Possible Pitfalls in the Analysis of Minerals and Loose Materials by Portable XRF, and How to Overcome Them



Abstract
1. Introduction
- On-site, with a simplified sample preparation or no preparation, the results being used for sample screening or for orientating further site investigations,
- On-site or off-site, with the best available sample preparation, and using QA/QC procedures similar to a laboratory.
2. Materials and Methods
2.1. pXRF Instruments
2.2. pXRF Operating Conditions and Methods
2.3. Laboratory Analysis Methods
3. Results
3.1. Analytical Modes in pXRF—Relevance and Applicability to Geochemical Investigations
3.1.1. The Different Analytical Modes
3.1.2. Compton Normalisation
Principle
Limitations of the Method
- The device software subtracts a background noise signal equivalent to a matrix containing 99% SiO2 (also roughly equivalent to 99% carbonates). The matrix density is therefore assumed to be of the order of 1.8 g·cm−3. If this mode is used for different matrices, the background noise is poorly taken into account, and lower or negative values can be obtained (i.e., real background noise above the subtracted noise, and negative background residue), or overestimated (i.e., actual background noise below the subtracted noise, a positive residue being added to the potentially present peak).
- The extraction is done only on the peak height (centre strip) and not on the shape (which would be more precise with a deconvolution of line profile). Moreover, the ratio between the lines of the same pure element is unfortunately known to be not constant, depending on matrix density.
- The extracted peak intensities are converted into concentrations by:
- (a)
- the relationship between the intensity of the element peak with the intensity of the Compton peak of the tube, or a region of interest assumed to bear no peak (depending on the filter used, because the low filter cannot measure the Compton peak).
- (b)
- a calibration of this ratio with soil standards entered in the device.
- (c)
- no additional correction (unlike fundamental parameters).
3.1.3. Fundamental Parameters (FP)
Principle
- sample homogeneity;
- completeness, which implies that all the elements are measured or estimated by stoichiometry or by complement to 100%.
Limitations of the Method
3.1.4. Empirical Method
Principle
Limitations of the Method
3.1.5. Matching Library Method (PMI)
Principle
Limitations of the Method
3.2. Impact of the Composition of Windows and Other Film Products Used for Protection in pXRF, and Sample Bag Applications
3.2.1. Spectrometer Window
