
In Situ X-ray Diffraction at High Temperatures: Formation of Ca2SiO4 and Ternesite in Recycled Autoclaved Aerated Concrete


Abstract
:1. Introduction
2. Materials and Methods
2.1. Raw Material Analysis
2.2. High-Temperature XRD
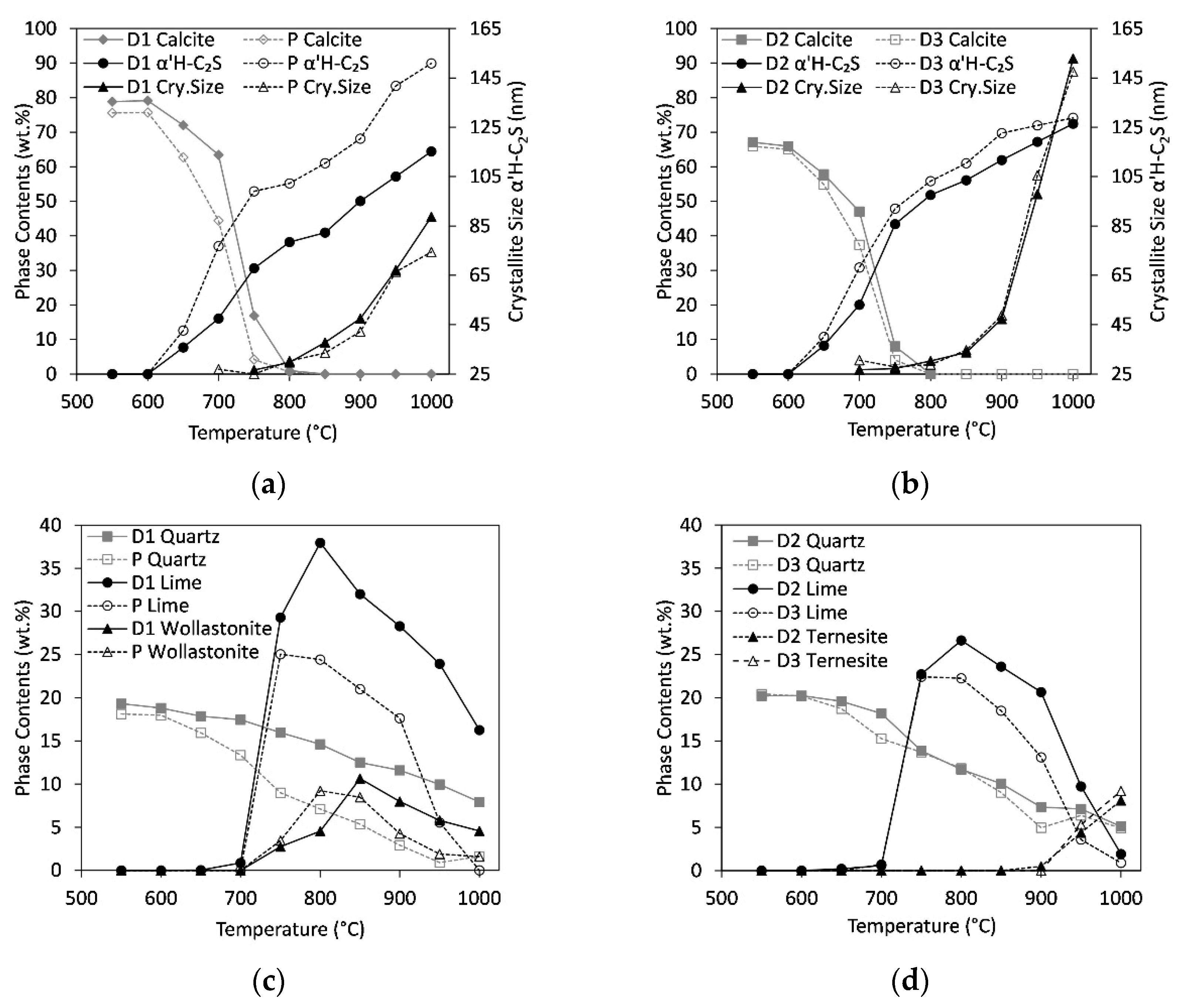
3. Results
3.1. Raw Material Composition
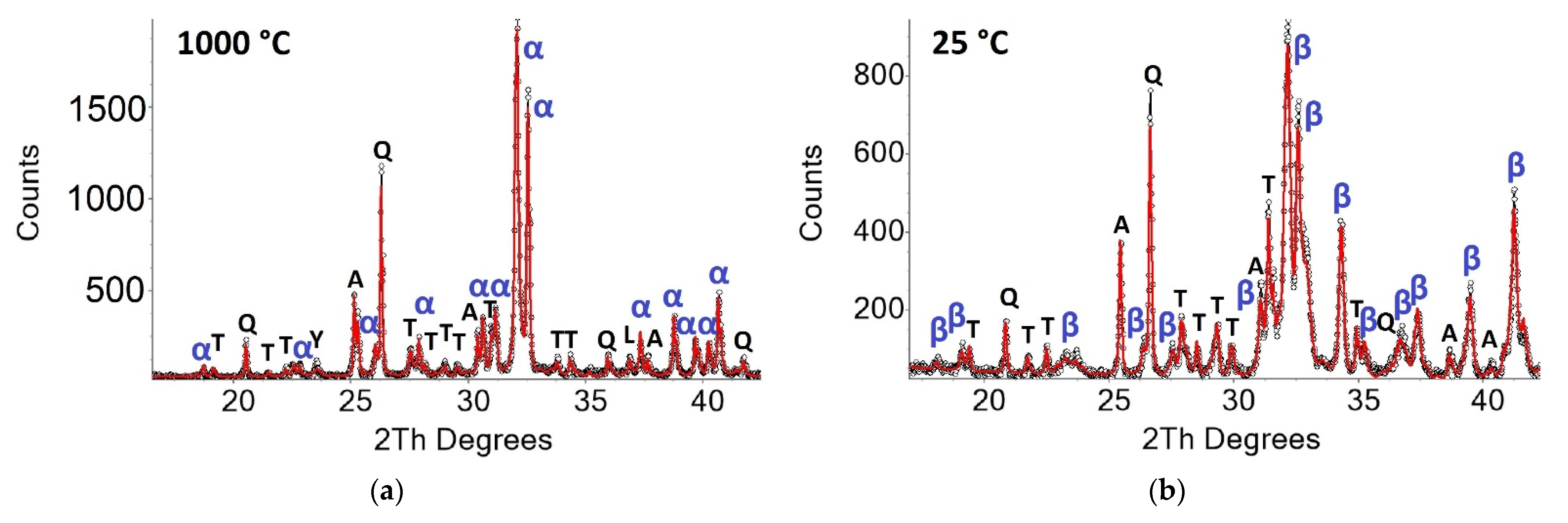
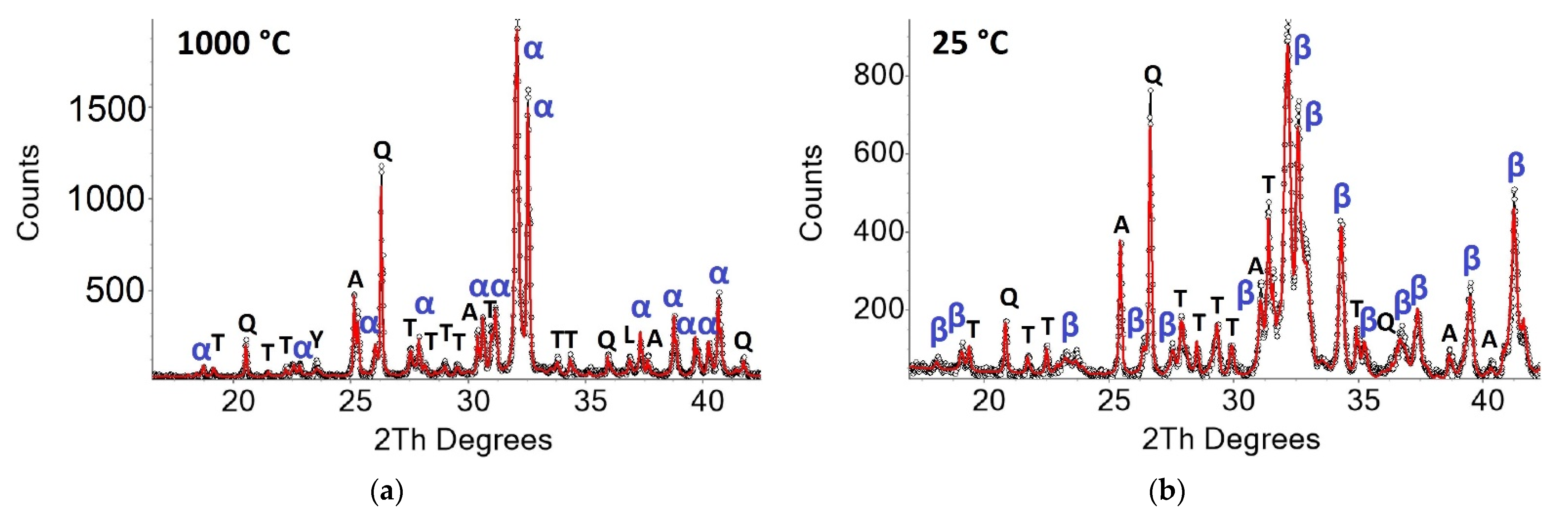
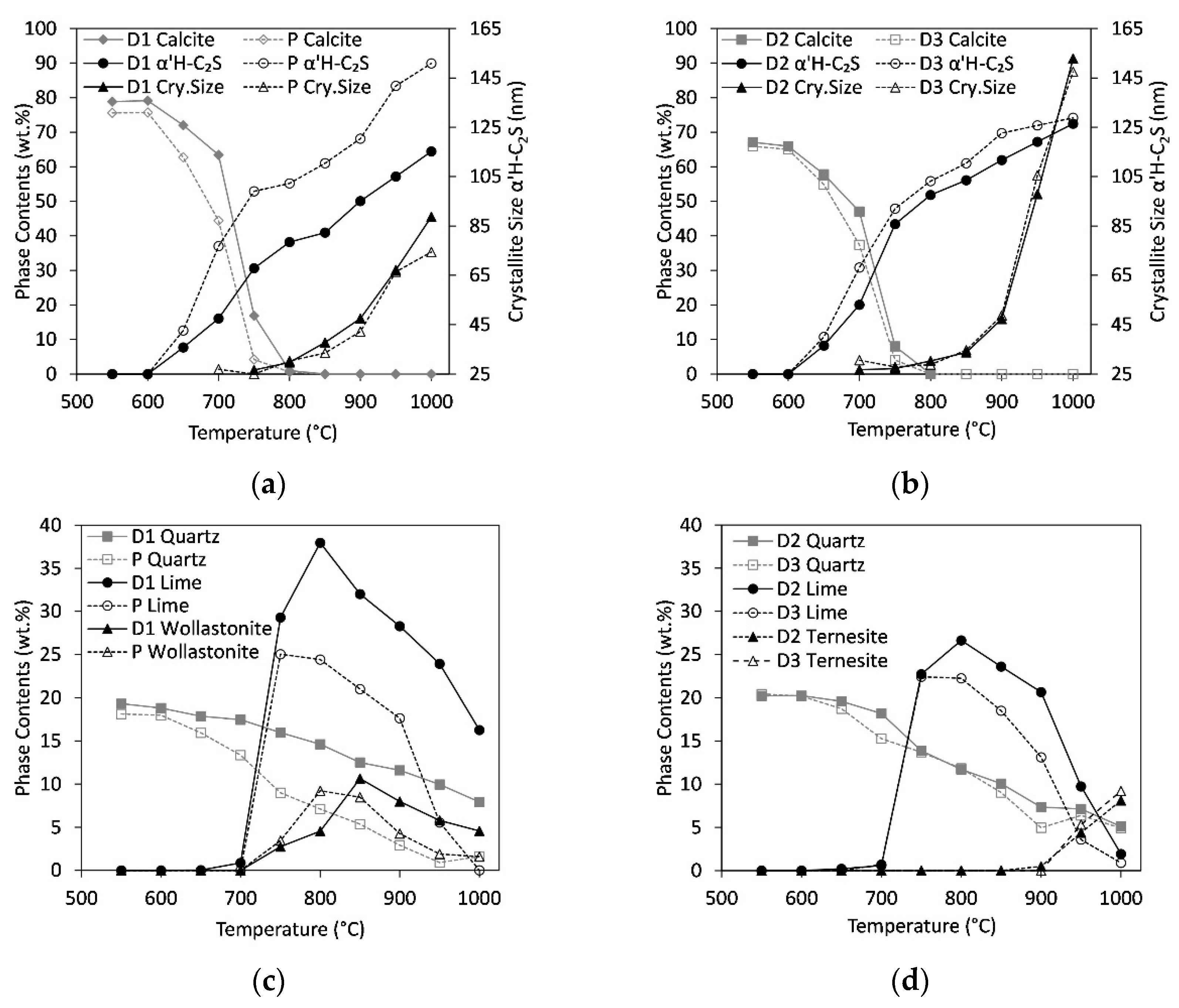
3.2. Qualitative Phase Contents In Situ at 1000 °C and 25 °C after Heating Experiments
