
Silicon-Oxygen Region Infrared and Raman Analysis of Opals: The Effect of Sample Preparation and Measurement Type


Abstract
1. Introduction
- A low-energy band at approximately 470 cm−1: ascribed to rocking of the Si-O-Si bridge.
- A medium-energy band at approximately 790 cm−1: ascribed to a motion where the bridging oxygen in Si-O-Si moves perpendicular to the line joining the silicon atoms. This has been described as both a bending motion and a symmetric Si-O-Si stretch.
- A higher-energy band at approximately 1100 cm−1: ascribed to an antisymmetric Si-O-Si stretch.
2. Materials and Methods
3. Overview of Previous Silica IR Studies
4. Results
4.1. SEM Examination
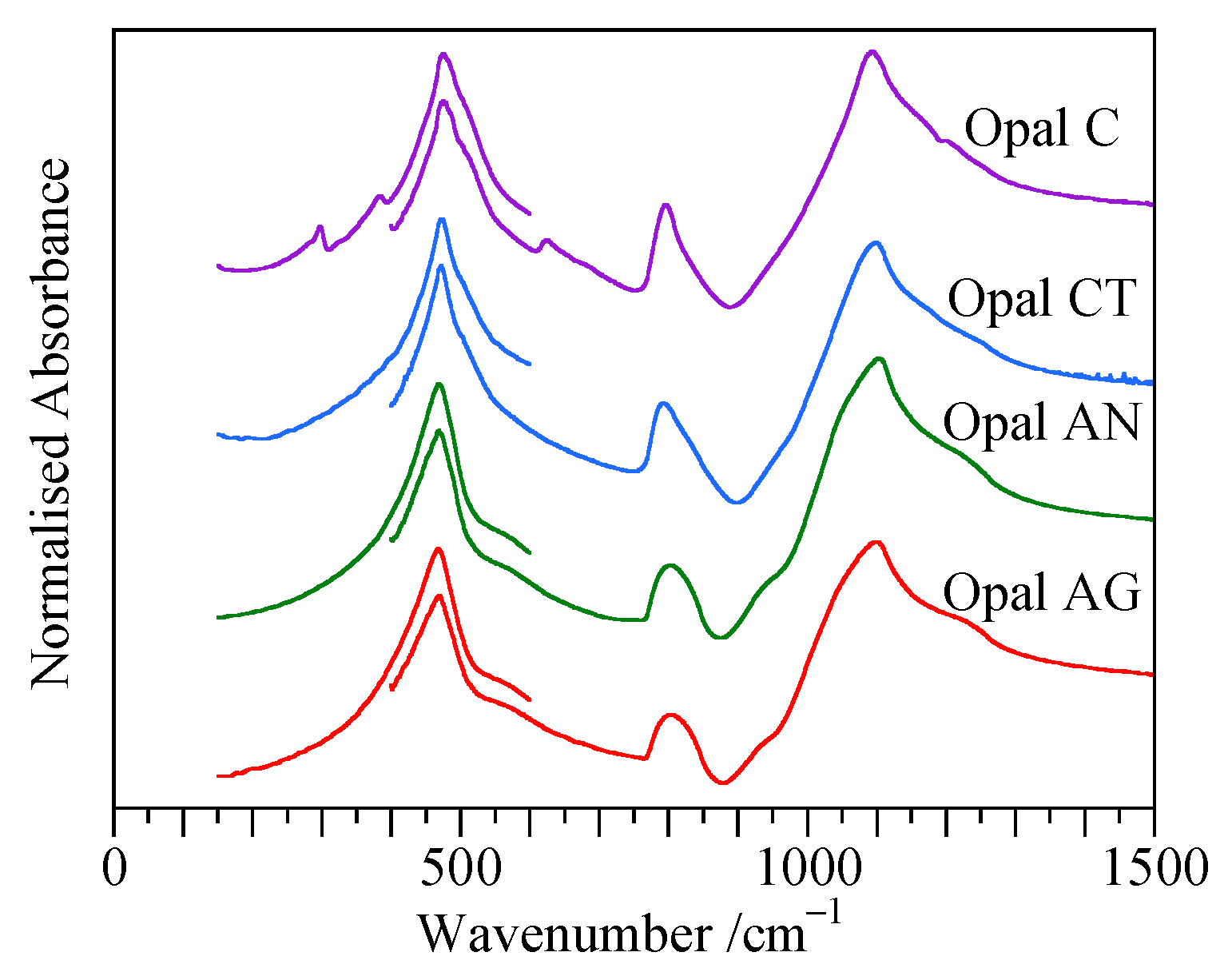
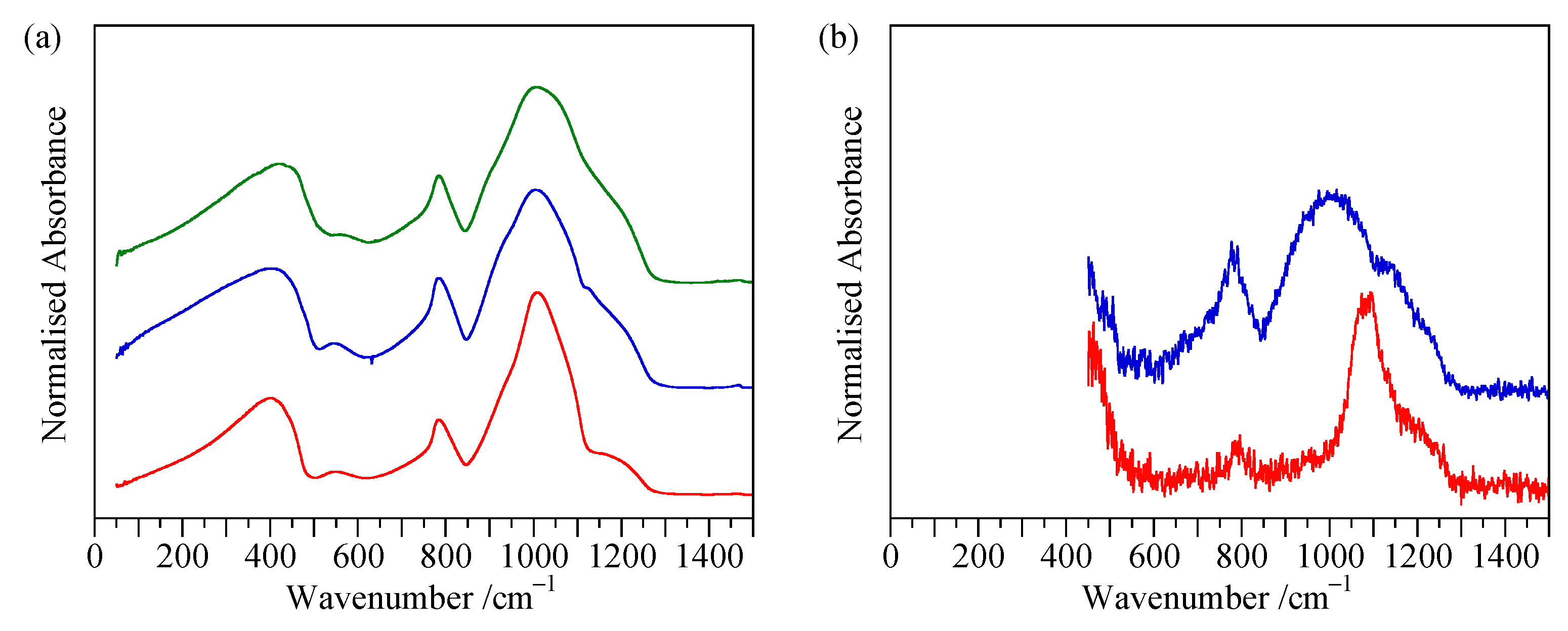
4.2. Transmission Spectra
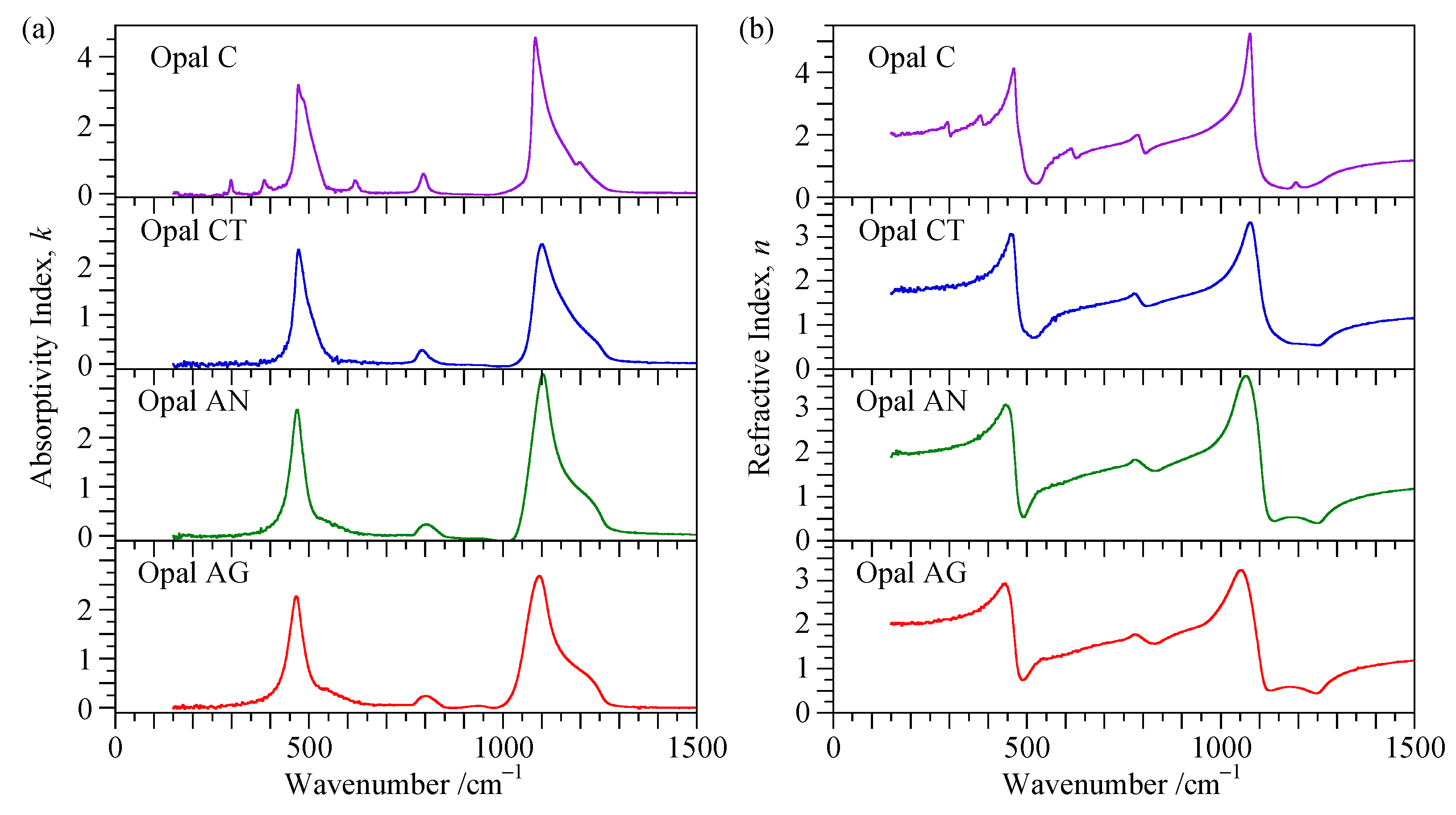
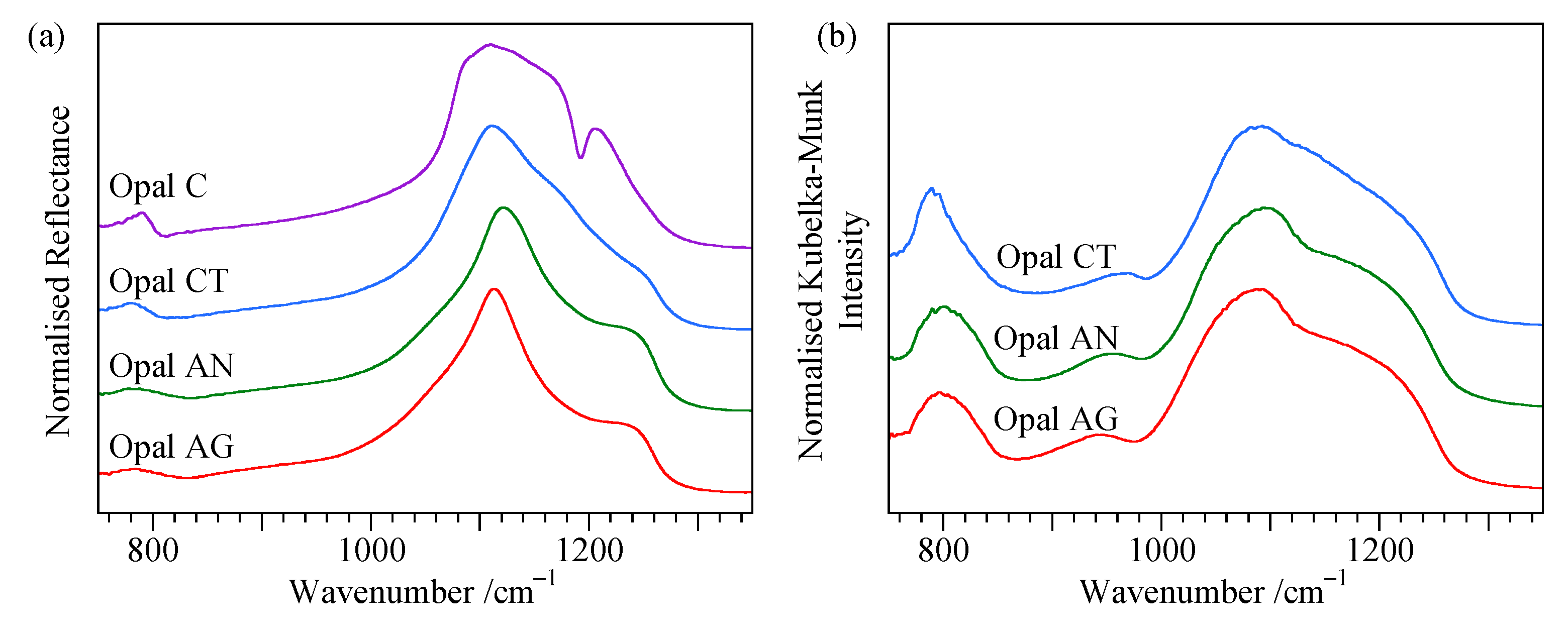
4.3. Reflectance Spectra
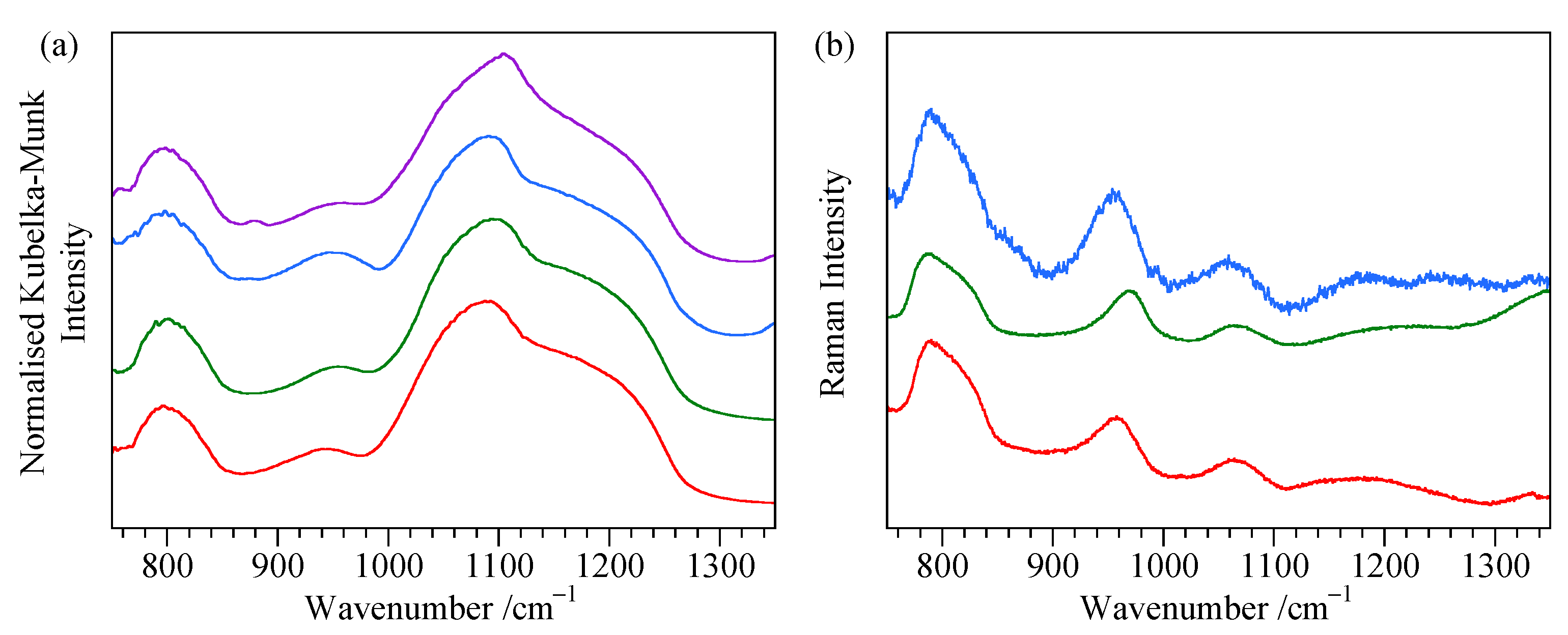
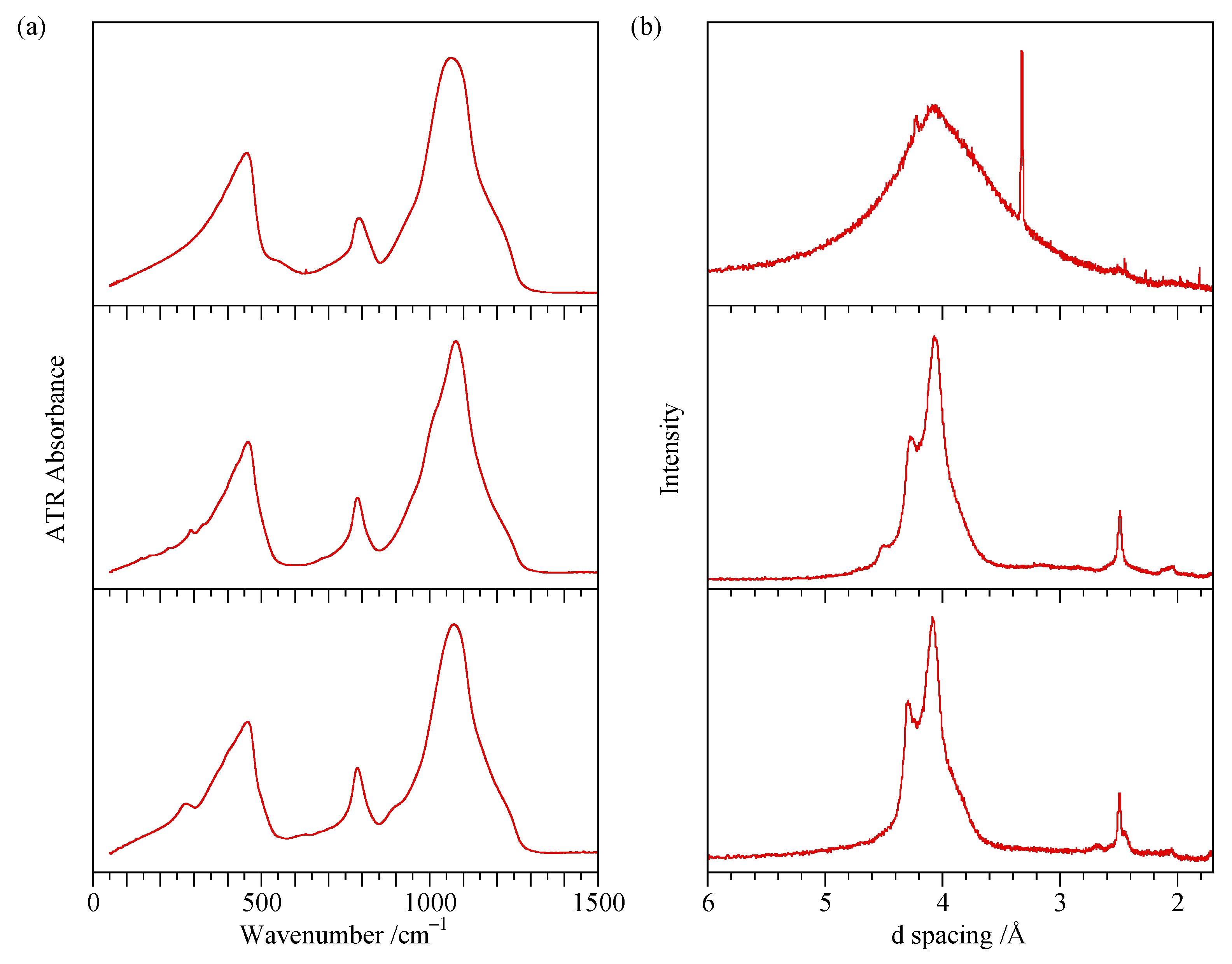
4.4. ATR Measurements of Ground Samples
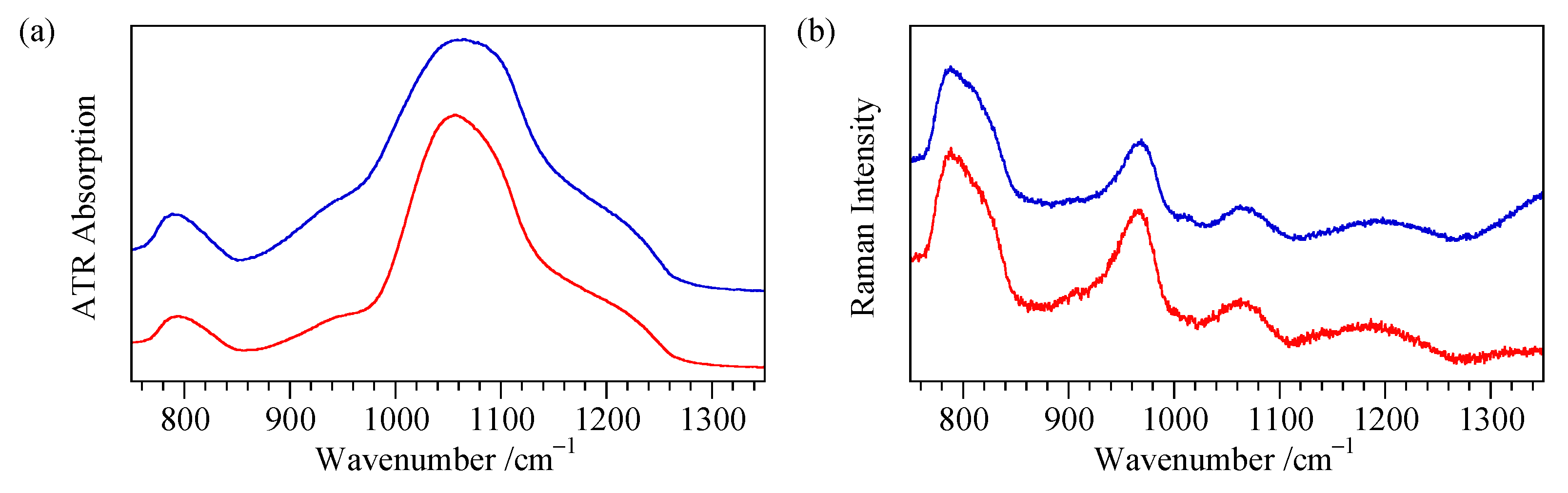
4.5. ATR Measurement of Flat Surfaces
4.6. Calculation of Si-O-Si Bond Angles
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
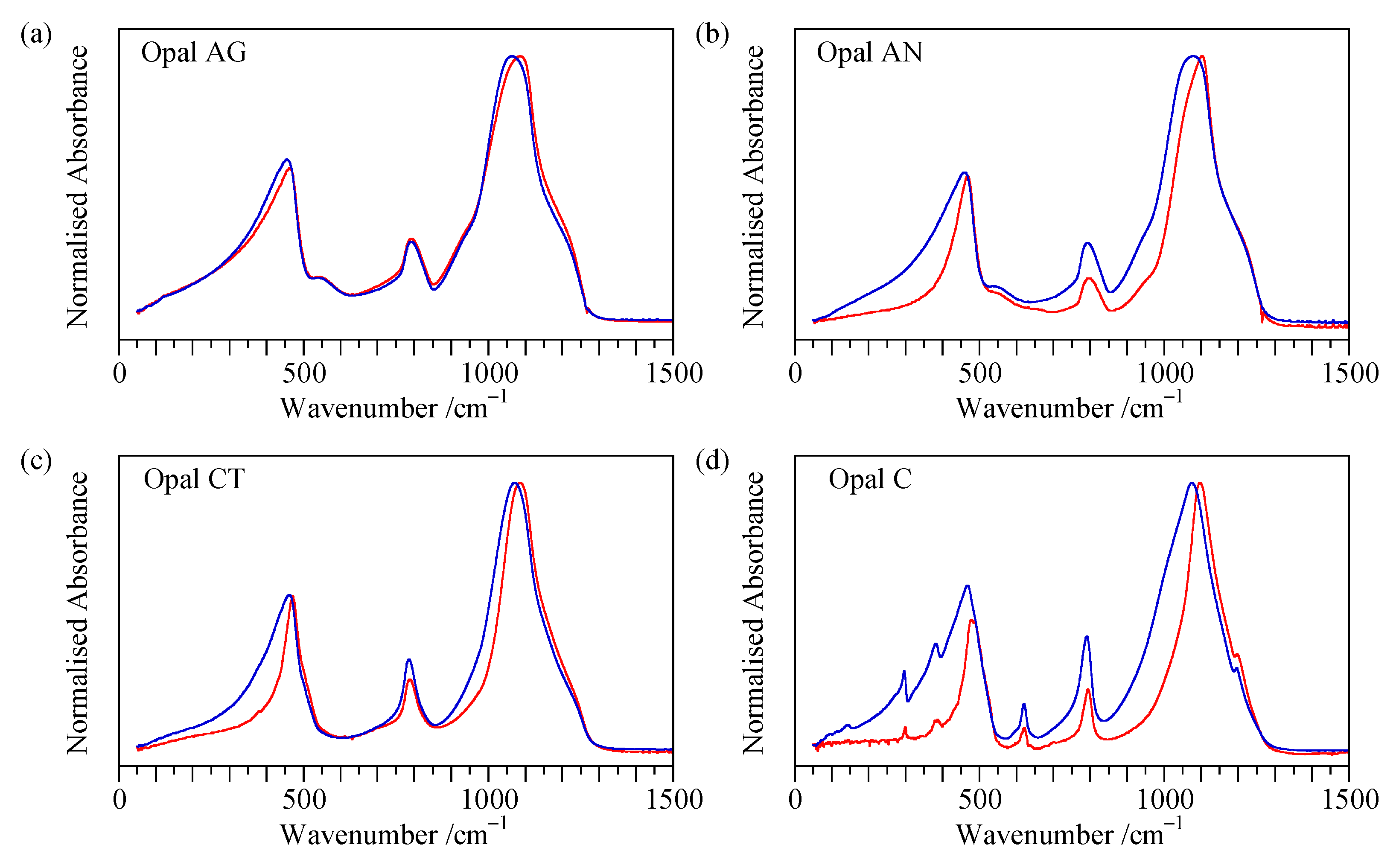
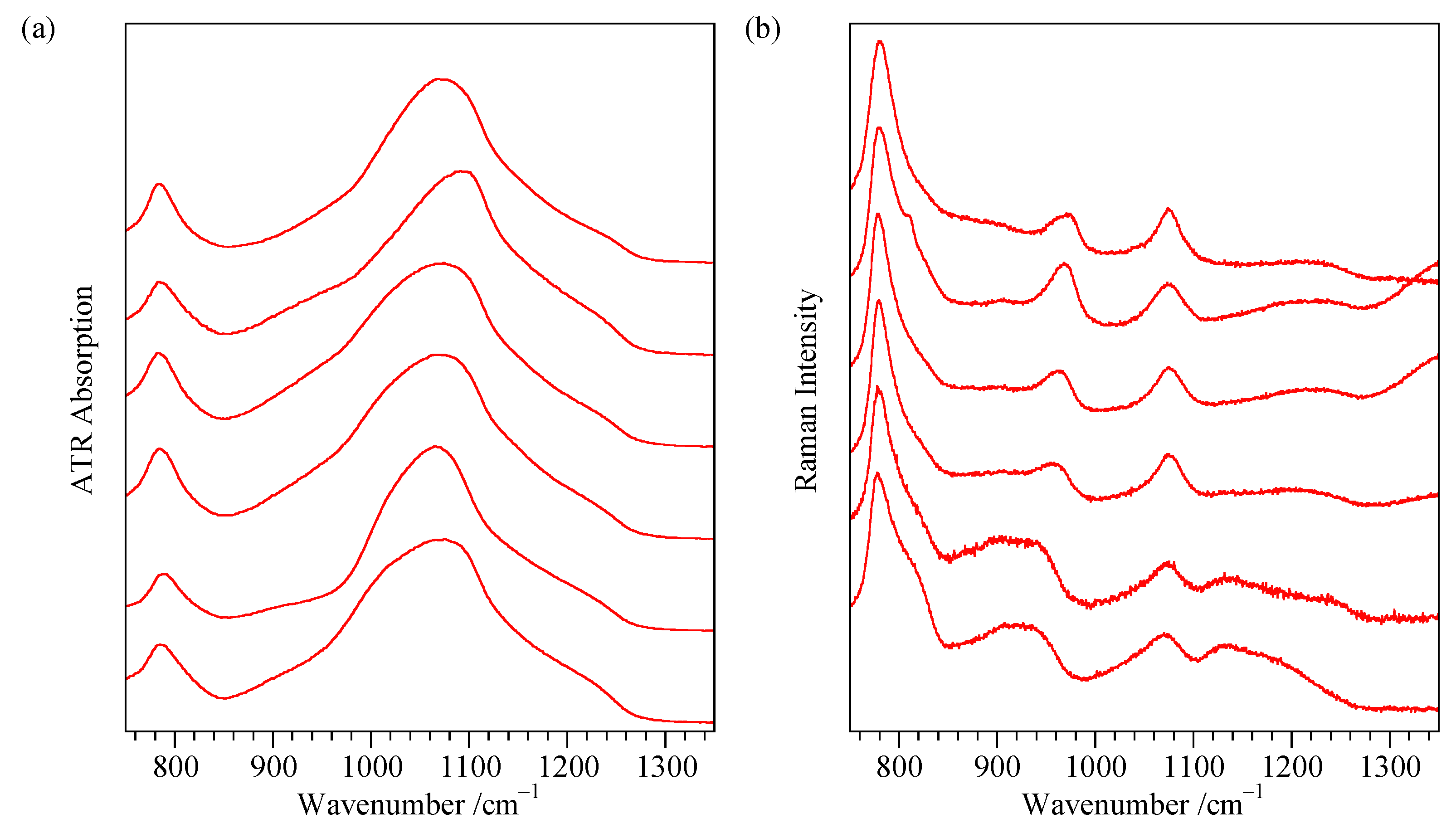
Appendix A. Additional Flat Sample ATR Spectra

Appendix B. Study of Impurities


References
- Jones, J.B.; Segnit, E.R. The nature of opal I. Nomenclature and constituent phases J. Geol. Soc. Aust. 1971, 18, 57–68. [Google Scholar] [CrossRef]
- Sanders, J.V. Colour of precious opal. Nature 1964, 204, 1151–1153. [Google Scholar] [CrossRef]
- Sanders, J.V. Close-packed structures of spheres of two different sizes I. Observations of natural opal. Philos. Mag. A 1980, 42, 705–720. [Google Scholar] [CrossRef]
- Gaillou, E.; Fritsch, E.; Aguilar-Reyes, B.; Rondeau, B.; Post, J.; Barreau, A.; Ostroumov, M. Common gem opal: An investigation of micro- to nano-structure. Am. Mineral. 2008, 93, 1865–1873. [Google Scholar] [CrossRef]
- Liesegang, M.; Milke, R. Australian sedimentary opal-A and its associated minerals: Implications for natural silica sphere formation. Am. Mineral. 2014, 99, 1488–1499. [Google Scholar] [CrossRef]
- Floerke, O.W.; Jones, J.B.; Segnit, E.R. The genesis of hyalite. Neues Jahrb. Mineral. Mon. 1973, 2, 82–89. [Google Scholar]
- Curtis, N.J.; Gascooke, J.R.; Johnston, M.R.; Pring, A. A review of the classification of opals with reference to recent new localities. Minerals 2019, 9, 299. [Google Scholar] [CrossRef]
- Wilson, M.J. The structure of opal-CT revisited. J. Non-Cryst. Solids 2014, 405, 68–75. [Google Scholar] [CrossRef]
- Fröhlich, F. The opal-CT nanostructure. J. Non-Cryst. Solids 2020, 533, 119938. [Google Scholar]
- Gaines, R.V.; Skinner, H.C.W.; Foord, E.E.; Mason, B.; Rozenzweig, A. Dana’s New Mineralogy; John Wiley and Sons: New York, NY, USA, 1997. [Google Scholar]
- Bell, R.J.; Bird, N.F.; Dean, P. The vibrational spectra of vitreous silica, germania and beryllium fluoride. J. Phys. C Solid State Phys. 1968, 1, 299–303. [Google Scholar] [CrossRef]