- Thick membranes (6 to 7 µm in polypropylene, Mylar™ or Kapton™) for analysing mainly heavy elements (e.g., Pb, Zn, and Cu). For Niton spectrometers (Xlt, Xl2t, Xl3t, and Xl5), Olympus (Delta), Vanta: Mylar, or Kapton, Bruker (Kapton), Hitachi (mylar), SciAps (Kapton);
- Thin membranes (4 µm) allowing the analysis of light elements (Niton Xl3t and Xl5: Prolene™), Olympus (Delta and Vanta: Prolene™), Bruker (UltraleneTM), Hitachi (polypropylene), SciAps (Prolene™).
3.2.2. Spectrometer Protection Shield
3.2.3. Films Used for the Cups
3.2.4. Sample Bags and Other Films Used for Packaging
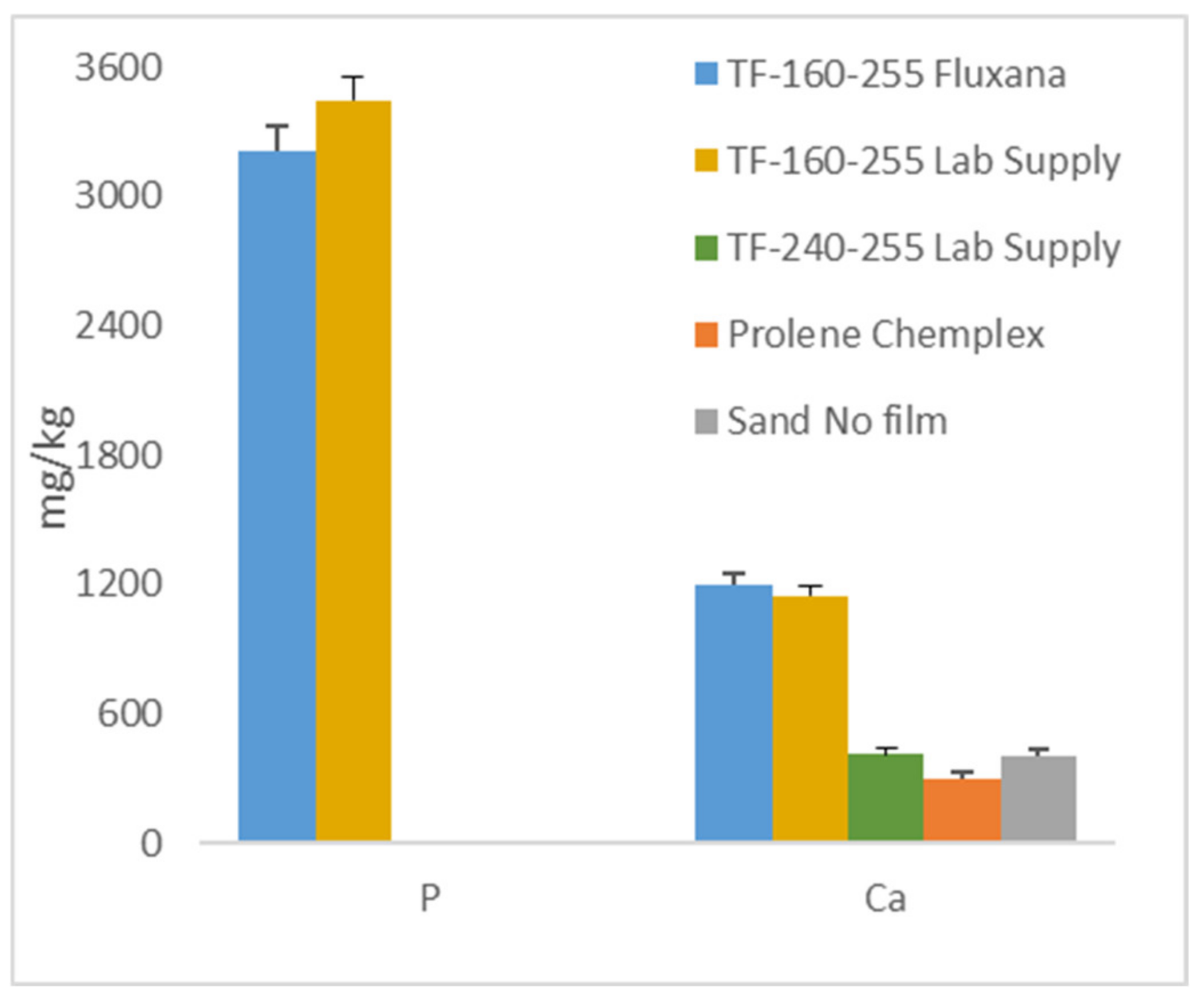
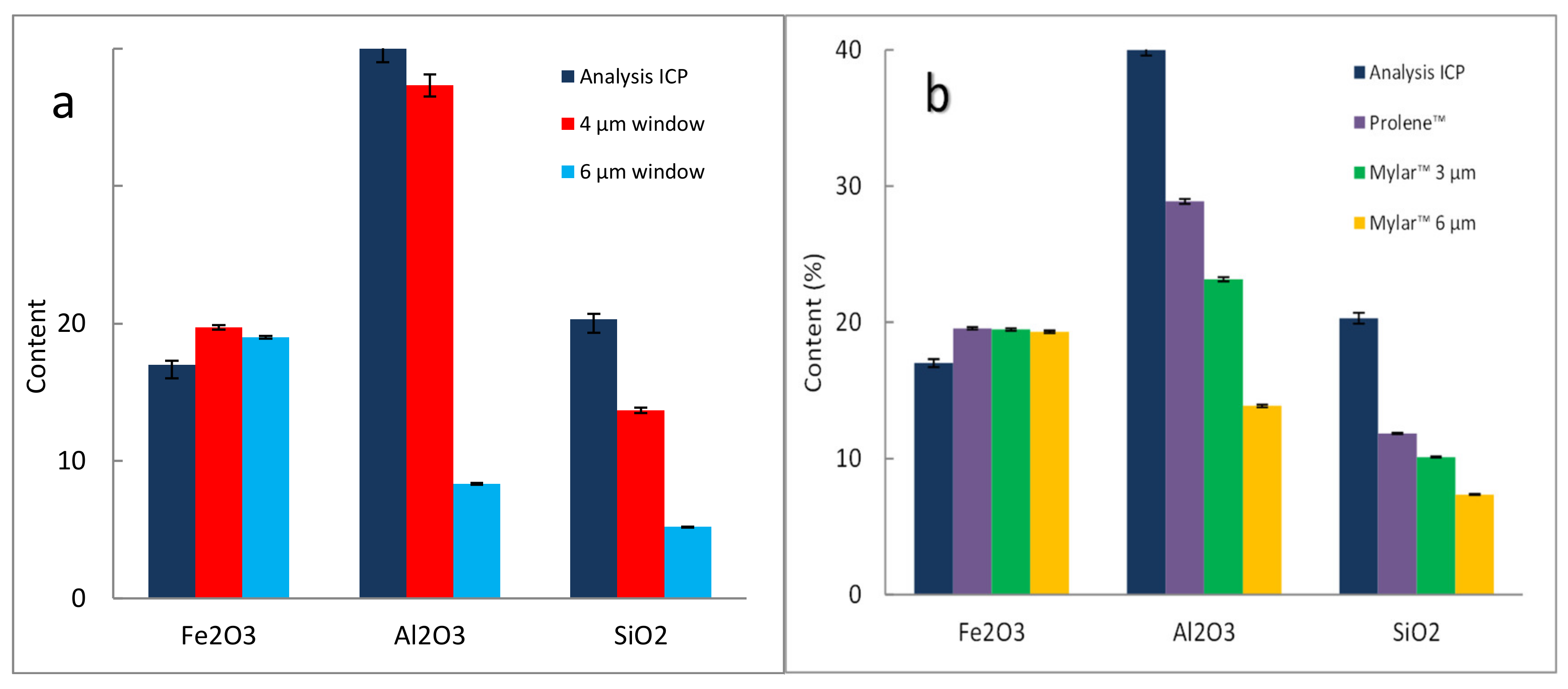
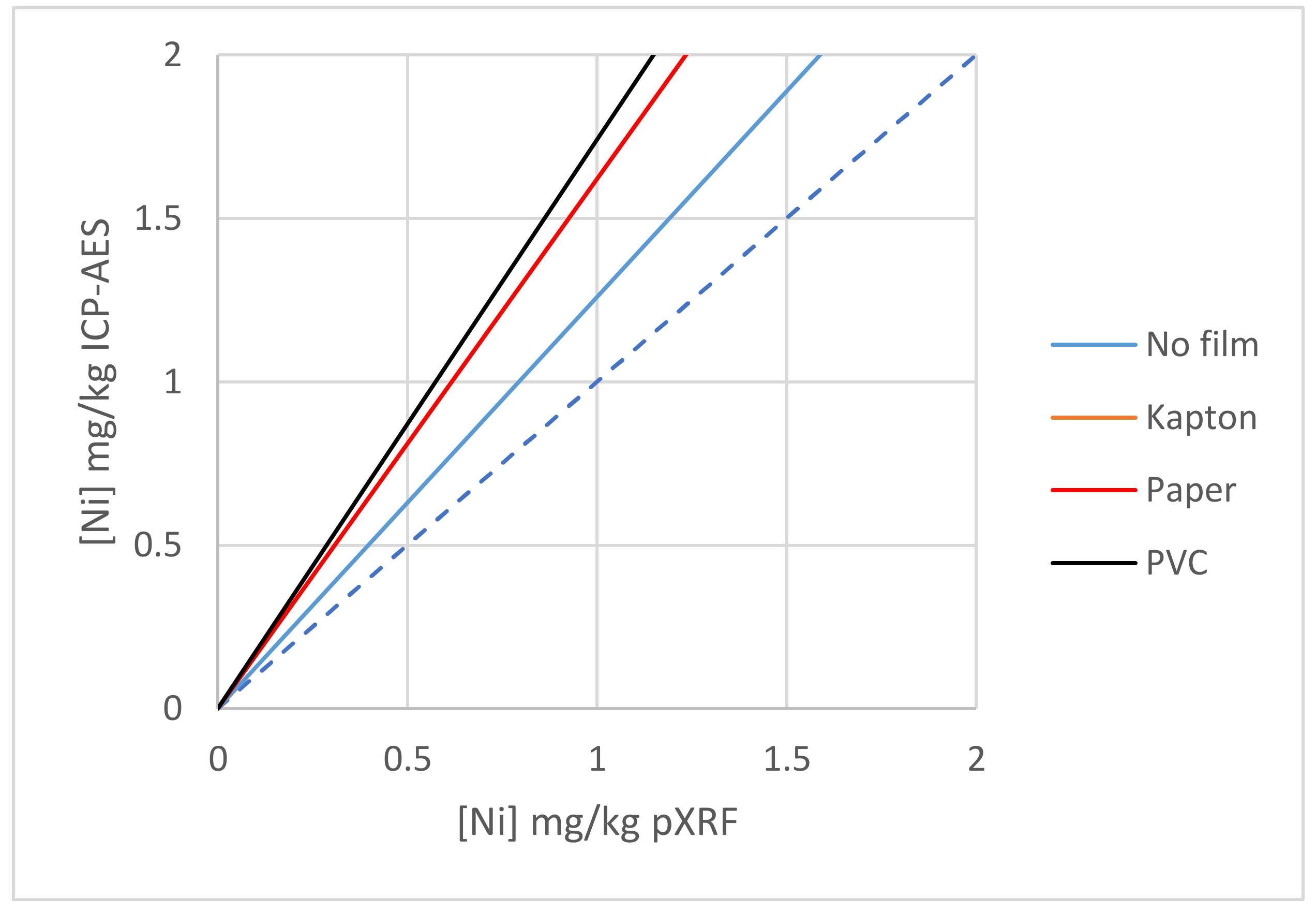
3.2.5. Effect of Films, Example 1
3.2.6. Effect of Films, Example 2
3.2.7. Effect of Films, Example 3 on Laterite Samples:
3.3. Light Element Analysis in pXRF
3.3.1. Difficulties Analysing Light Elements
- The fluorescence yield is very poor, as X-ray emission produces Auger electrons. In the Auger effect, an atom excited by ionisation, in the K layer for example, does not necessarily emit a photon of the K series outside the atom. Indeed, the emitted photon can be absorbed by the atom itself and expels a second electron from the upper layers (L, M, etc., in our example). This phenomenon is called the Auger effect and constitutes a non-radiative transition or internal conversion.
- The energy of the lines is low, so the photons are easily absorbed and generate a weak signal (Table 3).
- Light elements are only quantified from the K lines (lines 12 to 19 in Table 3), these are close to each other but also to the L lines of the heavier elements (lines 31 to 49 in Table 3). It is therefore difficult to distinguish them from each other. The energy resolution of pXRF devices is approximately 0.15 keV.
- In theory, on the basis of the energies of fluorescence (Table 1), the elements with atomic number Z < 20 should be considered as “light”. However, on the basis of the numerous studies published since 1995:
- Elements with Z < 12 cannot currently be analysed by pXRF, but only with state-of-the art laboratory equipment, or with another portable technique as LIBS [33];
- Elements with 18 < Z < 20 (K, Ca) are analysed in a generally satisfactory manner, despite the characteristics of their K lines. This is to be linked with their usual ranges, respectively 0.02 to 10% and 0.1 to 40%;
- Elements with 12 < Z < 18 exhibit significant difficulties and require high-performance devices and suitable preparation techniques. These are the elements that are the subject of this section.