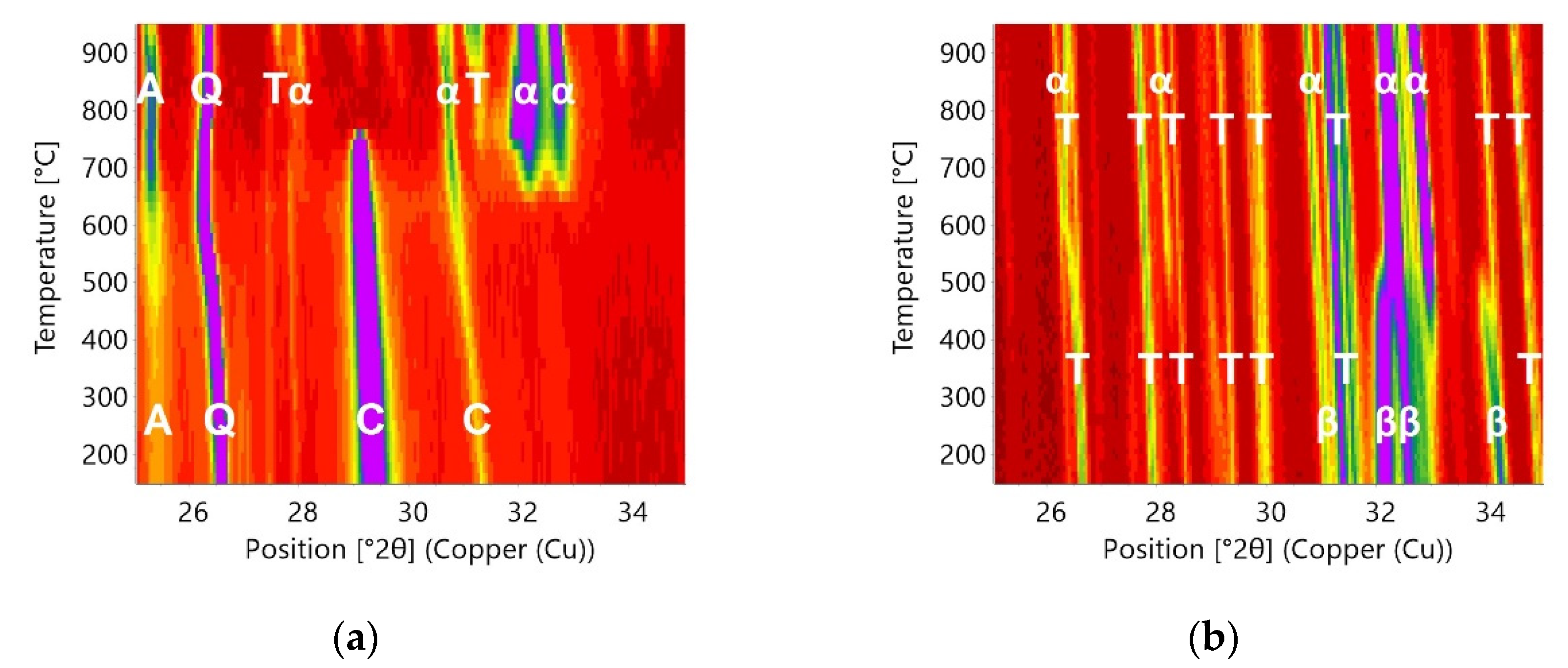
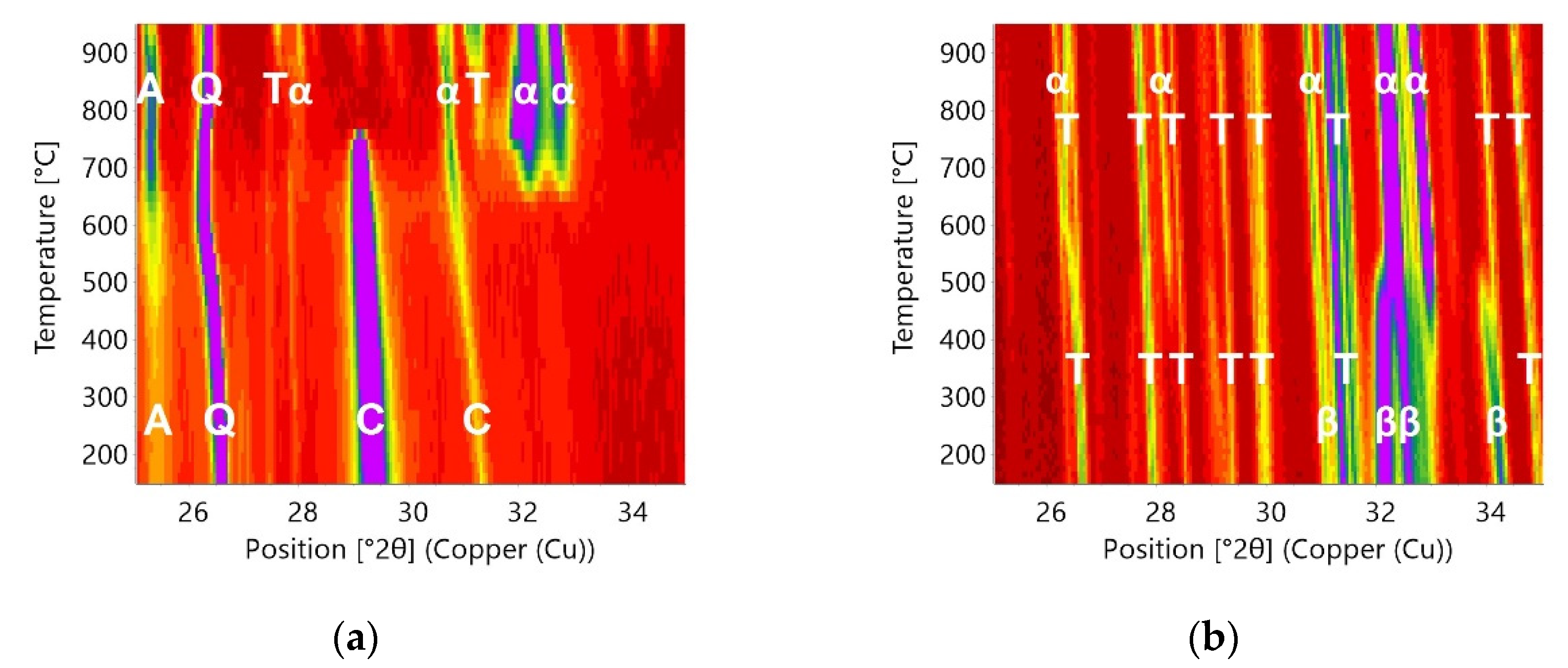
3.3. In Situ Measurements during Heating
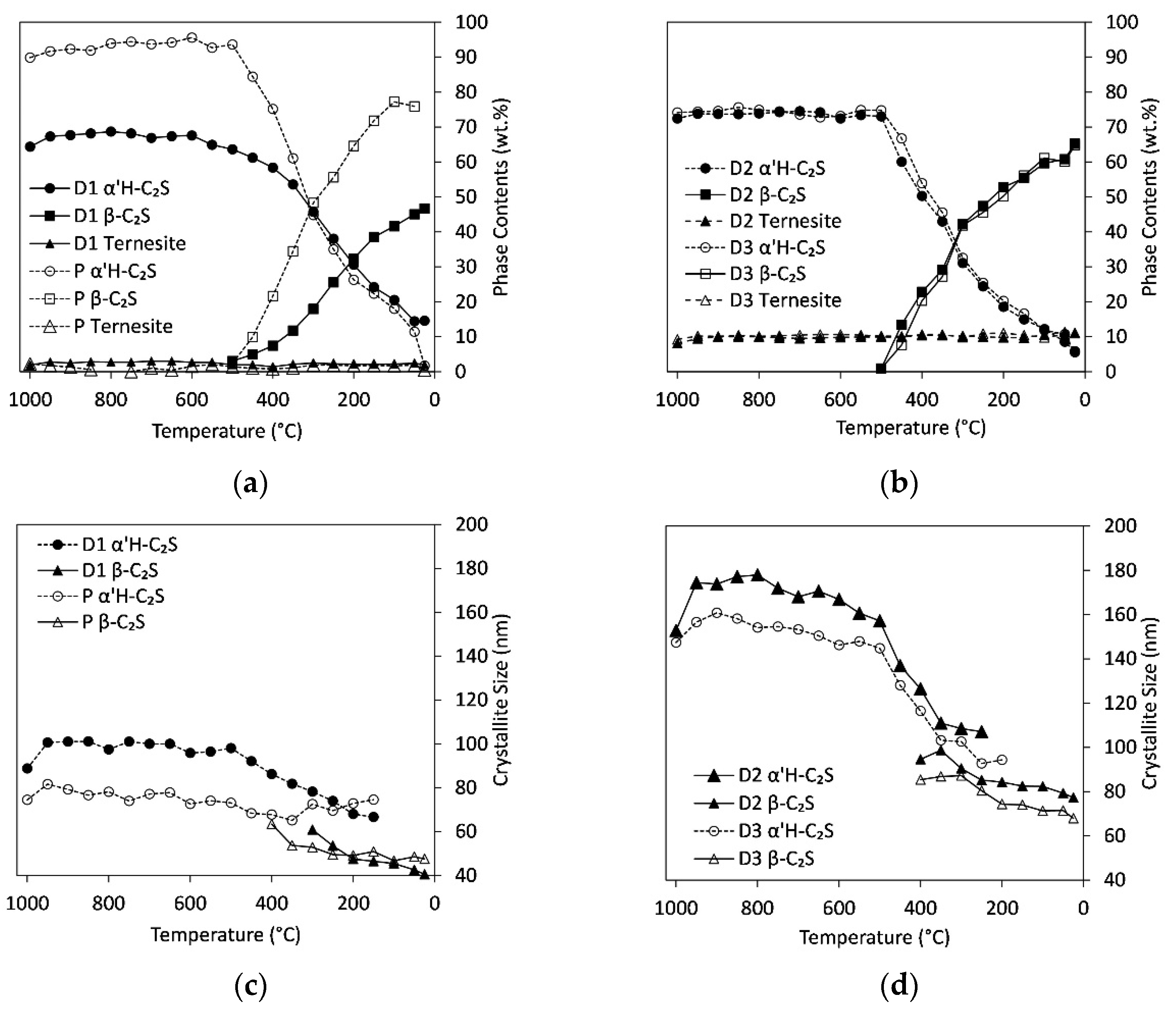
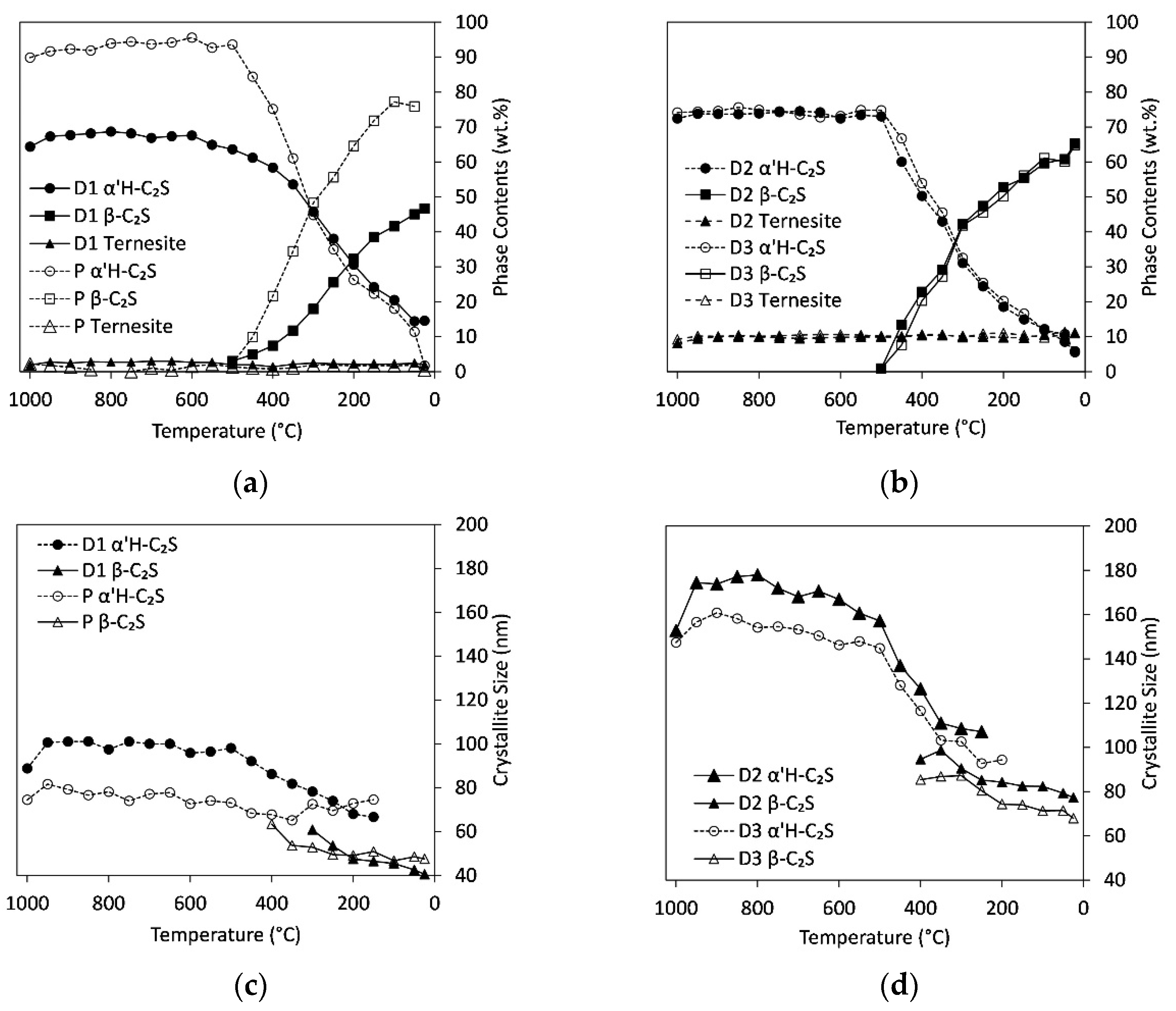
3.4. In Situ Measurements during Cooling
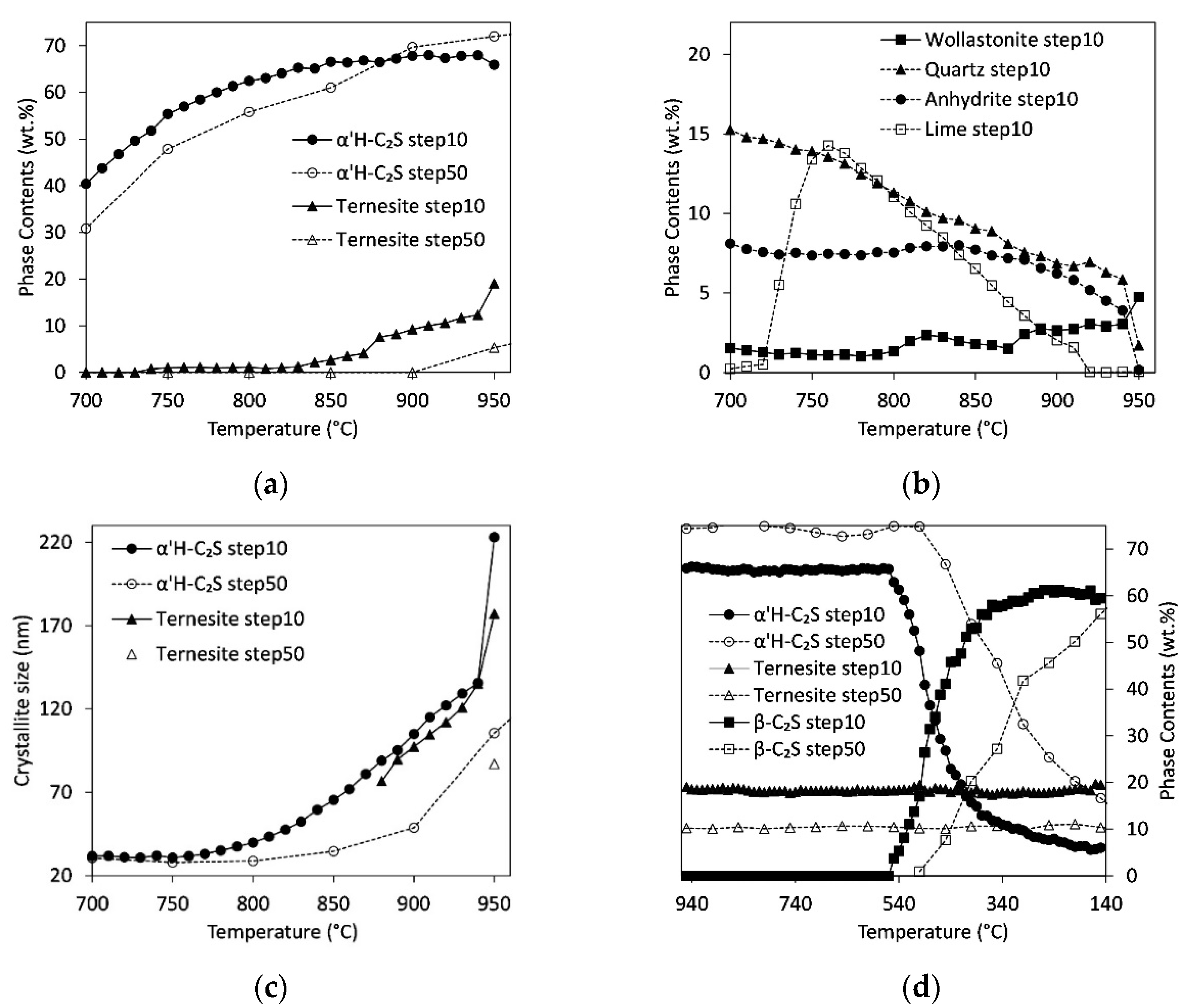
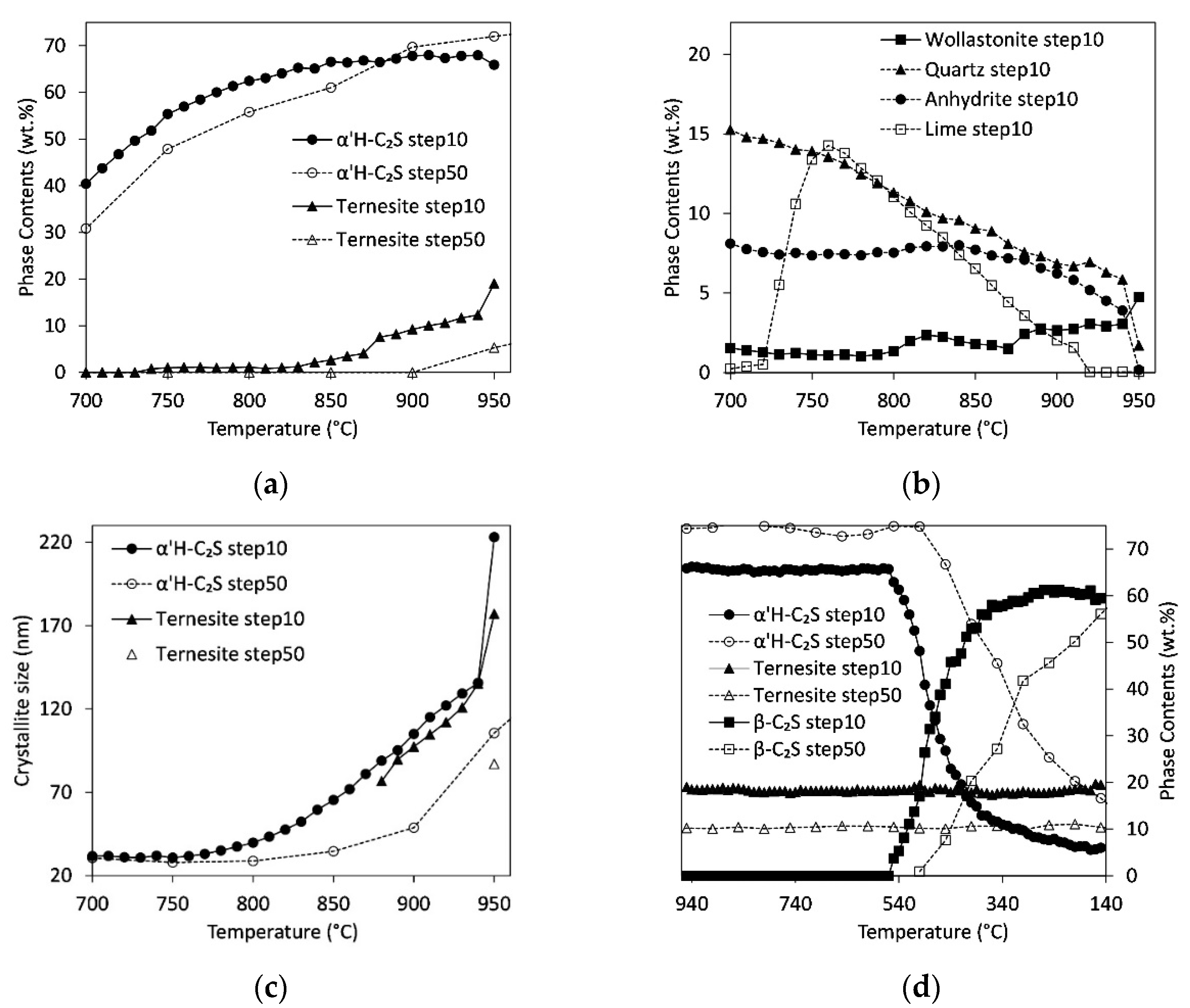
3.5. Influence of Heating Rate/Residence Time
3.6. Increase in C/S-Ratio
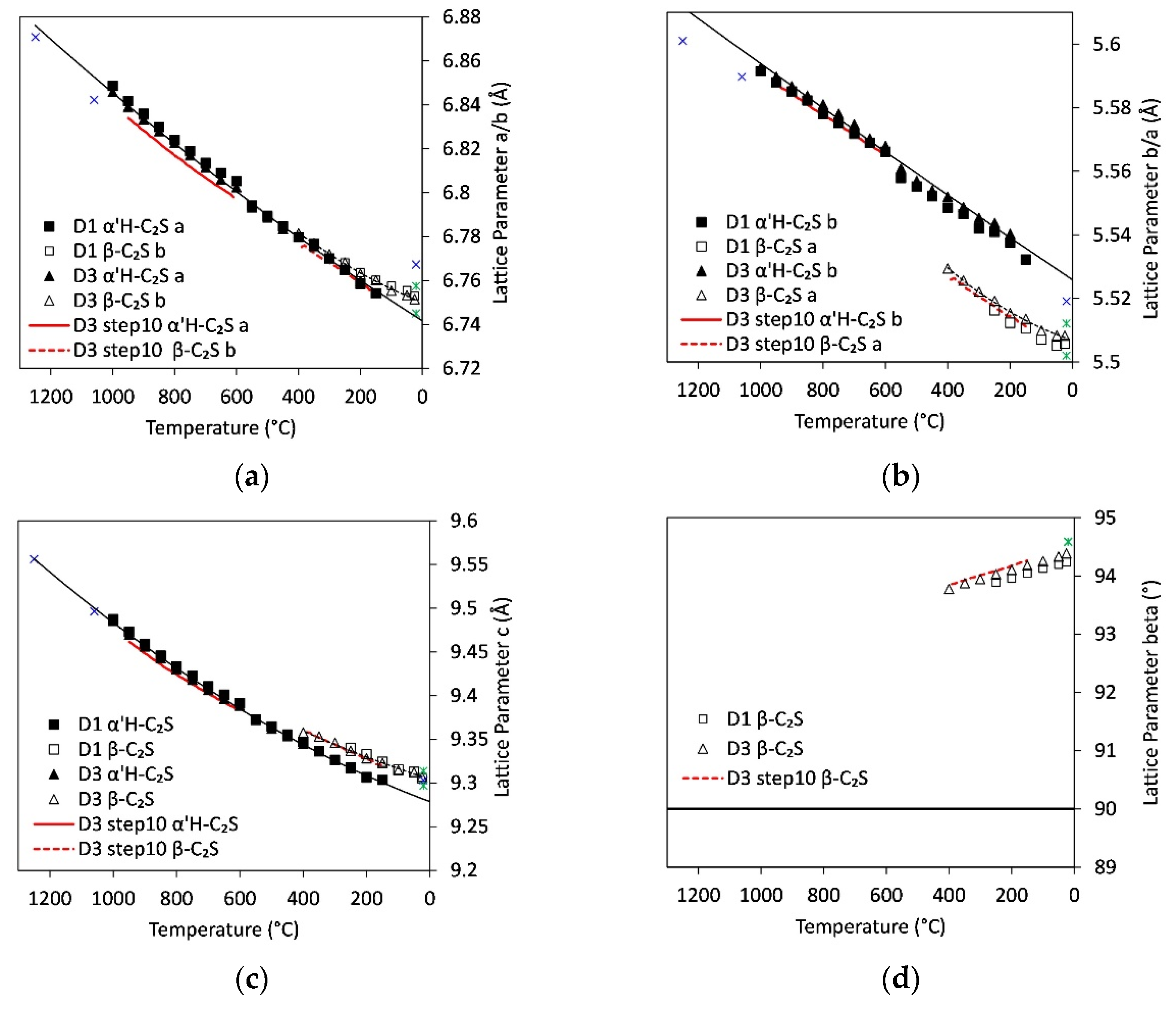
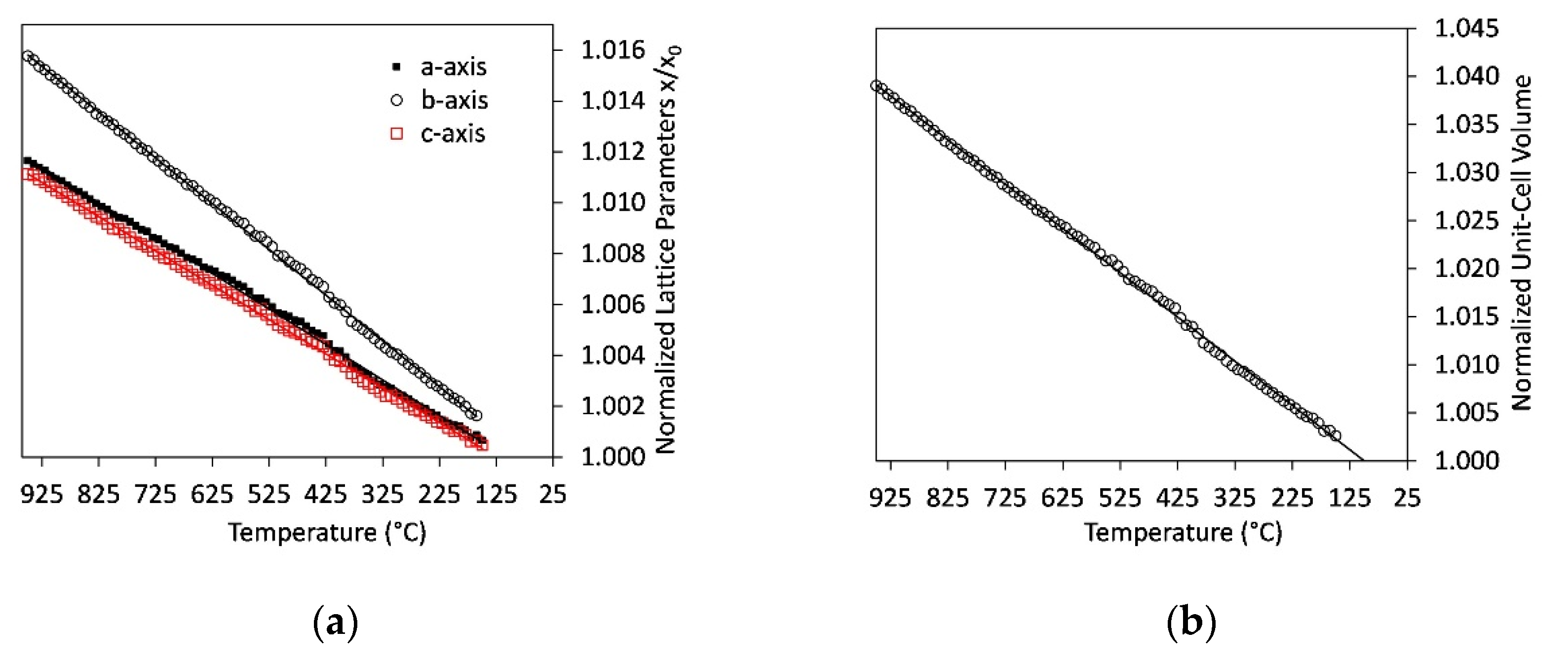
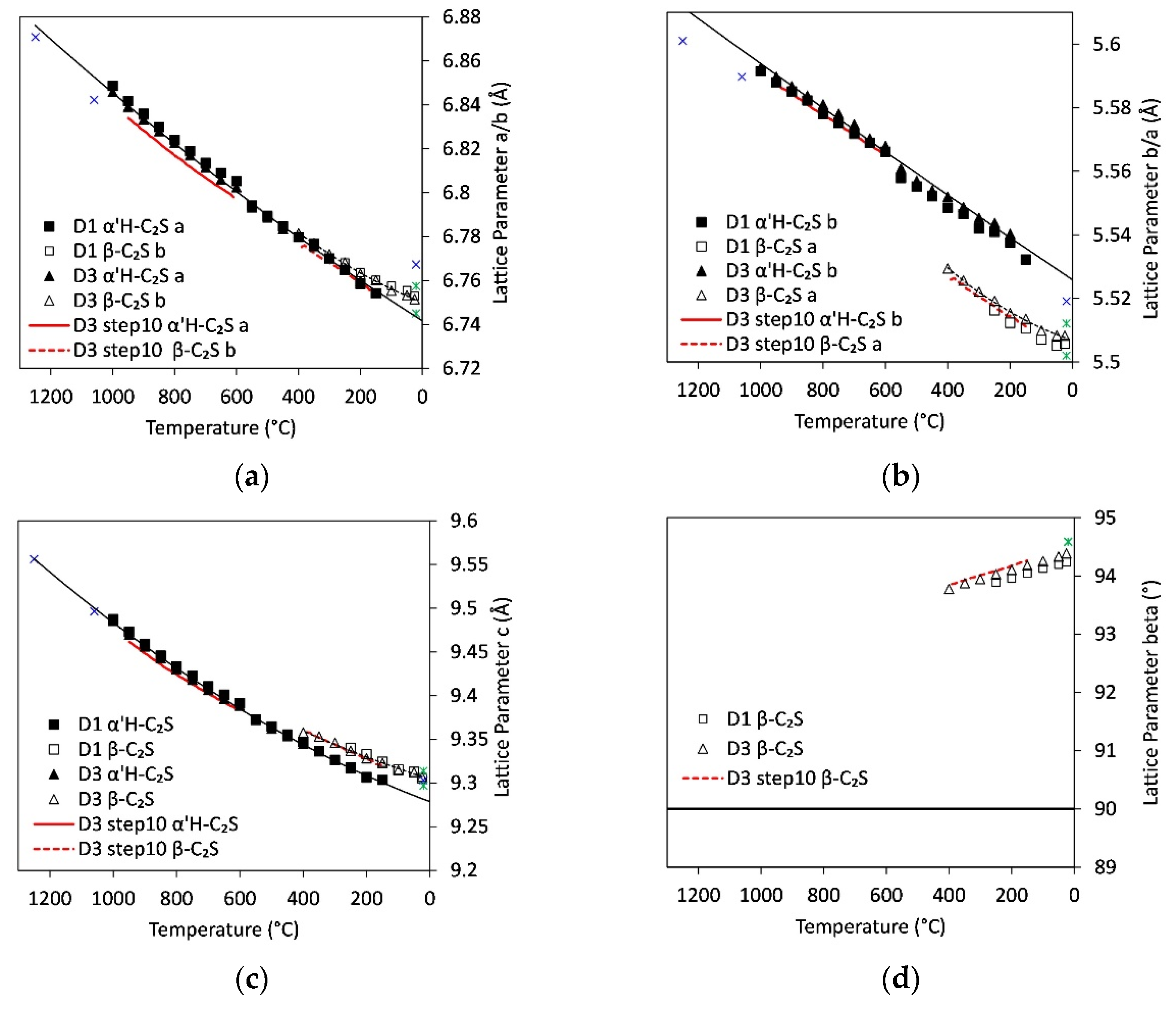
3.7. Lattice Parameters
4. Discussion