- Galeener, F.L. Band limits and the vibrational spectra of tetrahedral glasses. Phys. Rev. B 1979, 19, 4292–4297. [Google Scholar] [CrossRef]
- Galeener, F.L.; Leadbetter, A.J.; Stringfellow, M.W. Comparison of the neutron, Raman, and infrared vibrational spectra of vitreous SiO2, GeO2, and BeF2. Phys. Rev. B 1983, 27, 1052–1078. [Google Scholar] [CrossRef]
- Almeida, R.M. Detection of LO modes in glass by infrared reflection spectroscopy at oblique incidence. Phys. Rev. B 1992, 45, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Innocenzi, P. Infrared spectroscopy of sol–gel derived silica-based films: A spectra-microstructure overview. J. Non-Cryst. Solids 2003, 316, 309–319. [Google Scholar] [CrossRef]
- Goryniuk, M.C.; Rivard, B.A.; Jones, B. The reflectance spectra of opal-A (0.5–25 μm) from the Taupo Volcanic Zone: Spectra that may identify hydrothermal systems on planetary surfaces. Geophys. Res. Lett. 2004, 31, L24701. [Google Scholar] [CrossRef]
- Tomozawa, M.; Hong, J.-W.; Ryu, S.-R. Infrared (IR) investigation of the structural changes of silica glasses with fictive temperature. J. Non-Cryst. Solids 2005, 351, 1054–1060. [Google Scholar] [CrossRef]
- Froehlich, F.; Poupeau, G.; Badou, A.; Bourdonnec, F.X.L.; Sacquin, Y.; Dubernet, S.; Bardintzeff, J.M.; Eran, M.V.; Smith, D.C.; Diemer, E. Libyan desert glass: New field and Fourier transform infrared data. Meteorit. Planet. Sci. 2013, 48, 2517–2530. [Google Scholar] [CrossRef]
- Elzea, J.M.; Rice, S.B. TEM and X-Ray diffraction evidence for cristobalite and tridymite stacking sequences in opal. Clays Clay Miner. 1996, 44, 492–500. [Google Scholar] [CrossRef]
- Graetsch, H.; Gies, H.; Topalovic, I. NMR, XRD and IR study on microcrstalline opal. Phys. Chem. Miner. 1994, 21, 166–175. [Google Scholar] [CrossRef]
- Adamo, I.; Ghisoli, C.; Caucia, F. A contribution to the study of FTIR spectra of opals. Neues Jahrb. Mineral.-Abh. 2010, 187, 63–68. [Google Scholar] [CrossRef]
- Ostrooumov, M. A Raman, infrared and XRD analysis of the instability in volcanic opals from Mexico. Spectrochim. Acta Part A 2007, 68, 1070–1076. [Google Scholar] [CrossRef]
- Rice, S.B.; Freund, H.; Huang, W.-L.; Clouse, J.A.; Isaacs, C.M. Application of Fourier transform infrared spectroscopy to silica diagenis: The opal-A to opal-CT transformation. J. Sediment. Res. 1995, A65, 639–647. [Google Scholar]
- Auslender, M.; Ivry, J.; Pinhas, N.; Hava, S. Optical properties of silica opal templates in the visible and infrared. Opt. Mater. 2008, 30, 1735–1738. [Google Scholar] [CrossRef]
- Eckert, J.; Gourdon, O.; Jacob, D.E.; Meral, C.; Monteiro, P.J.M.; Vogel, S.C.; Wirth, R.; Wenk, H.-R. Ordering of water in opals with different microstructures. Eur. J. Mineral. 2015, 27, 203–213. [Google Scholar] [CrossRef]
- Caucia, F.; Marinoni, L.; Bordini, V.; Ghisoli, C.; Adamo, I. Physical and chemical properties of some Italian opals. Period. Mineral. 2012, 81, 93–106. [Google Scholar]
- Fritsch, E.; Gaillou, E.; Ostroumov, M.; Rondeau, B.; Devouard, B.; Barreau, A. Relationship between nanostructure and optical absorption in fibrous pink opals from Mexico and Peru. Eur. J. Mineral. 2004, 16, 743–752. [Google Scholar] [CrossRef]
