 (32) | 2.24 (39) |
| MgO | 0.88 (13) | 1.10 (11) | 1.02 (18) | 0.59 (13) | 1.25 (13) |
| CaO | 1.19 (19) | 1.09 (14) | 0.74 (24) | 0.26 (7) | 2.01 (32) |
| H2O | 18.50 | 18.50 | 18.50 | 18.50 | 22.00 |
| Total | 100 | 100.00 | 100.00 | 100.00 | 100.00 |
| Atoms | |||||
| Si | 28.58 (18) | 28.43 (35) | 28.33 (17) | 28.62 (14) | 9.03 (5) |
| Al | 7.42 (18) | 7.57 (35) | 7.67 (17) | 7.38 (14) | 2.97 (5) |
| Na | 1.45 (29) | 1.61 (39) | 3.06 (72) | 3.75 (68) | 1.13 (16) |
| K | 2.98 (22) | 2.86 (27) | 2.08 (18) | 2.21 (18) | 0.48 (9) |
| Mg | 0.63 (12) | 0.79 (8) | 0.73 (13) | 0.42 (9) | 0.31 (3) |
| Ca | 0.61 (8) | 0.56 (7) | 0.38 (12) | 0.13 (13) | 0.36 (6) |
| O | 71.74 (23) | 71.80 (17) | 71.84 (27) | 71.85 (24) | 24.61 (16) |
| H2O | 29.49 (14) | 29.56 (15) | 29.49 (27) | 29.55 (17) | 12.30 (8) |
| R | 0.794 | 0.790 | 0.787 | 0.795 | 0.753 |
| M/(M + D) | 0.782 | 0.768 | 0.823 | 0.914 | 0.706 |
| E% | 7.6 | 5.5 | 4.3 | 4.3 | 0.5 |
| Sample | O | Si | Al | Fe | Na | K | Ca | Mg |
|---|---|---|---|---|---|---|---|---|
| Pristine a | 61.6 (1) | 26.4 (4) | 6.0 (2) | 0.9 (2) | 1.8 (3) | 2.1 (2) | 0.9 (1) | 0.30 (0.04) |
| E-1h | 60.3 (6) | 27.0 (9) | 5.8 (4) | 0.7 (1) | 4.6 (6) | 1.4 (1) | n.d. | 0.16 (0.08) |
| E-24h | 59.8 (1) | 27.3 (9) | 5.8 (1) | 0.5 (1) | 4.9 (2) | 1.5 (1) | n.d. | 0.2 (0.1) |
| E-1W | 60 (3) | 27 (2) | 5.6 (8) | 0.7 (2) | 5.4 (1) | 1.4 (1) | n.d. | n.d. |
| E-1M | 59.7 (6) | 27.1 (5) | 4.8 (2) | 0.7 (1) | 6.1 (2) | 1.6 (1) | n.d. | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacella, A.; Ballirano, P.; Fantauzzi, M.; Rossi, A.; Viti, C.; Arrizza, L.; Nardi, E.; Caprioli, R.; Montereali, M.R. Surface and Bulk Modifications of Fibrous Erionite in Mimicked Gamble’s Solution at Acidic pH. Minerals 2021, 11, 914. https://doi.org/10.3390/min11090914
Pacella A, Ballirano P, Fantauzzi M, Rossi A, Viti C, Arrizza L, Nardi E, Caprioli R, Montereali MR. Surface and Bulk Modifications of Fibrous Erionite in Mimicked Gamble’s Solution at Acidic pH. Minerals. 2021; 11(9):914. https://doi.org/10.3390/min11090914
Chicago/Turabian StylePacella, Alessandro, Paolo Ballirano, Marzia Fantauzzi, Antonella Rossi, Cecilia Viti, Lorenzo Arrizza, Elisa Nardi, Raffaela Caprioli, and Maria Rita Montereali. 2021. "Surface and Bulk Modifications of Fibrous Erionite in Mimicked Gamble’s Solution at Acidic pH" Minerals 11, no. 9: 914. https://doi.org/10.3390/min11090914
APA StylePacella, A., Ballirano, P., Fantauzzi, M., Rossi, A., Viti, C., Arrizza, L., Nardi, E., Caprioli, R., & Montereali, M. R. (2021). Surface and Bulk Modifications of Fibrous Erionite in Mimicked Gamble’s Solution at Acidic pH. Minerals, 11(9), 914. https://doi.org/10.3390/min11090914