- Light elements are, more than the others, subject to the phenomena of absorption of X-rays of fluorescence by air. On some devices, a light gas (usually helium) is used to purge the head of the device, which improves the quantification of light elements starting from the magnesium (Z = 12).
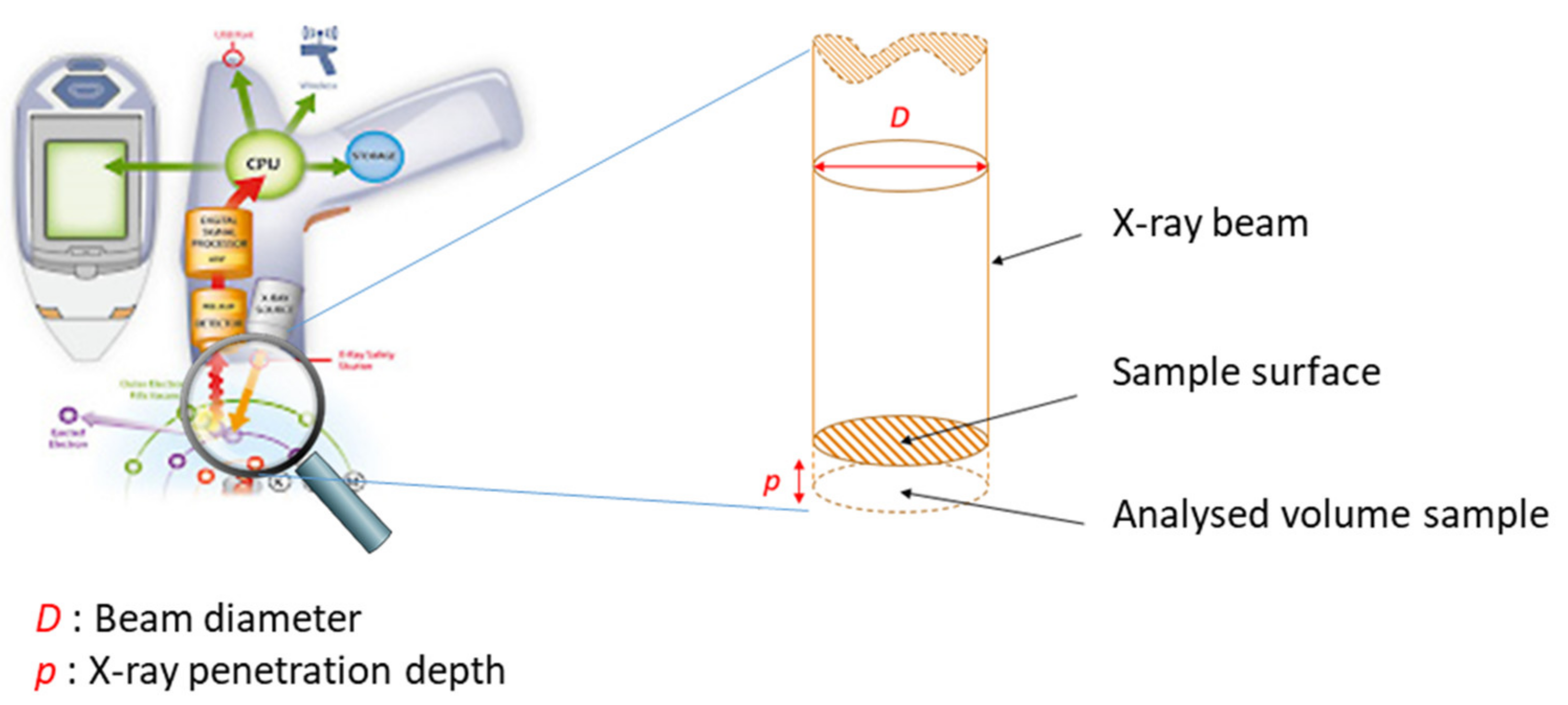
3.3.2. Geometry to Be Observed for Excitation of the Sample and Detection of X Photons
3.3.3. Geometry to Be Respected for the Analysis of Light Elements
- pelletising with a hammer shock tube,
- pelletising with the press, possibly with the addition of a binder, kaolin, talc, or wax,
- compacting the surface of the material and measuring over it, the nose of the device simply placed, without pressing.
3.3.4. Other Points to Observe for the Analysis of Light Elements
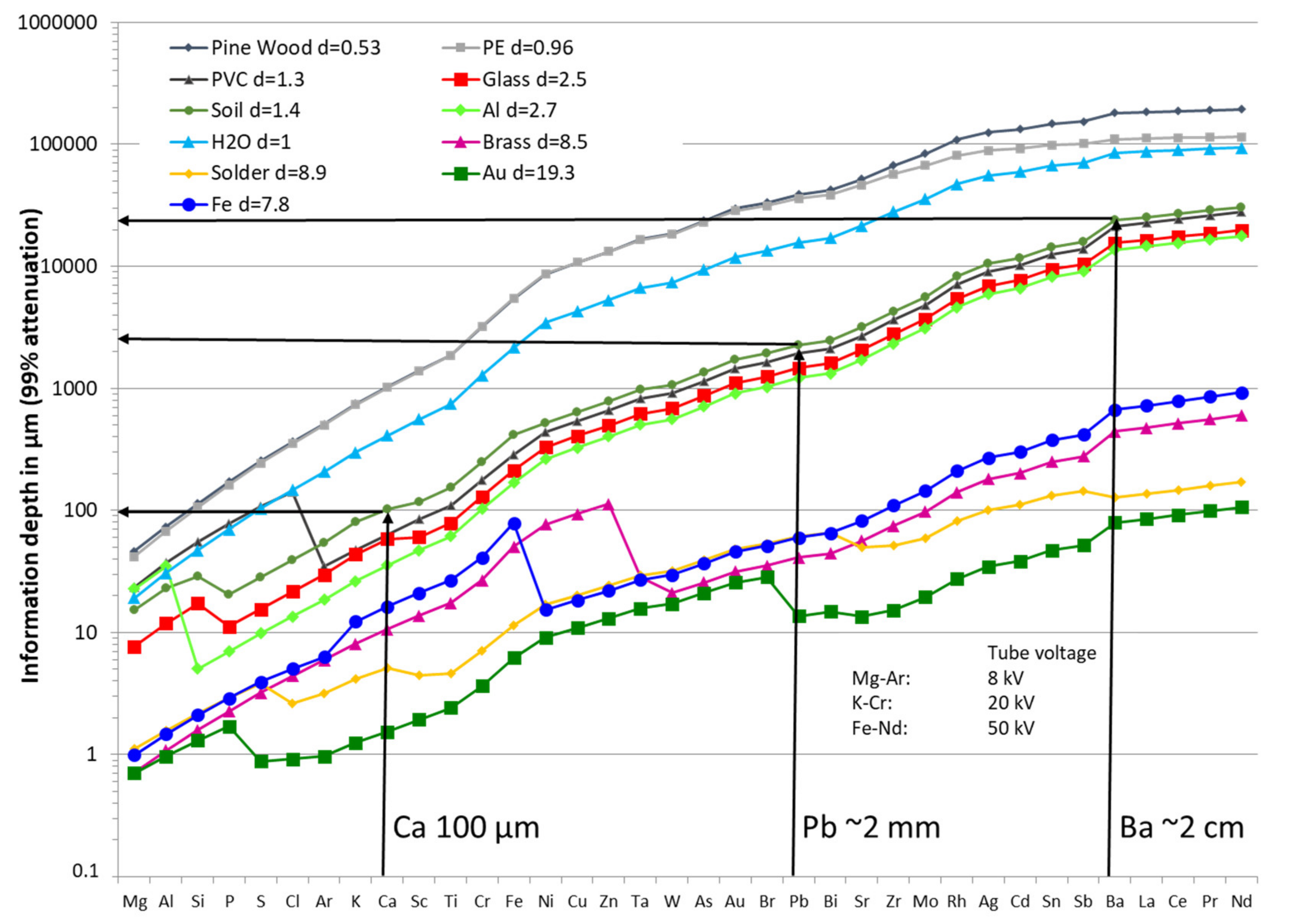
- For a low energy line with a shallow penetration depth, the surface layer must therefore be representative of the sample, which makes sample preparation critical (this problem being similar to that of sampling);
- For a very energetic line, which penetrates deeper, the received signal is an average over a large volume, but if the sample is too thin, the hypothesis of “infinite thickness” is no longer valid, and there may be interference from the bottom support material.