- Graetsch, H. Structural characteristics of opaline and microcrystalline silica minerals. Rev. Mineral. Geochem. 1994, 29, 209–232. [Google Scholar]
- Zhao, K.; Bai, F. Crystallinity and play-of-colour in gem opal with digit patterns from Wegel Tena, Ethiopia. Minerals 2020, 10, 625. [Google Scholar] [CrossRef]
- Ivanov, V.G.; Reyes, B.A.; Fritsch, E.; Faulques, E. Vibrational states in opal revisited. J. Phys. Chem. C 2011, 115, 11968–11975. [Google Scholar] [CrossRef]
- Durman, R.; Favre, P.; Jayasooriya, U.A.; Kettle, S.F.A. Longitudinal optical-transverse optical (L.O.-T.O.) splitting on internal modes in the Raman spectra of noncentric crystals. J. Crystallogr. Spectrosc. Res. 1987, 17, 431–484. [Google Scholar] [CrossRef]
- Armanda, P.; Hermet, P.; Bantignies, J.-L.; Haidoux, A.; Maurin, D.; Ménaert, B.; Peña, A.; Papet, P. Optical properties in the infrared range of the birefringent α-GeO2 single crystal. Mater. Res. Bull. 2020, 129, 110881. [Google Scholar] [CrossRef]
- Harbecke, B.; Heinz, B.; Grosse, P. Optical properties of thin films and the Berreman effect. Appl. Phys. A 1985, 38, 263–267. [Google Scholar] [CrossRef]
- Wang, T.B.; Liu, Z.G.; Tan, C.Z. Relationship between the frequency of the main LO mode of silica glass and angle of incidence. J. Chem. Phys. 2003, 119, 505–508. [Google Scholar] [CrossRef]
- Lippincott, E.R.; Valkenburg, A.v.; Weir, C.E.; Bunting, E.N. Infrared studies of polymorphs of silicon dioxide and germaniun dioxide. J. Res. Natl. Bur. Stand. 1958, 61, 61–70. [Google Scholar] [CrossRef]
- Etchepare, J.; Merian, M.; Kaplan, P. Vibrational normal modes of SiO2, II Cristobalite and tridymite. J. Chem. Phys. 1978, 68, 1531–1537. [Google Scholar] [CrossRef]
- Bates, J.B. Raman spectra of α and β cristobalite. J. Chem. Phys. 1972, 57, 4042–4047. [Google Scholar] [CrossRef]
- Michalski, J.R.; Kraft, M.D.; Diedrich, T.; Sharp, T.G.; Christensen, P.R. Thermal emission spectroscopy of the silica polymorphs and considerations for remote sensing of Mars. Geophys. Res. Lett. 2003, 30, PLA 2-1–PLA 2-4. [Google Scholar] [CrossRef]
- Almeida, R.M.; Panto, C.G. Structural investigation of silica gel films by infrared spectroscopy. J. Appl. Phys. 1990, 68, 4225–4332. [Google Scholar] [CrossRef]
- McMillan, P.F.; Remmele, R.L. Hydroxyl sites in SiO2 glass: A note on infrared and Raman spectra. Am. Mineral. 1986, 71, 772–778. [Google Scholar]
- Capeletti, L.B.; Zimnoch, J.H. Fourier transform infrared and Raman characterization of silica-based materials. In Applications of Molecular Spectroscopy to Current Research in the Chemical and Biological Sciences; Stauffer, M.T., Ed.; IntechOpen: Rijeka, Croatia, 2016. [Google Scholar]
- Fidalgo, A.; Ilharco, L.M. Chemical tailoring of porous silica xerogels: Local structure by vibrational spectroscopy. Chem. Eur. J. 2004, 10, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Lichvar, P.; Liska, M.; Galusek, D. What is the true Kramers-Kronig transform? Ceramics 2002, 46, 25–27. [Google Scholar]