4.1. Formation Mechanisms of C2S and Ternesite
4.2. Quantitative Results: Influence of Heating Rate, C/S-Ratio, and Sulfate Content
4.3. Foreign Ion Incorporation in C2S
4.4. Influence of Cooling Rate and Reaction Progress on Quench Product
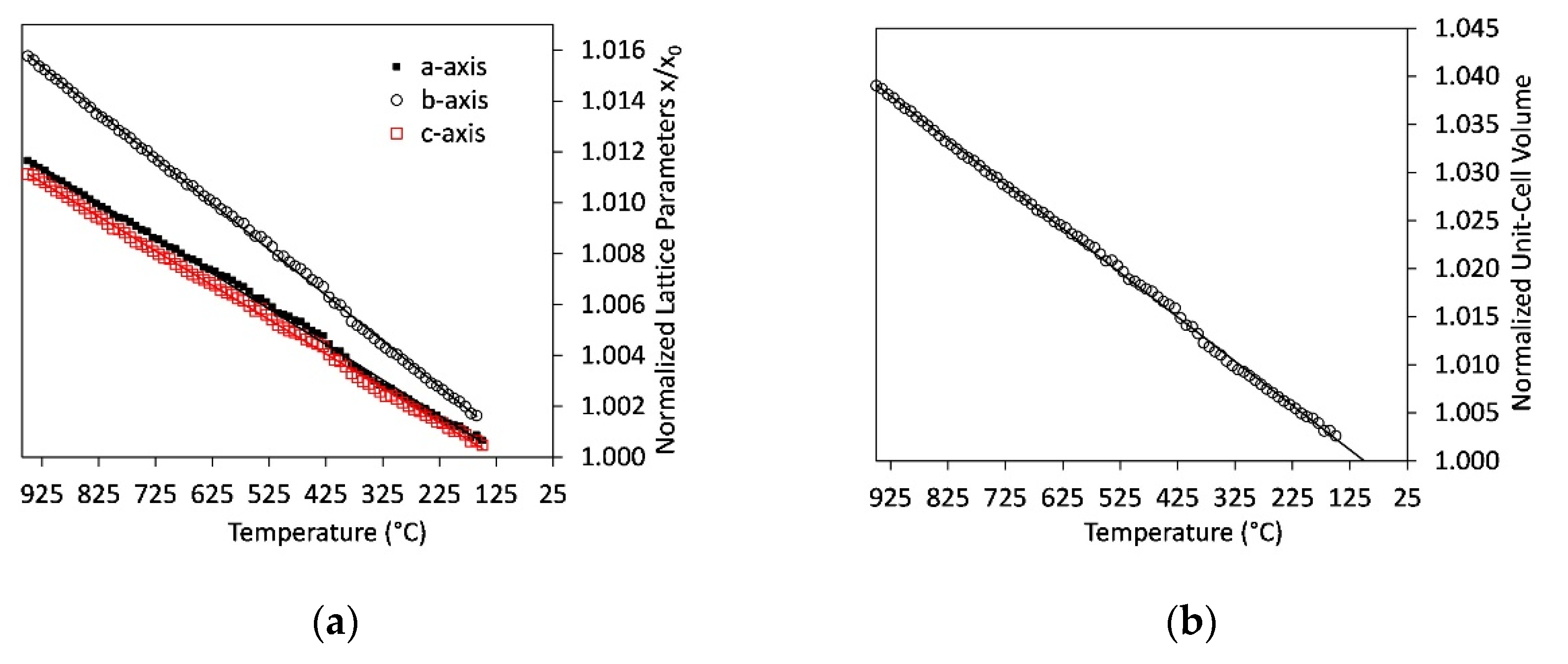
4.5. Thermal Expansion of Ternesite
5. Conclusions
- Increased residence time during heating increases the ternesite content but promotes also the formation of high crystalline α’H-C2S;
- The sulfate content has a positive effect on the formation of α’H-C2S (phase content and crystallinity);
- The C/S-ratio has to be adapted carefully to the sulfate content, otherwise the formation of ternesite takes place at the expense of C2S;
- The ternesite stability during quench depends on the C/S-ratio and cooling rate: Ternesite stays stable or even increases in amount in experiments with residual quartz and lime. In the case of a low C/S-ratio where only residual quartz is present, a decrease in ternesite amount during very slow cooling was observed;
- The temperature and range of the α’H-C2S→β-C2S transformation depend strongly on the cooling rate. The first formation of β-C2S was observed at 540 °C (slow quench) and 450 °C (fast quench);
- Thermal expansion coefficients of ternesite are similar to literature data of larnite. However, the incorporation of CaSO4 modules in the structure switches the direction of maximum compression.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Activity Report 2019. Cembureau 2020. Available online: https://cembureau.eu/media/clkdda45/activity-report-2019.pdf (accessed on 25 March 2021).