3.4. Analytical Interferences in pXRF, and Taking Them into Account in Geochemical Investigations
3.4.1. Principle
- Physical effects of the matrix (particle size, homogeneity, surface effect);
- Humidity and porosity of the sample;
- Chemical effects of the matrix (decrease or increase in the intensity of a line of an element caused by the presence of another element);
- Interference between the Kα/Kβ lines;
- Interference between the K/L, K/M and L/M lines.
3.4.2. Chemical Effects of the Matrix
Matrix Effect: Absorption
Matrix Effect: Excitation
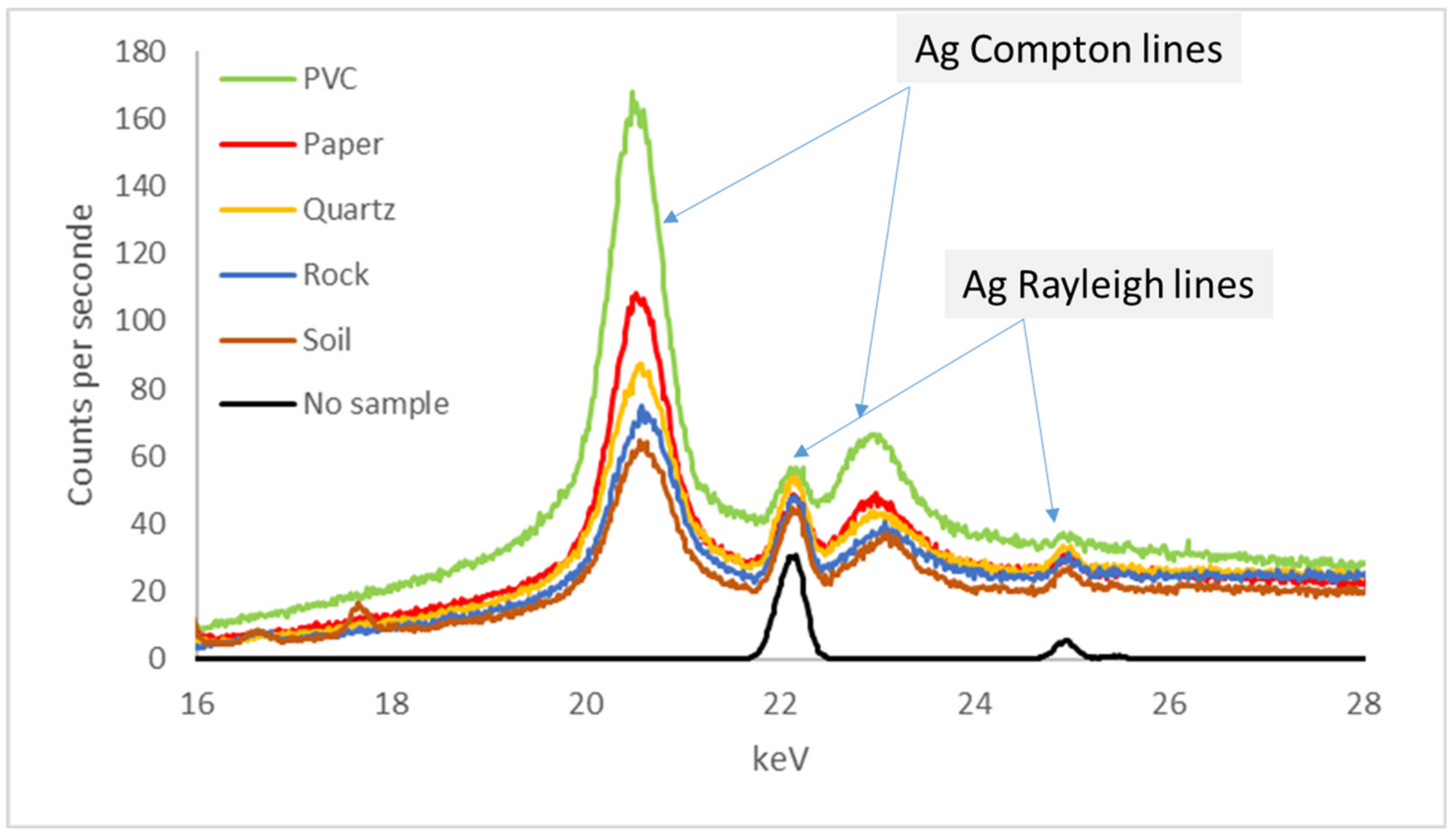
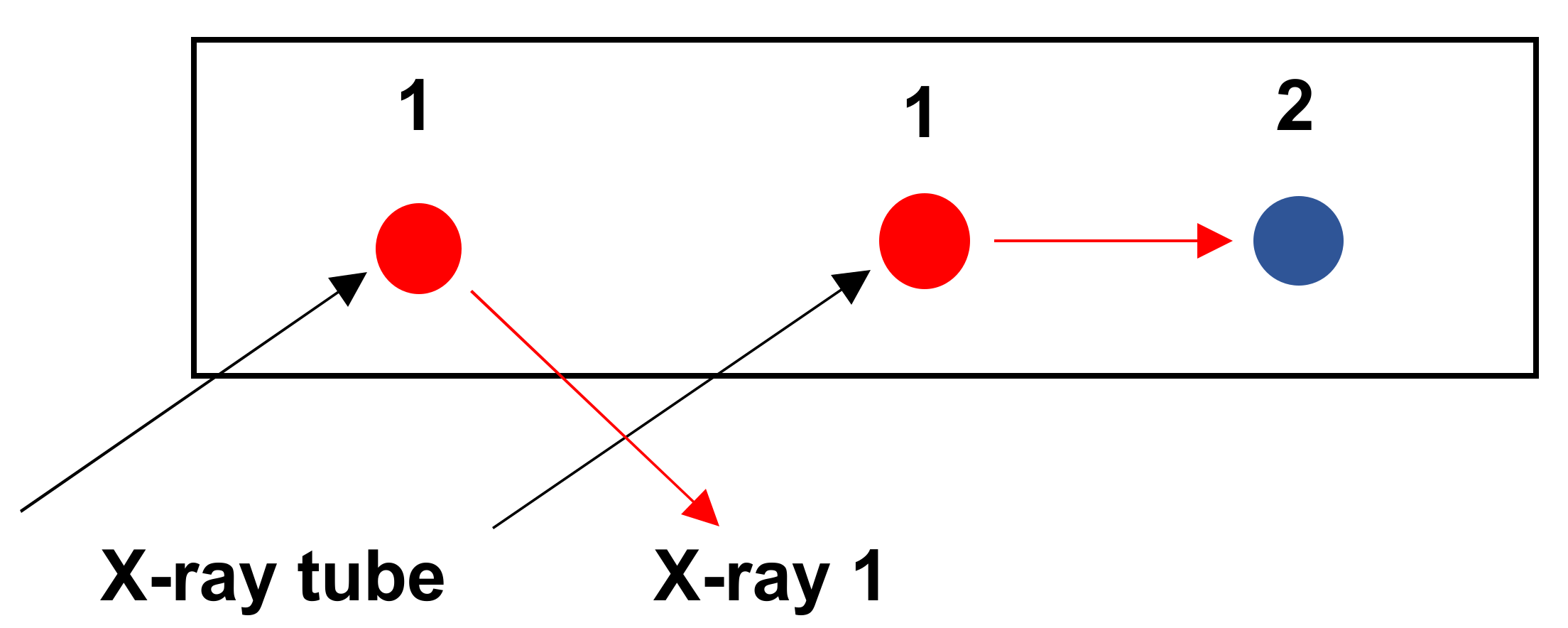
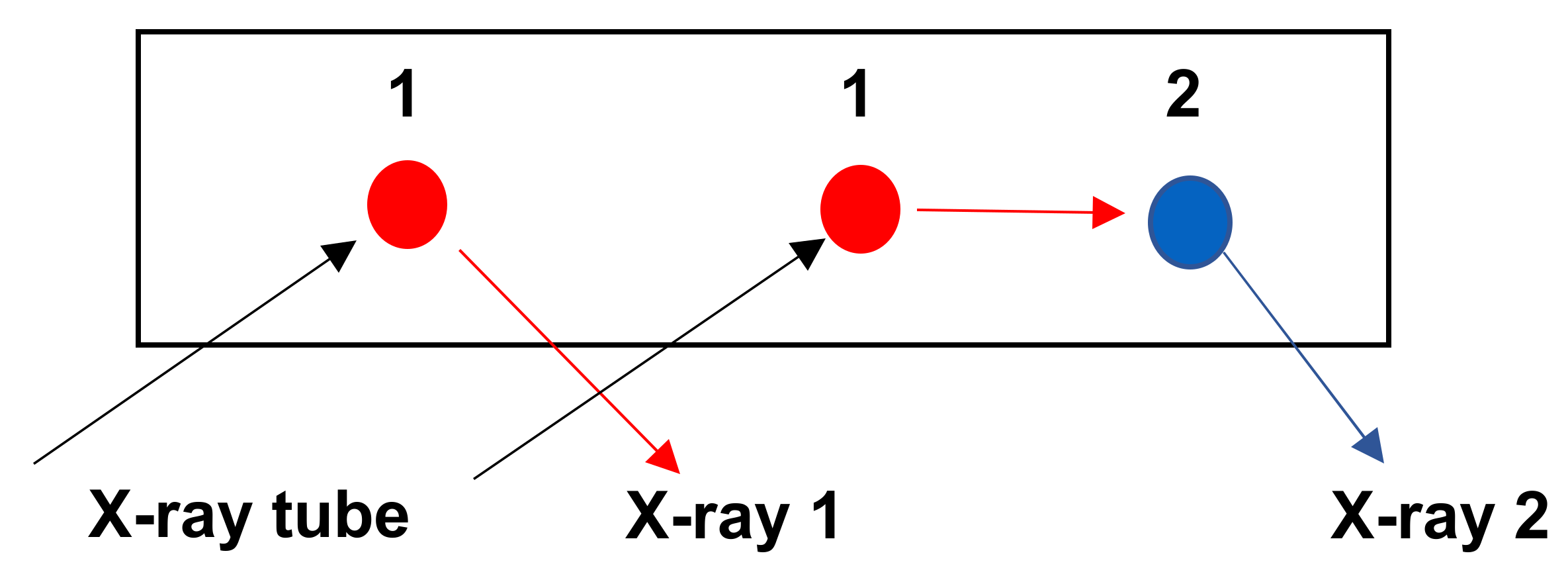
3.4.3. Interference between K /Kβ Lines
3.4.4. Interference between K/L, K/M and L/M Lines
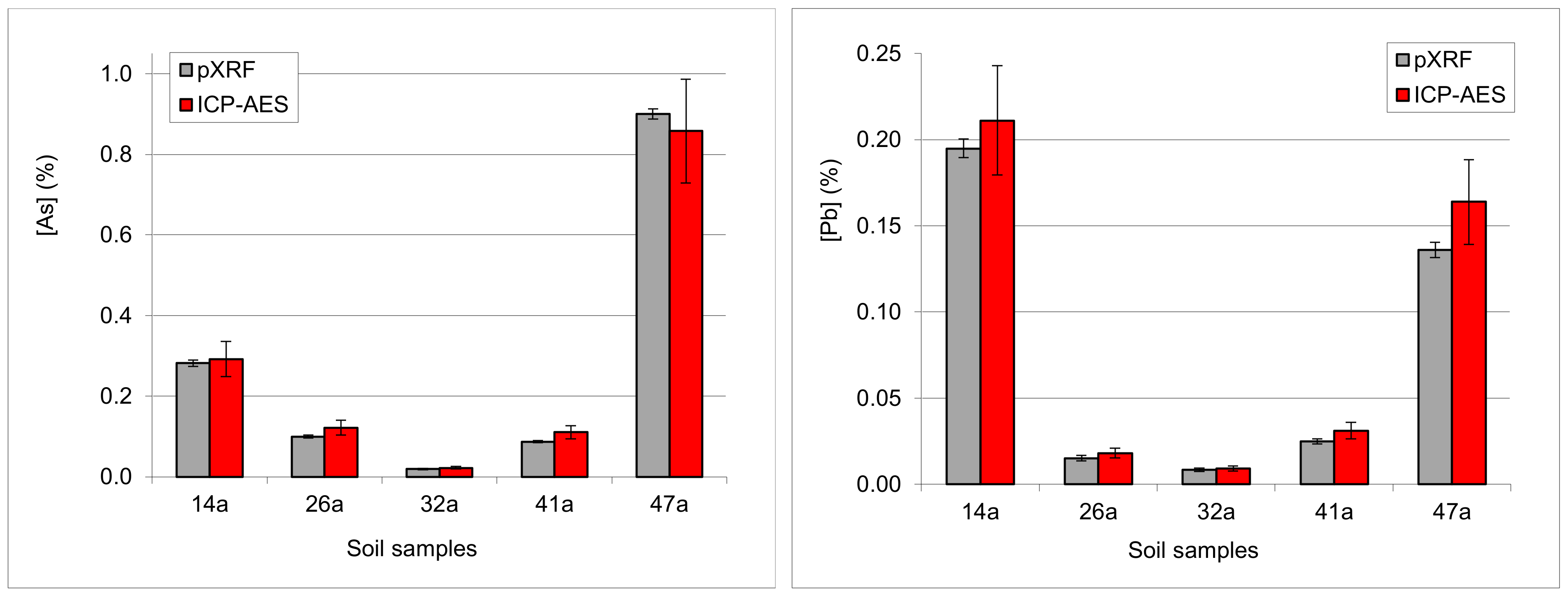
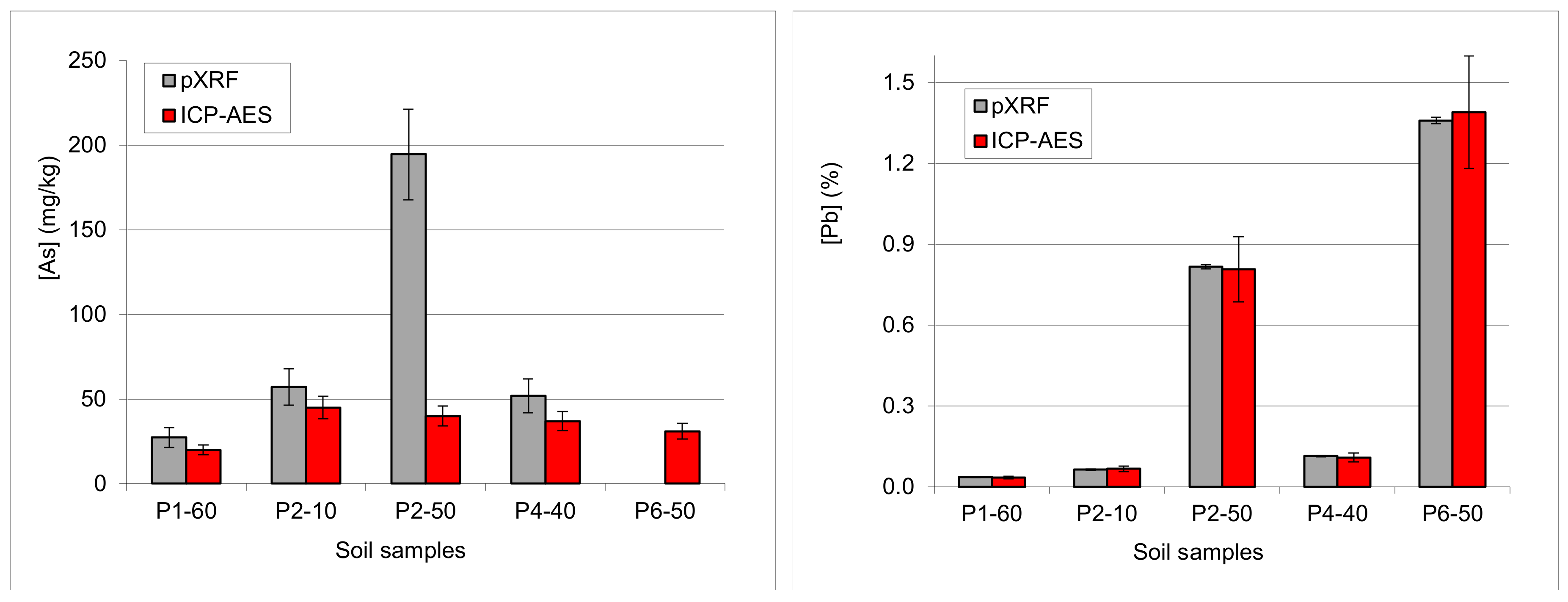
3.4.5. Examples: As-Pb Interference
3.5. Physical Effects of the Matrix
3.5.1. Physical Effects of the Matrix (Homogeneity, Particle Size, and Surface Effect)
3.5.2. Effects of Moisture in Fine-Grained Samples