- Kingma, K.J.; Hemley, R.J. Raman spectroscopic study of microcrystalline silica. Am. Mineral. 1994, 79, 269–273. [Google Scholar]
- Ilieva, A.; Mihailova, B.; Tsintov, Z.; Petrov, O. Structural state of microcrystalline opals: A Raman spectroscopic study. Am. Mineral. 2007, 92, 1325–1333. [Google Scholar] [CrossRef]
- Galeener, F.L.; Lucovsky, G. Longitudinal optical vibrations in glasses: GeO2 and SiO2. Phys. Rev. Lett. 1976, 37, 1474–1477. [Google Scholar] [CrossRef]
- Mueller, C.M.; Molinelli, A.; Karlowatz, M.; Aleksandrov, A.; Orlando, T.; Mizaikoff, B. Infrared attenuated total reflection spectroscopy of quartz and silica micro- and nanoparticulate films. J. Phys. Chem. C 2011, 116, 37–43. [Google Scholar] [CrossRef]
- Fritsch, E.; Gaillou, E.; Rondeau, B.; Barreau, A.; Albertini, D.; Ostroumov, M. The nanostructure of fire opal. J. Non-Cryst. Solids 2006, 352, 3957–3960. [Google Scholar] [CrossRef]
- Ruff, S.W.; Farmer, J.D.; Calvin, W.M.; Herkenhoff, K.E.; Johnson, J.R.; Morris, R.V.; Rice, M.S.; Arvidson, R.E.; Bell, J.F., III; Christensen, P.R.; et al. Characteristics, distribution, origin, and significance of opaline silica observed by the Spirit rover in Gusev crater, Mars. J. Geophys. Res. 2011, 116, E00F23. [Google Scholar] [CrossRef]
- Luo, J.; Smith, N.J.; Pantanob, C.G.; Kima, S.H. Complex refractive index of silica, silicate, borosilicate, and boroaluminosilicate glasses—Analysis of glass network vibration modes with specular-reflection IR spectroscopy. J. Non-Cryst. Solids 2018, 494, 94–103. [Google Scholar] [CrossRef]
- Su, Q.; Xia, Y.; Wang, L.; Liu, J.; Shi, W. Optical and electrical properties of different oriented CVD diamond films. Appl. Surf. Sci. 2006, 252, 8239–8242. [Google Scholar] [CrossRef]
- Frondel, C. The System of Mineralogy, Volume III Silica Materials; John Wiley and Sons: New York, NY, USA; London, UK, 1962. [Google Scholar]
- Rondeau, B.; Fritz, E.; Mazzero, F.; Gauthier, J.-P.; Cencki-Tok, B.; Bekele, E.; Gaillou, E. Play-of-color opal from Wegel Tena, Wollo Province, Ethiopia. Gems Gemol. 2010, 46, 90–105. [Google Scholar] [CrossRef]
- Kitamura, R.; Pilon, L.; Jonasz, M. Optical constants of silica glass from extreme ultraviolet to far infrared at near room temperature. Appl. Opt. 2007, 46, 8118–8133. [Google Scholar] [CrossRef]
- Amma, S.-i.; Luo, J.; Pantano, C.G.; Kim, S.H. Specular reflectance (SR) and attenuated total reflectance (ATR) infrared (IR) spectroscopy of transparent flat glass surfaces: A case study for soda lime float glass. J. Non-Cryst. Solids 2015, 428, 189–196. [Google Scholar] [CrossRef]
- Ishikawa, K.; Suzuki, K.; Okamura, S. Asymmetric peak line shape of infrared dielectric function spectra for thermally grown silicon dioxide films. J. Appl. Phys. 2000, 88, 7150–7156. [Google Scholar] [CrossRef]
- Li, Y.; Yan, H.; Yang, K.; Yao, C.; Wang, Z.; Zou, X.; Yan, C.; Yuan, X.; Ju, X.; Yang, L. Surface molecular structure defects and laser-induced damage threshold of fused silica during a manufacturing process. Sci. Rep. 2017, 7, 17870. [Google Scholar] [CrossRef] [PubMed]