- Robbie, M.A. Global CO2 emissions from cement production, 1928–2018. Earth Syst. Sci. Data 2019, 11, 1675–1710. [Google Scholar]
- Kreft, O. REPOST Recycling-Cluster Porenbeton 2019. Available online: https://innovative-produktkreislaeufe.de/Projekte/REPOST/_/REPOST_Vortrag_ReziProK_Kick-off.pdf (accessed on 25 March 2021).
- Chatterjee, A.K. High-belite cements—Present status and future technological options: Part I. Cem. Concr. Res. 1996, 26, 1213–1225. [Google Scholar] [CrossRef]
- Chatterjee, A.K. Future technological options: Part II. Cem. Concr. Res. 1996, 26, 1227–1237. [Google Scholar] [CrossRef]
- Chatterjee, A.K. High-belite Portland cement: An update on development characterization and applications. In Proceedings of the 11th International Congress on the Chemistry of Cement (ICCC), Durban, South Africa, 11–16 May 2003; pp. 31–40. [Google Scholar]
- Serpell, R.; Zunino, F. Recycling of hydrated cement pastes by synthesis of α’H-C2S. Cem. Concr. Res. 2017, 100, 398–412. [Google Scholar] [CrossRef]
- Cuberos, A.J.M.; De la Torre, Á.G.; Martín-Sedeño, M.C.; Moreno-Real, L.; Merline, M.; Ordónez, L.M.; Aranda, M.A.G. Phase development in conventional and active belite cement pastes by Rietveld analysis and chemical constraints. Cem. Concr. Res. 2009, 39, 833–842. [Google Scholar] [CrossRef]
- Stanĕk, T.; Sulovský, P. Active low-energy belite cement. Cem. Concr. Res. 2015, 68, 203–210. [Google Scholar] [CrossRef]
- Sorensen, A.; Thomas, R.; Maguire, M.; Quezada, I. Calcium Sulfoaluminate (CSA) Cement: Benefits and Applications. Concr. Int. 2018, 40, 65–69. [Google Scholar]
- Hanein, T.; Galan, I.; Elhoweris, A.; Khare, S. Production of belite calcium sulphoaluminate cement using sulfur as a fuel and as a source of clinker sulfur trioxide: Pilot kiln trial. Adv. Cem. Res. 2016, 28, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Dienemann, W.; Schmitt, D.; Bullerjahn, F.; Haha, M.B. Belite-Calciumsulfoaluminate-Ternesite (BCT)—A new low-carbon clinker Technology. Cem. Int. 2013, 11, 100–109. [Google Scholar]
- Hanein, T.; Galan, I.; Glasser, F.P.; Skalamprinos, S.; Elhoweris, A.; Imabi, M.S.; Bannermann, M.N. Stability of ternesite and the production at scale of ternesite-based clinkers. Cem. Concr. Res. 2017, 98, 91–100. [Google Scholar] [CrossRef]
- Bullerjahn, F.; Schmitt, D.; Haha, M.B. Effect of raw mix design and of clinkering process on the formation and mineralogical composition of (ternesite) belite calcium sulphoaluminate ferrite clinker. Cem. Concr. Res. 2014, 59, 87–95. [Google Scholar] [CrossRef]
- Bullerjahn, F.; Schmitt, D.; Haha, M.B. CSA raw mix design: Effect on clinker formation and reactivity. Mater. Struct. 2015, 48, 3895–3911. [Google Scholar] [CrossRef]
- Haha, M.B.; Bullerjahn, F.; Zajac, M. On the reactivity of ternesite. In Proceedings of the 14th International Congress on the Chemistry of Cement, Beijing, China, 13–16 October 2015. [Google Scholar]
- Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In Situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures. Minerals 2020, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Serpell, R.; Lopez, M. Properties of mortars with reactivated cementitious materials. Cem. Concr. Res. 2015, 64, 16–26. [Google Scholar] [CrossRef]
- Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In Situ Raman imaging of high-temperature solid-state reactions in the CaSO4-SiO2 system. Int. J. Coal Sci. Technol. 2019, 6, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Garbev, K.; Beuchle, G.; Schweike, U.; Merz, D.; Dregert, O.; Stemmermann, P. Preparation of a Novel Cementitious Material from Hydrothermally Synthesized C-S-H Phases. J. Am. Ceram. Soc. 2014, 97, 2298–2307. [Google Scholar] [CrossRef]
- Stemmermann, P.; Schweike, U.; Garbev, K.; Beuchle, G.; Möller, H. Celitement—A sustainable prospect fort the cement industry. Cem. Int. 2010, 8, 52–67. [Google Scholar]
- Beuchle, G.; Stemmermann, P.; Schweike, U.; Garbev, K. Single-Phase Hydraulic Binder, Methods for the Production Thereof and Structural Material Produced Therewith. U.S. Patent 8,382,892, 26 February 2013; U.S. Patent 8,226,763, 24 July 2012; U.S. Patent 8,226,764, 24 July 2012.