3.5.3. Moisture and Grain Size
3.5.4. Depth of Analytical Investigations
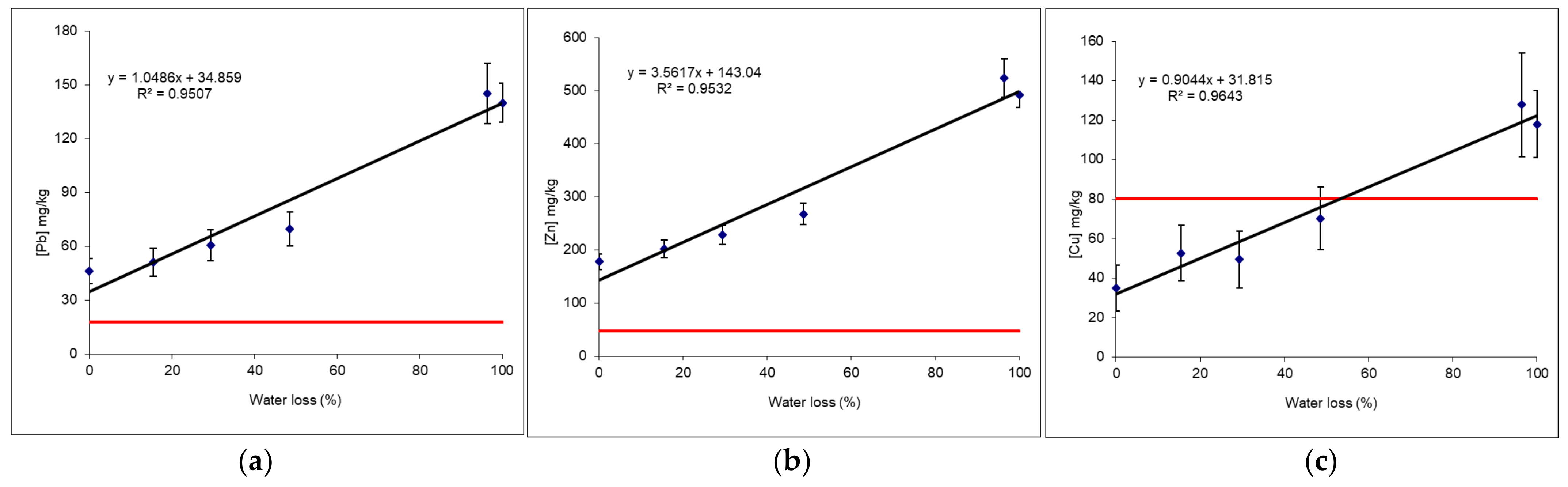
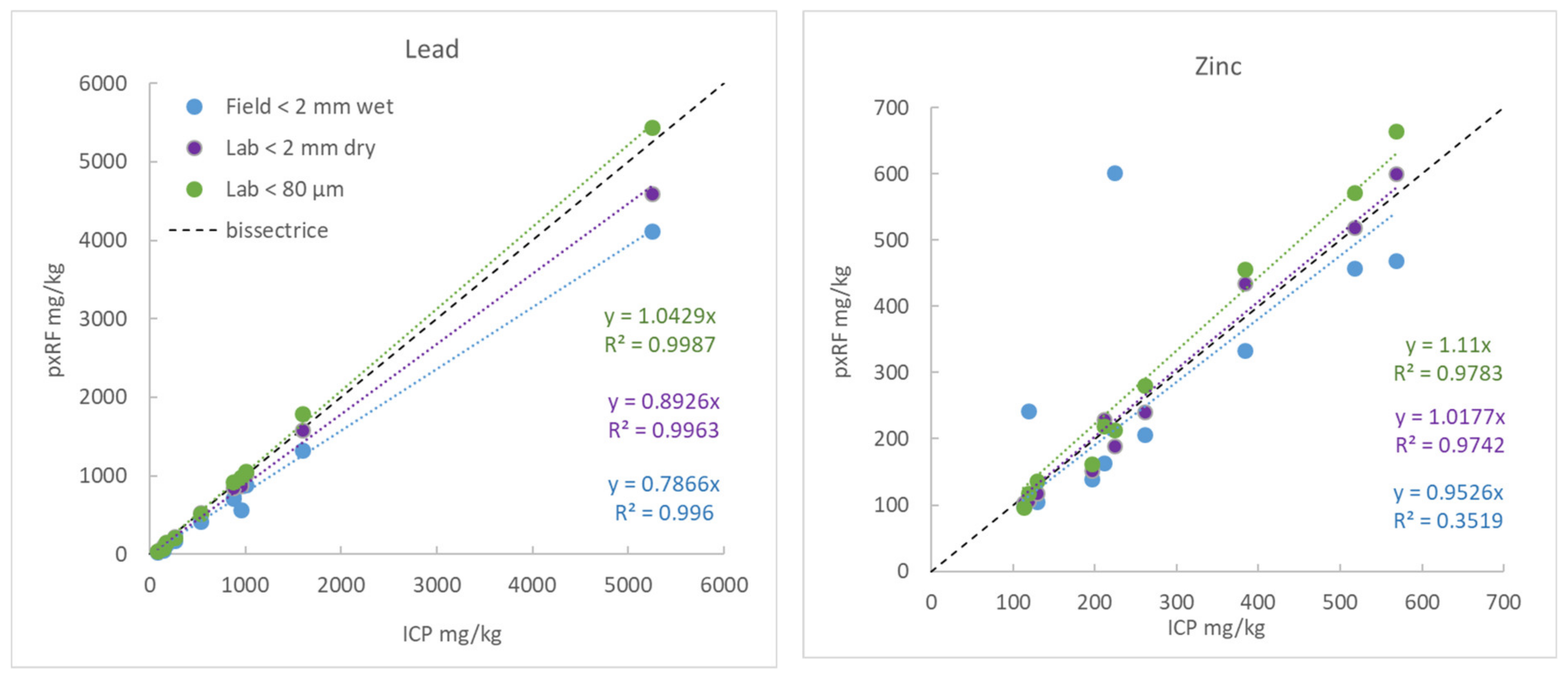
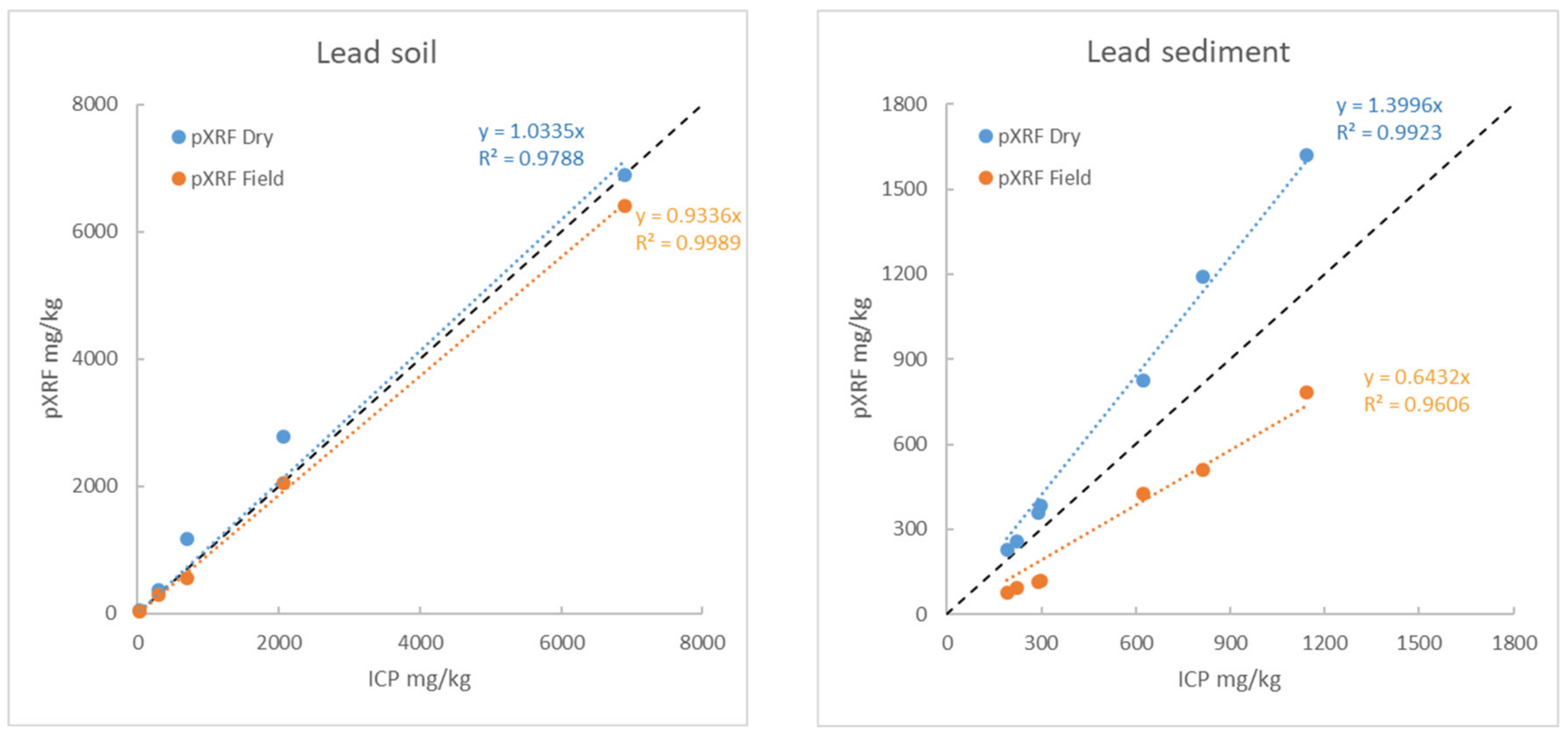
3.5.5. Example 1: Behaviour of Pb and Zn as a Function of Humidity and Article Size
- Raw samples, that is to say sieved to 2 mm but still wet (measurements on-site “Wet ground” with an average humidity rate of 20%),
- Raw samples, that is to say sieved to 2 mm and dried in a lab oven at 40 °C (“Lab < 2 mm dry”),
- Samples dried and ground to 80 µm (“Lab < 80 µm”).
3.5.6. Example 2: Impact of Density on the Measurement
3.5.7. Other Potential Issues with pXRF
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gałuszka, A.; Migaszewski, Z.M.; Namieśnik, J. Moving your laboratories to the field—Advantages and limitations of the use of field portable instruments in environmental sample analysis. Environ. Res. 2015, 140, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Huang, B.; Weindorf, D.C.; Chen, Y. Metals Analysis of Agricultural Soils via Portable X-ray Fluorescence Spectrometry. Bull. Environ. Contam. Toxicol. 