- Mozzi, R.L.; Warren, B.E. The structure of vitreous silica. J. Appl. Crystallogr. 1969, 2, 164–172. [Google Scholar] [CrossRef]
- Yuan, X.; Cormack, A.N. Si–O–Si bond angle and torsion angle distribution in vitreous silica and sodium silicate glasses. J. Non-Cryst. Solids 2003, 319, 31–43. [Google Scholar] [CrossRef]
- Jones, J.B.; Segnit, E.R. Genesis of cristobalite and tridymite at low temperature. J. Geol. Soc. Aust. 1972, 18, 419–422. [Google Scholar] [CrossRef]
- Takada, A.; Glaser, K.J.; Bell, R.G.; Catlow, C.R.A. Molecular dynamics study of tridymite. IUCrJ 2018, 5, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.K. Opal, cristobalite and tridymite: Noncrystallinity versus crystallinity, nomenclature of the silica minerals and bibliography. Powder Diffr. 1998, 13, 2–19. [Google Scholar] [CrossRef]
- Konnert, J.H.; Appleman, D.E. The crystal structure of low tridymite. Acta Crystallogr. 1978, B34, 391–403. [Google Scholar] [CrossRef]
- Dutkiewicz, A.; Landgrebe, T.C.W.; Rey, P.F. Origin of silica and fingerprinting of Australian sedimentary opals. Gondwana Res. 2015, 27, 786–795. [Google Scholar] [CrossRef]
- Gaillou, E.; Delaunay, A.; Rondeau, B.; Bouhnik-le-Coz, M.; Fritsch, E.; Corren, G.; Monnier, C. The geochemistry of gem opals as evidence of their origin. Ore Geol. Rev. 2008, 34, 113–126. [Google Scholar] [CrossRef]
- Udvardi, B.; Kovács, I.J.; Fancsik, T.; Kónya, P.; Bátori, M.; Stercel, F.; Falus, G.; Szalai, Z. Effects of particle size on the attenuated total reflection spectrum of minerals. Appl. Spectrosc. 2017, 71, 1157–1168. [Google Scholar] [CrossRef]
- Karlowatz, M.; Aleksandrov, A.; Orlando, T.; Cathcart, J.M.; Mizaikoff, B. The influence of wetting and drying cycles on mid-infrared attenuated total reflection spectra of quartz: Understanding spectroscopy of disturbed soil. Proc. SPIE Int. Soc. Opt. Eng. 2004, 5415, 629–637. [Google Scholar]
- Campbell, K.A.; Guido, D.M.; Gautret, P.; Foucher, F.; Ramboz, C.; Westall, F. Geyserite in hot-spring siliceous sinter: Window on Earth’s hottest terrestrial (paleo)environment and its extreme life. Earthe Sci. Rev. 2015, 148, 44–64. [Google Scholar] [CrossRef]
- Jones, B.; Renaut, R.W. Microstructural changes accompanying the opal-A to opal-CT transition: New evidence from the siliceous sinters of Geysir, Haukadalur, Iceland. Sedimentology 2007, 54, 921–948. [Google Scholar] [CrossRef]
- Urusov, V.S.; Shvanskaya, L.V.; Bychkov, A.Y.; Mokhov, A.V.; Labutova, E.A. Microstructure investigations of Kamchatka geyserites. Mosc. Univ. Geol. Bull. 2008, 63, 311–319. [Google Scholar] [CrossRef]
- Lynne, B.Y.; Campbell, K.A.; Moore, J.N.; Browne, P.R.L. Diagenesis of 1900-year-old siliceous sinter (opal-A to quartz) at Opal Mound, Roosevelt Hot Springs, Utah, U.S.A. Sediment. Geol. 2005, 179, 249–278. [Google Scholar] [CrossRef]
- Segnit, E.R.; Stevens, T.J.; Jones, J.B. The role of water in opals. J. Geol. Soc. Aust. 1965, 12, 211–226. [Google Scholar] [CrossRef]
- Bobon, M.; Christy, A.A.; Kluvanec, D.; Illasova, L. State of water molecules and silanol groups in opal minerals: A near infrared spectrscopic study of opals from Slovakia. Phys. Chem. Miner. 2011, 38, 809–818. [Google Scholar] [CrossRef]