- Hunsinger, H.; Beuchle, G.; Stemmermann, P.; Schweike, U.; Giziewicz, K.; Garbev, K. Method for Producing Dicalcium Silicate. U.S. Patent 975,177,1B2, 5 September 2017. [Google Scholar]
- Irran, E.; Tillmanns, E.; Hentschel, G. Ternesite, Ca5(SiO4)2SO4, a new mineral from the Ettringer Bellerberg/Eifel, Germany. Mineral. Petrol. 1997, 60, 121–132. [Google Scholar] [CrossRef]
- Skalamprinos, S.; Galan, I.; Hanein, T.; Glasser, F. Enthalpy of formation of ye’elimite and ternesite. J. Therm. Anal. Calor. 2018, 313, 2345–2359. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Varela, M.T.; Carmona-Quiroga, P.M. Ternesite as a component of sulfobelitic cements. MATEC Web Conf. 2018, 149, 01011. [Google Scholar] [CrossRef]
- Galuskin, E.; Galuskina, I.O.; Gfeller, F.; Krüger, B.; Kusz, J.; Vapnik, Y.; Dulski, M.; Dzierżanowski, P. Silicocarnotite, Ca5[(SiO4)(PO4)](PO4), a new “old” mineral from the Negev Desert, Israel, and the ternesite-silicocarnotite solid solution: Indicators of high-temperature alteration of pyrometamorphic rocks of the Hatrurim Complex, Southern Levan. Eur. J. Mineral. 2015, 28, 105–123. [Google Scholar] [CrossRef]
- Dickens, B.; Brown, W.E. The crystal structure of Ca5(PO4)2(SiO4) (silico-carnotite). Tschermaks Min. Petr. Mitt. 1971, 16, 1–27. [Google Scholar] [CrossRef]
- Brotherton, P.D.; Epstein, J.M.; Pryce, M.W.; White, A.H. Crystal structure of ‘calcium sulphosilicate’ Ca5(SiO4)2SO4. Aust. J. Chem. 1974, 27, 657–660. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Amer. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Toraya, H.; Yamazaki, S. Simulated annealing structure solution of a new phase of dicalcium silicate Ca2SiO4 and the mechanism of structural changes from α-dicalcium silicate hydrate to α’L-dicalcium silicate via the new phase. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 2002, 58, 613–621. [Google Scholar] [CrossRef]
- Kriven, W.M. Possible alternative transformation tougheners to zirconia: Crystallographic aspects. J. Am. Ceram. Soc. 1988, 71, 1021–1030. [Google Scholar] [CrossRef]
- Ishida, H.; Yamazaki, S.; Sasaki, K.; Okade, Y.; Mitsuda, T. α-Dicalcium silicate hydrate: Praparation, decomposed phase, and its hydration. J. Am. Ceram. Soc. 1993, 76, 1707–1712. [Google Scholar] [CrossRef]
- Yamnova, N.A.; Zubkova, N.V.; Eremin, N.N.; Zadov, A.E.; Gazeev, V.M. Crystal structure of larnite β-C2S and specific features of polymorphic transitions in dicalcium orthosilicate. Kristallografiya 2011, 56, 235–245. [Google Scholar]
- Mumme, W.G.; Hill, R.J.; Bushnell, G.W.; Segnit, E.R. Rietveld crystal structure refinements, crystal chemistry and calculated powder diffraction data for the polymorphs of dicalcium silicate and related phases. Neues Jahrb. Mineral. 1995, 169, 35–68. [Google Scholar]
- Wright, A.F.; Lehmann, M.S. The structure of quartz at 25 and 590 °C determined by neutron diffraction. J. Sol. State Chem. 1981, 36, 371–380. [Google Scholar] [CrossRef]
- Le Page, Y.; Donnay, G. Refinement of the crystal structure of quartz. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 2456–2459. [Google Scholar] [CrossRef]
- Ackermann, R.J.; Sorrell, C.A. Thermal expansion and the high-low transformation in quartz. I. High-temperature X-ray studies. J. Appl. Cryst. 1974, 7, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Mumme, W.G.; Cranswick, L.; Chakoumakos, B. Rietveld crystal structure refinement from high temperature neutron powder diffraction data for the polymorphs of dicalcium silicate. Neues Jahrb. Fuer Mineral. Abh. 1996, 170, 171–188. [Google Scholar]
- Jost, K.H.; Ziemer, B.; Deydel, R. Redetermination of beta-dicalcium silicate. Acta Cryst. B 1977, 33, 1696–1700. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.; Fentiman, C.H.; Jeffery, J.W. Structurally related dicalcium silicate phases. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1980, 36, 353–356. [Google Scholar] [CrossRef]
- Yamnova, N.A.; Yu, K.; Egorow-Tismanko, E.R.; Gobechiya, E.R.; Zadov, A.E.; Gazeev, V.M. New data on polymorphs of anhydrous dicalcium orthosilicate. New Data Miner. 2008, 43, 54–71. [Google Scholar]
- Merlini, M.; Gemmi, M.; Cruciani, G.; Artioli, G. High-temperature behaviour of melilite: In situ X-ray diffraction study of gehlenite-åkermanite-Na melilite solid solution. Phys. Chem. Miner. 2008, 35, 147–155. [Google Scholar] [CrossRef]
- Hauke, K.; Kehren, J.; Böhme, N.; Zimmer, S.; Geisler, T. In Situ Hyperspectral Raman Imaging: A new method to investigate sintering processes of ceramic material at high temperature. Appl. Sci. 2019, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Fierens, P.; Picquet, P. Kinetic studies of the thermal synthesis of calcium silicates above 1400 °C: I, Dynamic thermal synthesis of Ca2SiO4. J. Am. Ceram. Soc. 1975, 58, 50–51. [Google Scholar] [CrossRef]
- Rashid, A.R.; Shamsudin, R.; Abdul Hamid, M.A.; Jalar, A. Low temperature production of wollastonite from limestone and silica sand through solid-state reaction. J. Asian Ceram. Soc. 2014, 2, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Schwiete, H.E.; Krönert, W.; Deckert, K. Existenzbereiche und Stabilisierung von Hochtemperaturmodifikationen des Dicalciumsilicates. Zem. Kalk Gips 1986, 9, 359–366. [Google Scholar]
- Lai, G.-C.; Nojiri, T.; Nakano, K. Studies of the stability of β-Ca2SiO4 doped by minor ions. Cem. Concr. Res. 1992, 22, 743–754. [Google Scholar] [CrossRef]
- Pöhler, K. Fremdionenstabilisierte Dicalciumsilicate: Synthese und Hydraulische Reaktion. Ph.D. Thesis, Martin-Luther-Universität Halle-Wittenberg, Halle, Germany, 2016. [Google Scholar]
- Zhao, Y.; Lu, L.; Wang, S.; Gong, C.; Huang, Y. Modification of Dicalcium Silicates Phase Composition by BaO, SO3, and MgO. J. Inorg. Organomet. Polym. 2013, 23, 930–936. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Hong, S.-H. Influence of Minor Ions on the Stability and Hydrations Rates of β-Dicalcium Silicate. J. Am. Ceram. Soc. 2004, 87, 900–905. [Google Scholar] [CrossRef]
- Fukuda, K.; Maki, I.; Ito, S. Thermoelastic Behavior in Ca2SiO4 Solid Solutions. J. Am. Ceram. Soc. 1996, 79, 2925–2928. [Google Scholar] [CrossRef]
- Fukuda, K.; Ito, S. Highly reactive Remelted Belite. J. Am. Ceram. Soc. 1999, 82, 637–640. [Google Scholar] [CrossRef]
- Garbev, K. Struktur, Eigenschaften und Quantitative Rietveldanalyse von Hydrothermal Kristallisierten Calciumsilikathydraten (C-S-H-Phasen). Ph.D. Thesis, Forschungszentrum Karlsruhe Bericht FZKA 6877 2004. Forschungszentrum Karlsruhe, Karlsruhe, Germany, 2003. [Google Scholar]
- Remy, C.; Andrault, D.; Madon, M. High-Temperature, High-Pressure X-ray Investigation of Dicalcium Silicate. J. Am. Ceram. Soc. 1997, 80, 851–860. [Google Scholar] [CrossRef]
- Forest, J. Connaissance de l’Orthosilicate de Calcium (Knowledge of Dicalcium Silicate). Bull. Soc. Fr. Mineral. Cristallogr. 1971, 94, 118–137. [Google Scholar]
- Xiong, Z.; Liu, X.; Shieh, S.R.; Wang, S.; Chang, L.; Tang, J.; Hong, X.; Zhang, Z.; Wang, H. Some thermodynamic properties of larnite (β-Ca2SiO4) constrained by high T/P experiment and/or theoretical simulation. Am. Min. 2016, 101, 277–288. [Google Scholar] [CrossRef]
- Ishizawa, N.; Miyata, T.; Minato, I.; Marumo, F.; Iwai, S.I. Acta Cryst. 1980, 36, 228–230. [CrossRef]
- Chessin, H.; Hamilton, W.C. Position and thermal parameters of oxygen atoms in calcite. Acta Cryst. 1965, 18, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Merlino, S.; Bonaccorsi, E.; Armbruster, T. The real structure of tobermorite 11 A: Normal and anomalous forms, OD. character and polytypic modifications. Eur. J. Mineral. 2001, 13, 577–590. [Google Scholar] [CrossRef] [Green Version]
- De Villiers, J.P.R. Crystal structures of aragonite, strontianite, and witherite. Am. Min. 1971, 56, 758–766. [Google Scholar]