2014, 92, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Weindorf, D.C.; Wang, D.; Chakraborty, S. Characterizing soils via portable X-ray fluorescence spectrometer: 4. Cation exchange capacity (CEC). Geoderma 2015, 239–240, 130–134. [Google Scholar] [CrossRef]
- Lemière, B. A review of applications of pXRF (field portable X-ray fluorescence) for applied geochemistry. J. Geochem. Explor. 2018, 188, 350–363. [Google Scholar] [CrossRef]
- Tighe, M.; Rogan, G.; Wilson, S.; Grave, P.; Kealhofer, L.; Yukongdi, P. The potential for portable X-ray fluorescence determination of soil copper at ancient metallurgy sites, and considerations beyond measurements of total concentrations. J. Environ. Manag. 2018, 206, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Weindorf, D.C.; Bakr, N.; Zhu, Y. Advances in Portable X-ray Fluorescence (PXRF) for Environmental, Pedological, and Agronomic Applications. Adv. Agron. 2014, 128, 1–45. [Google Scholar] [CrossRef]
- Ravansari, R.; Wilson, S.C.; Tighe, M. Portable X-ray fluorescence for environmental assessment of soils: Not just a point and shoot method. Environ. Int. 2020, 134, 105250. [Google Scholar] [CrossRef]
- Kilbride, C.; Poole, J.; Hutchings, T. A comparison of Cu, Pb, As, Cd, Zn, Fe, Ni and Mn determined by acid extraction/ICP–OES and ex situ field portable X-ray fluorescence analyses. Environ. Pollut. 2006, 143, 16–23. [Google Scholar] [CrossRef]
- Radu, T.; Diamond, D. Comparison of soil pollution concentrations determined using AAS and portable XRF techniques. J. Hazard. Mater. 2009, 171, 1168–1171. [Google Scholar] [CrossRef]
- Hall, G.; Buchar, A.; Bonham-Carter, G. Quality Control Assessment of Portable XRF Analysers: Development of Standard Operating Procedures, Performance on Variable Media and Recommended Uses. Canadian Mining Industry Research Organization (CAMIRO) Exploration Division, Project 10E01 Phase I Report. 2012. Available online: https://www.appliedgeochemists.org/index.php/publications/other-publications/2-uncategorised/106-portable-xrf-for-the-exploration-and-mining-industry (accessed on 17 December 2013).