- Day, R.; Jones, B. Variations in water content in opal-A and opal-CT from geyser discharge aprons. J. Sediment. Res. 2008, 78, 301–315. [Google Scholar] [CrossRef]
- Liesegang, M.; Milke, R. Silica colloid ordering in a dynamic sedimentary environment. Minerals 2018, 8, 12. [Google Scholar] [CrossRef]
- Schmidt, P.; Bellot-Gurlet, L.; Sciau, P. Moganite detection in silica rocks using Raman and inrared spectroscopy. Eur. J. Mineral. 2013, 25. [Google Scholar]
- Thomas, P.; Chauviré, B.; Flower-Donaldson, K.; Aldridge, L.; Smallwood, A.; Liu, B. FT-NIR and DSC characterisation of water in opal. Ceram. Int. 2020, 46, 29443–29450. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Opal Type | Locality | Comments |
|---|---|---|---|
| CP-2 | AG | Coober Pedy, South Australia, Australia | |
| G1419 | AN | Mt Cora, New South Wales, Australia | |
| G1442 | AG | William Creek, South Australia, Australia | |
| G7532 | AG | Queensland | |
| G7975 | CT | Mexico | |
| G8608 | AG | White Cliffs, New South Wales, Australia | |
| G8877 | AN | Dalby, Queensland, Australia | |
| G9260 | AG | Australia | |
| G9590 | AG | Coober Pedy, South Australia, Australia | |
| G9594 | AG | Coober Pedy, South Australia, Australia | |
| G9812 | AG | Coober Pedy, South Australia, Australia | |
| G9942 | CT | Angaston, South Australia, Australia | |
| G9887 | CT | Curdimurka, South Australia, Australia | |
| G13740 | CT | Monto, Queensland, Australia | |
| G13743 | CT | Siebengebirge, Germany | |
| G13767 | AG | Lightning Ridge, New South Wales, Australia | Mix of samples |
| G13771 | AG | White Cliffs, New South Wales, Australia | |
| G14581 | AG to CT | Andamooka, South Australia, Australia | Transitional form |
| G15584 | AG | White Cliffs, New South Wales, Australia | |
| G21471 | A | Rotorua, New Zealand | Geyserite |
| G25374 | CT | Somalia (possibly Ethiopia) | |
| G32226 | CT | St Austell, England | |
| G32925 | CT | Zelinograd, Qazaqstan | |
| G32740 | AN | Valec, Czechia | |
| G32752 | CT | Afar, Ethiopia | Mix of samples |
| G33912 | CT | Acari, Peru | |
| G34475 | A | Dubnik, Slovakia | Hydrophane |
| G NEW01 | A | Caldes de Malvella, Spain | Menilite |
| G NEW04 | CT | Mt Lyobo, Tanzania | |
| G NEW18 | CT to C | Oregon, USA | Transitional form |
| G NEW22 | AG | Lambina, South Australia, Australia | |
| G NEW23 | A | Caldes de Malvella, Spain | Menilite |
| M5081 | C | Iceland | |
| MS-1 | AN | Mt Squaretop, Queensland, Australia | |
| MS-4 | AN | Mt Squaretop, Queensland, Australia | |
| OOC4 | AG to CT | Mazaron, Spain | Transitional form |
| T1664 | C | Guanajuanto Mexico | |
| T1665 | A | Rotorua, New Zealand | Geyserite |
| T2222 | CT | Mt Barker, South Australia, Australia | |
| T22824 | AG to CT | Megyasro, Hungary | Transitional form |
| Frequency (cm−1) | Description | Substrate | References |
|---|---|---|---|
| 457–490 | TO1: Si-O-Si rock | Silica, opal-A and glass | [13,15,16,17] |
| 507 | LO1 Si-O-Si rock | Silica and glass | [13,15] |
| 784–810 | TO2; Si-O-Si symmetric stretch | Silica and glass | [13,15,17] |