- Kamhi, S.R. On the structure of vaterite, CaCO3. Acta Cryst. 1963, 16, 770–772. [Google Scholar] [CrossRef]
- Hawthorne, F.C.; Ferguson, R.B. Anhydrous Sulfates II. Refinement of the Crystal Structure of Anhydrite. Can. Min. 1975, 13, 289–292. [Google Scholar]
- Bezou, C.; Norlund Christensen, A.; Cox, D.; Lehmann, M.; Nonat, A. Structures cristallines de Ca SO4 * 0.5(H2O) et CaSO4 * 0.6(H2O). C. R. Seances l’Acad. Sci. 1991, 312, 43–48. [Google Scholar]
- Allan, D.R.; Angel, R.J. A high-pressure structural study of microcline (KAlSi3O8) to 7 GPa. Eur. J. Min. 1997, 9, 263–275. [Google Scholar] [CrossRef]
- Winter, J.K.; Okamura, F.P.; Ghose, S. A high temperature structural study of high albite, monalbite, and the analbite-monalbite phase transition. Am. Min. 1979, 64, 409–423. [Google Scholar]
- Huang, Q.; Chmaissem, O.; Caponi, J.J.; Chaillout, C.; Marezio, M.; Tholence, J.L.; Santoro, A. Neutron powder diffraction study of the crystal structure of Hg Ba2Ca4Cu5O12+d at room temperature and at 10K. Physica C 1994, 227, 1–9. [Google Scholar] [CrossRef]
- Hesse, K.F. Refinement of the crystal structure of wollastonite-2M (parawollastonite). Zeitschr. Kristallogr. 1984, 168, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.V. Reexamination of the crystal structure of melilite. Am. Min. 1953, 38, 643–661. [Google Scholar]
- Louisnathan, S.J. Refinement of the crystal structure of a natural gehlenite, Ca2Al(Al,Si)2O7. Can. Min. 1971, 10, 822–837. [Google Scholar]
- Grice, J.-D. The structure of spurrite, tilleyite and scawtite, and relationships to other silicate-carbonate minerals. Can. Min. 2005, 43, 1489–1500. [Google Scholar] [CrossRef]
- Colville, A.A.; Geller, S. The crystal structure of brownmillerite, Ca2FeAlO5. Acta Cryst. 1971, 27, 2311–2315. [Google Scholar] [CrossRef]
- Cuesta, A.; de la Torre, A.G.; Losilla, E.R.; Santacruz, I.; Aranda, M.A.G. Pseudocubic crystal structure and phase transition in doped ye’elimite. Cry. Growth Design 2014, 14, 5158–5163. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | LoI (wt.%) | SiO2 (wt.%) | CaO (wt.%) | SO3 (wt.%) | Al2O3 (wt.%) | Fe2O3 (wt.%) | MgO (wt.%) | K2O (wt.%) | Na2O (wt.%) |
|---|---|---|---|---|---|---|---|---|---|
| SM(P) | 6.68 | 49.2 | 31.4 | 4.20 | 3.14 | 1.29 | 0.96 | 0.75 | 0.75 |
| SM(D1) | 11.6 | 56.1 | 24.7 | 1.44 | 2.00 | 0.63 | 0.40 | 0.60 | 0.60 |
| SM(D2) | 9.90 | 47.0 | 26.3 | 10.7 | 2.28 | 0.84 | 0.48 | 0.58 | 0.58 |
| SM(D3) | 14.3 | 43.3 | 25.7 | 9.42 | 2.84 | 1.12 | 0.57 | 0.60 | 0.60 |
| Sample | Amorphous (wt.%) | Quartz (wt.%) | Calcite (wt.%) | Vaterite (wt.%) | Aragonite (wt.%) | Toberm. (wt.%) | Feldspars (wt.%) | Anhydrite (wt.%) | Bassanite (wt.%) |
|---|---|---|---|---|---|---|---|---|---|
| SM(P) | 64.7(1.0) | 13.2(2) | 3.20(16) | 1.16(11) | 0.21(6) | 13.3(3) | 0.5(3) | 2.38(14) | 1.36(13) |
| SM(D1) | 52.0(6) | 25.9(2) | 3.95(11) | 2.82(11) | 4.97(17) | 5.33(17) | 2.7(4) | 0.48(10) | 1.87(12) |
| SM(D2) | 60.5(6) | 22.2(2) | 4.25(13) | 2.30(13) | 1.80(14) | 2.4(2) | 5.00(13) | 1.59(16) | |
| SM(D3) | 60.4(6) | 19.64(2) | 4.01(12) | 2.64(12) | 2.69(14) | 3.5(2) | 3.37(12) | 3.75(15) |
| Sample | Weight Loss 30–950 °C (wt.%) | H2O 30–450 °C (wt.%) | CO2 450–830 °C (wt.%) | SO3 830–1400 °C (wt.%) | Amorphous CaCO3 (wt.%) | Amorphous CaSO4 (wt.%) | Amorphous C-S-H (wt.%) |
|---|---|---|---|---|---|---|---|
| SM(P) | 6.3 | 2.9 | 2.4 | 4.9 | 1.4 | 5.6 | 59.1 |
| SM(D1) | 13.9 | 3.7 | 9.7 | 1.6 | 9.0 | 1.4 | 41.2 |
| SM(D2) | 10.2 | 2.0 | 7.6 | 10.9 | 8.6 | 12.5 | 39.6 |
| SM(D3) | 15.9 | 2.5 | 9.9 | 11.3 | 11.8 | 13.3 | 34.9 |
| Sample and Temperature (°C) | Amorph | α’H-C2S | β-C2S | Ternesite | Quartz | Calcite | Lime | Anh. | Wollast. |
|---|---|---|---|---|---|---|---|---|---|
| P 600 | 18.0(8) | 75.7(1.2) | 2.7(4) | ||||||
| P 700 | 37.0(1.0) | 13.4(6) | 44.4(1.1) | 2.8(3) | |||||
| P 800 | 55.2(1.0) | 7.1(4) | 0.6(2) | 24.4(5) | 1.84(15) | 9.2(4) | |||
| P 1000 | 89.9(8) | 2.2(4) | 1.6(2) | 0.03(2) | 1.5(3) | ||||
| P 600 | 95.6(7) | 1.5(3) | 0.25(9) | ||||||
| P 500 | 93.6(1.0) | 2.3(8) | 1.4(3) | 0.26(10) | |||||
| P 400 | 75.2(1.0) | 21.6(8) | 0.8(2) | 0.32(15) | 0.5(3) | ||||
| P 25 | 1.7(9) | 87.4(1.3) | 0.5(3) | 1.3(2) | 1.1(3) | 1.0(3) | |||
| (P)Std | - | 90.0(6) | 0.44(13) | 2.52(7) | 0.63(11) | 0.14(5) | |||
| D1 600 | 18.8(6) | 79.1(7) | 0.52(16) | ||||||
| D1 700 | 16.1(7) | 17.5(2) | 63.4(9) | 0.9(2) | 0.42(14) | ||||
| D1 800 | 38.2(6) | 14.6(4) | 1.0(2) | 38.0(5) | 0.25(8) | 4.6(4) | |||
| D1 1000 | 64.4(5) | 1.9(2) | 7.9(2) | 16.26(17) | 0.21(6) | 4.6(2) | |||
| D1 600 | 67.6(7) | 2.7(2) | 8.3(3) | 4.02(13) | 11.44(17) | 2.09(19) | |||
| D1 500 | 63.6(7) | 3.1(4) | 1.9(2) | 9.8(5) | 3.9(2) | 10.28(17) | 2.3(3) | ||
| D1 400 | 58.4(7) | 7.4(6) | 1.40(17) | 10.0(4) | 4.3(2) | 10.59(18) | 2.5(2) | ||
| D1 25 | 14.6(8) | 46.7(1.6) | 1.95(18) | 9.4(4) | 6.7(3) | 12.0(4) | 0.52(14) | 2.46(19) | |
| (D1)Std | 10.2(1.7) | 4.2(3) | 47.2(9) | 1.17(11) | 8.76(16) | 7.02(15) | 12.07(17) | 4.32(12) | |
| D2 600 | 20.3(6) | 65.9(1.0) | 10.6(9) | ||||||
| D2 700 | 20.0(1.0) | 18.2(6) | 47.0(1.1) | 0.6(2) | 11.6(9) | ||||
| D2 800 | 51.8(1.0) | 11.7(4) | 26.7(6) | 5.9(4) | 2.9(4) | ||||
| D2 1000 | 72.5(7) | 8.1(2) | 5.1(3) | 1.93(6) | 6.90(17) | 1.13(14) | |||
| D2 600 | 72.5(6) | 9.8(3) | 4.87(18) | 0.37(10) | 5.1(2) | 0.44(17) | |||
| D2 500 | 73.0(7) | 0.8(3) | 9.9(2) | 4.4(4) | 0.65(6) | 4.7(2) | 0.57(17) | ||
| D2 400 | 50.3(6) | 22.8(5) | 10.4(3) | 4.9(4) | 0.3(2) | 0.66(6) | 5.0(2) | 0.6(2) | |
| D2 25 | 5.9(5) | 65.2(9) | 11.0(3) | 3.7(2) | 0.84(16) | 1.10(7) | 4.47(17) | 0.86(18) | |
| (D2)Std | 4.8(1.3) | 1.08(1.7) | 62.6(6) | 11.36(17) | 5.62(12) | 1.49(11) | 2.52(5) | 4.50(9) | 0.64(10) |
| D3 600 | 20.2(6) | 65.0(1.0) | 11.2(9) | ||||||
| D3 700 | 30.8(9) | 15.3(5) | 37.4(1.0) | 0.7(2) | 13.3(8) | ||||
| D3 800 | 55(2) | 11.8(6) | 22.3(9) | 7.3(5) | 1.9(5) | ||||
| D3 1000 | 74.2(6) | 9.2(3) | 4.9(2) | 0.90(10) | 5.1(2) | 1.2(2) | |||
| D3 600 | 73.2(7) | 10.6(3) | 4.7(3) | 0.25(5) | 3.8(2) | 0.7(3) | |||
| D3 500 | 74.8(8) | 0.9(3) | 10.2(3) | 3.0(4) | 0.25(6) | 3.6(29 | 0.74(19) | ||
| D3 400 | 53.9(7) | 20.3(5) | 10.6(3) | 3.2(4) | 1.0(3) | 0.23(6) | 3.6(2) | 0.9(3) | |
| D3 25 | 5.6(5) | 64.7(1.0) | 11.1(3) | 4.1(2) | 0.77(16) | 0.50(7) | 4.00(17) | 1.06(18) | |
| (D3)Std | - | 1.35(18) | 65.4(6) | 8.31(16) | 5.57(13) | 1.23(12) | 1.84(5) | 7.46(11) | 0.99(11) |
| D3_step10 950 | 65.9(5) | 19.0(2) | 1.71(9) | 0.04(2) | 0.18(7) | 4.7(3) | |||
| D3_step10 25 | 1.1(2) | 67.3(5) | 14.10(15) | 0.45(10) | 0.41(9) | 0.16(7) | 4.43(10) | ||
| D3_2.5 950 | 62.9(5) | 4.5(3) | 6.0(2) | 15.6(2) | 6.2(2) | 0.9(2) | |||
| D3_2.5 1000 | 66.7(5) | 11.8(2) | 3.14(18) | 6.77(9) | 5.61(15) | 1.17(18) | |||
| D3_2.5 25 | 10.2(5) | 49.9(1.1) | 18.6(5) | 1.6(29 | 1.83(15) | 4.68(12) | 4.23(15) | 0.90(17) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullrich, A.; Garbev, K.; Bergfeldt, B. In Situ X-ray Diffraction at High Temperatures: Formation of Ca2SiO4 and Ternesite in Recycled Autoclaved Aerated Concrete. Minerals 2021, 11, 789. https://doi.org/10.3390/min11080789
Ullrich A, Garbev K, Bergfeldt B. In Situ X-ray Diffraction at High Temperatures: Formation of Ca2SiO4 and Ternesite in Recycled Autoclaved Aerated Concrete. Minerals. 2021; 11(8):789. https://doi.org/10.3390/min11080789
Chicago/Turabian StyleUllrich, Angela, Krassimir Garbev, and Britta Bergfeldt. 2021. "In Situ X-ray Diffraction at High Temperatures: Formation of Ca2SiO4 and Ternesite in Recycled Autoclaved Aerated Concrete" Minerals 11, no. 8: 789. https://doi.org/10.3390/min11080789
APA StyleUllrich, A., Garbev, K., & Bergfeldt, B. (2021). In Situ X-ray Diffraction at High Temperatures: Formation of Ca2SiO4 and Ternesite in Recycled Autoclaved Aerated Concrete. Minerals, 11(8), 789. https://doi.org/10.3390/min11080789