| 820 | LO2; Si-O-Si symmetric stretch | Silica and glass | [13,15] |
| 870 | H-bonded Si-OH stretch | Opal-A | [16] |
| 870 | Non-bridging oxygen vibration | Silica | [47] |
| 950–985 | Isolated Si-OH stretch | Opal-A | [9,16,40,41,42] |
| 1040–1260 | TO3: Si-O-Si antisymmetric stretch | Xerogel, glass, opal-A, silica | [13,15,16,17,34,41,42] |
| 1111 | Antisymmetric stretch (Reststrahlen) | Opal-A | [16] |
| 1130–1286 | LO3: Si-O-Si antisymmetric stretch | Xerogel, glass | [13,15,16,17,34,41,42] |
| 1170 | LO4: disorder-induced mode | Silica | [15] |
| 1200 | TO4: disorder-induced mode | Silica | [15] |
| 1266 | Antisymmetric stretch (Reststrahlen) | Opal-A | [16] |
| 1361 | Christiansen feature | Opal-A | [16] |
| Sample | Low-Energy Band (Synchrotron) /cm−1 | Low-Energy Band (Nicolet) /cm−1 | Medium-Energy Band /cm−1 | High-Energy Band /cm−1 |
|---|---|---|---|---|
| Opal-AG | 469 ± 1 | 470 ± 2 | 802 ± 1 | 1099 ± 2 |
| Opal-AN | 469 ± 1 | 469 ± 1 | 801 ± 1 | 1103 ± 2 |
| Opal-CT | 472 ± 1 | 472 ± 1 | 793 ± 2 | 1099 ± 2 |
| Opal-C | 475 ± 1 | 474 ± 2 | 795 ± 1 | 1095 ± 2 |
| Sample | Low-Energy Band /cm−1 | Medium-Energy Band /cm−1 | High-Energy Band /cm−1 |
|---|---|---|---|
| Opal-AG | 467 | 802 | 1094 |
| Opal-AN | 469 | 804 | 1103 |
| Opal-CT | 474 | 792 | 1101 |
| Opal-C | 472 | 795 | 1084 |
| Sample | Low-Energy Band TO1/LO1 /cm−1 | Medium-Energy Band TO2/LO2 /cm−1 | High-Energy Band TO3/LO3 /cm−1 |
|---|---|---|---|
| Opal-AG | 458/503 | 799/807 | 1080/1253 |
| Opal-AN | 463/505 | 800/810 | 1091/1258 |
| Opal-CT | 468/529 | 790/798 | 1090/1254 |
| Opal-C | 470/536 | 793/802 | 1080/1245 |
| Type | β (Nm−1) | α (Nm−1) | Si-O-Si Bond Angle (°) |
|---|---|---|---|
| Opal-AG | 99 | 592 | 146 |
| Opal-AN | 101 | 590 | 152 |
| Opal-CT | 103 | 566 | 167 |
| Opal-C | 104 | 570 | 156 |
| Feature | Technique | Opal-AG and Opal-AN | Opal-CT |
|---|---|---|---|
| Low-energy bands | Transmission mode (TO1) | Peak approximately 469 cm−1. | Peak at 472 cm−1 with variable shoulder at 510 cm−1. |
| Specular reflectance (absorptivity index) | Peak approximately 468 cm−1. | Peak at 474 cm−1. | |
| Specular reflectance (TO1) | Peak approximately 460 cm−1. | Peak at 470 cm−1. | |
| Specular reflectance (LO1) | Peak approximately 504 cm−1. Only appears with KK a transformation. | Peak at 536 cm−1. Appears in 75° incidence specular reflectance. Enhanced with p-polarisation. | |
| Powder ATR | Peak coincident with high refractive index. Ranges from a relatively sharp peak at 470 cm−1 to a broad peak at 450 cm−1 with a long tail to low energy. | Peak coincident with high refractive index. Ranges from a relatively sharp peak at 475 cm−1 to a broad peak at 460 cm−1 with a long tail to low energy. | |
| Flat sample ATR | Peak coincident with high refractive index. At approximately 410 cm−1 with a long tail to low energy. | Peak coincident with high refractive index. At approximately 410 cm−1 with a long tail to low energy. | |
| Medium-energy bands | Transmission mode (TO2) | At 802 cm−1. Broader than for opal-CT. | At 793 cm−1. Narrower than for opal-A. |
| Specular reflectance (absorptivity index) | Peak approximately 803 cm−1. | Peak approximately 792 cm−1. | |
| Specular reflectance (TO2) | Peak approximately 800 cm−1. Broader than for opal-CT. | Peak at 790 cm−1. Narrower than for opal-A. | |
| Specular reflectance (LO2) | Peak approximately 808 cm−1. Only resolved by KK transformation. | Peak at 798 cm−1. Only resolved by KK transformation. | |
| Diffuse reflectance | At approximately 799 cm−1. Broader than for opal-CT. | At approximately 789 cm−1. Narrower than for opal-A. | |
| Powder ATR | In the range 790–800 cm−1. More prominent than for specular reflectance. | In the range 780–790 cm−1. More prominent than for specular reflectance. | |
| Flat sample ATR | At 780 cm−1 and more prominent than for powder ATR. | At 780 cm−1 and more prominent than for powder ATR. | |
| Silanol bands | Transmission mode | Shoulder at approximately 945 cm−1 (opal-AG) and 950 cm−1 (opal-AN). Possibly coincident with Raman bands. | Not apparent. |
| Diffuse reflectance | At approximately 945 cm−1 (opal-AG) and 950 cm−1 (opal-AN). Possibly coincident with Raman bands. | At approximately 970 cm−1. Possibly coincident with Raman bands. | |
| High-energy bands | Transmission mode (TO3) | Approximately 1100 cm−1 with severe baseline asymmetry. | Approximately 1100 cm−1 with severe baseline asymmetry. |
| Specular reflectance (absorptivity) | Peak approximately 1100 cm−1. | Peak approximately 1100 cm−1. | |
| Specular reflectance (TO3) | Peak approximately 1085 cm−1. | Peak at 1090 cm−1. | |
| Specular reflectance (LO3) | Peak approximately 1255 cm−1. Enhanced relative to TO3 with p-polarisation. | Peak at 1254 cm−1. Enhanced relative to TO3 with p-polarisation. | |
| Diffuse reflectance | Peak approximately 1090 cm−1. Broad. | Peat at 1100 cm−1. Broad. | |
| Powder ATR | Peak coincident with high refractive index. In the range 1060–1100 cm−1. | Peak coincident with high refractive index. In the range 1065–1100 cm−1. | |
| Flat sample ATR | Peak coincident with high refractive index. In the range 1000–1010 cm−1 | Peak coincident with high refractive index. In the range 1000–1030 cm−1. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curtis, N.J.; Gascooke, J.R.; Pring, A. Silicon-Oxygen Region Infrared and Raman Analysis of Opals: The Effect of Sample Preparation and Measurement Type. Minerals 2021, 11, 173. https://doi.org/10.3390/min11020173
Curtis NJ, Gascooke JR, Pring A. Silicon-Oxygen Region Infrared and Raman Analysis of Opals: The Effect of Sample Preparation and Measurement Type. Minerals. 2021; 11(2):173. https://doi.org/10.3390/min11020173
Chicago/Turabian StyleCurtis, Neville J., Jason R. Gascooke, and Allan Pring. 2021. "Silicon-Oxygen Region Infrared and Raman Analysis of Opals: The Effect of Sample Preparation and Measurement Type" Minerals 11, no. 2: 173. https://doi.org/10.3390/min11020173
APA StyleCurtis, N. J., Gascooke, J. R., & Pring, A. (2021). Silicon-Oxygen Region Infrared and Raman Analysis of Opals: The Effect of Sample Preparation and Measurement Type. Minerals, 11(2), 173. https://doi.org/10.3390/min11020173