- Bourke, A.; Ross, P.-S. Portable X-ray fluorescence measurements on exploration drill-cores: Comparing performance on unprepared cores and powders for ‘whole-rock’ analysis. Geochem. Explor. Environ. Anal. 2015, 16, 147–157. [Google Scholar] [CrossRef]
- Rouillon, M.; Taylor, M.P. Can field portable X-ray fluorescence (pXRF) produce high quality data for application in environmental contamination research? Environ. Pollut. 2016, 214, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Evans, C.A.; Hodges, K.V.; Bleacher, J.E.; Graff, T.G. A review of the handheld X-ray fluorescence spectrometer as a tool for field geologic investigations on Earth and in planetary surface exploration. Appl. Geochem. 2016, 72, 77–87. [Google Scholar] [CrossRef]
- Laperche, V.; Hammade, V. Diagnostic Rapide sur site—Utilisation de Méthodes D’évaluation de la Teneur en Métaux de sols Pollués par Mesure de leur Susceptibilité Magnétique et par Fluorescence X; Report CNRSSP/02/08; CNRSSP: Douai, France, 2002. [Google Scholar]
- Laperche, V. Evaluation des Performances du Spectromètre Portable de Fluorescence X Niton XL723S (au Laboratoire et sur le Terrain). BRGM Report RP-53377-FR. 2005, 72p. Available online: http://infoterre.brgm.fr/rapports/RP-53377-FR.pdf (accessed on 23 December 2020).
- Liakopoulos, A.; Lemière, B.; Michael, K.; Crouzet, C.; Laperche, V.; Romaidis, I.; Drougas, I.; Lassin, A. Environmental impacts of unmanaged solid waste at a former base metal mining and ore processing site (Kirki, Greece). Waste Manag. Res. 2010, 28, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Lemiere, B.; Laperche, V.; Haouche, L.; Auger, P. Portable XRF and wet materials: Application to dredged contaminated sediments from waterways. Geochem. Explor. Environ. Anal. 2014, 14, 257–264. [Google Scholar] [CrossRef]
- Quiniou, T.; Laperche, V. An assessment of field-portable X-ray fluorescence analysis for nickel and iron in laterite ore (New Caledonia). Geochem. Explor. Environ. Anal. 2014, 14, 245–255. [Google Scholar] [CrossRef]
- Parsons, C.T.; Grabulosa, E.M.; Pili, E.; Floor, G.H.; Román-Ross, G.; Charlet, L. Quantification of trace arsenic in soils by field-portable X-ray fluorescence spectrometry: Considerations for sample preparation and measurement conditions. J. Hazard. Mater. 2013, 262, 1213–1222. [Google Scholar] [CrossRef]
- Markowicz, A.A. Quantification and Correction Procedures. In IAEA, In Situ Applications of X Ray Fluorescence Techniques; IAEA-TECDOC-1456 (ISBN:92-0-107105-1). 2005. Available online: http://www-pub.iaea.org/books/IAEABooks/7212/In-Situ-Applications-of-X-Ray-Fluorescence-Techniques (accessed on 15 September 2014).
- Adams, C.; Brand, C.; Dentith, M.; Fiorentini, M.L.; Caruso, S.; Mehta, M. The use of pXRF for light element geochemical analysis: A review of hardware design limitations and an empirical investigation of air, vacuum, helium flush and detector window technologies. Geochem. Explor. Environ. Anal. 2020, 20, 366–380. [Google Scholar] [CrossRef]
- Ravansari, R.; Lemke, L.D. Portable X-ray fluorescence trace metal measurement in organic rich soils: pXRF response as a function of organic matter fraction. Geoderma 2018, 319, 175–184. [Google Scholar] [CrossRef]
- Wilson, A.W.; Turner, D.C.; Robbins, A.A. Evaluation of Some Sample Support Films for Microsample X-Ray Analysis. Mater. Sci. 1999, 301–307. [Google Scholar]
- Hall, G.E.; Bonham-Carter, G.F.; Buchar, A. Evaluation of portable X-ray fluorescence (pXRF) in exploration and mining: Phase 1, control reference materials. Geochem. Explor. Environ. Anal. 2014, 14, 99–123. [Google Scholar] [CrossRef]
- Boon, K.A.; Ramsey, M.H. Judging the fitness of on-site measurements by their uncertainty, including the contribution from sampling. Sci. Total. Environ. 2012, 419, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Arne, D.C.; Jeffress, G.M. Sampling and Analysis for Public Reporting of Portable X-ray Fluorescence Data Under the 2012 Edition of the JORC Code. In Proceedings of the Sampling 2014, Perth, WA, USA, 29–30 July 2014. [Google Scholar]
- Morris, P.A. Field-Portable X-Ray Fluorescence Analysis and Its Application in GSWA. Geological Survey of Western Australia RECORD 2009/7. 2009. Available online: https://geodocs.dmirs.wa.gov.au/Web/documentlist/3/Combined/N09AD (accessed on 20 October 2020).
- Hall, G.E.; McClenaghan, M.B.; Pagé, L. Application of portable XRF to the direct analysis of till samples from various deposit types in Canada. Geochem. Explor. Environ. Anal. 2015, 16, 62–84. [Google Scholar] [CrossRef]
- Zhou, S.; Yuan, Z.; Cheng, Q.; Zhang, Z.; Yang, J. Rapid in situ determination of heavy metal concentrations in polluted water via portable XRF: Using Cu and Pb as example. Environ. Pollut. 2018, 243, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B. Heterogeneities in Samples of Contaminated Soil. Ph.D. Thesis, Waste Science and Technology, Department of Civil, Mining and Environmental Engineering, Luleå University of Technology, Luleå, Sweden, 2007; 52p. ISRN LTU-DT-07/52-SE. [Google Scholar]
- Weltje, G.J.; Tjallingii, R. Calibration of XRF core scanners for quantitative geochemical logging of sediment cores: Theory and application. Earth Planet. Sci. Lett. 2008, 274, 423–438. [Google Scholar] [CrossRef]
- Quiniou, T.; Laperche, V. Mesures In Situ des Teneurs; Programme CNRT, Rapport final, BRGM/RC-59436-FR; BRGM: Orléans, France, 2011; 183p. [Google Scholar]
- Harmon, R.S.; Russo, R.E.; Hark, R.R. Applications of laser-induced breakdown spectroscopy for geochemical and environmental analysis: A comprehensive review. Spectrochim. Acta Part B Atom. Spectrosc. 2013, 87, 11–26. [Google Scholar] [CrossRef]
- Lawrence Berkeley National Laboratory. X-Ray Data Booklet. LBNL/Pub-490 Rev. 3. Lawrence Berkeley National Laboratory, Centre for X-Ray Optics and Advanced Light Source, 2007. Available online: https://xdb.lbl.gov/xdb-new.pdf (accessed on 7 May 2020).
- Jenkins, R. An Introduction to X-Ray Spectrometry; Philips Electronic Instruments: Mahwah, NJ, USA; Heyden: London, UK, 1976; 163p. [Google Scholar]
- Bertin, E.P. Principles and Pratice of X-Ray Spectrometric Analysis. Plenum Press: New York, NY, USA, 1971; 679p. [Google Scholar]
- Jenkins, R.; De Vries, J.L. Practical X-Ray Spectrometry, 2nd ed.; Philips Technical Library, Springer Inc.: New York, NY, USA, 1968; 190p. [Google Scholar]
- Ebel, H. Quantitative X-Ray Fluorescence Analysis. Anal. Chim. Acta 1996, 320, 293. [Google Scholar] [CrossRef]
- US-EPA Method 6200. 2008. Available online: https://www.epa.gov/sites/production/files/2015-12/documents/6200.pdf (accessed on 23 December 2020).
- Gallhofer, D.; Lottermoser, B.G. The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals 2018, 8, 320. [Google Scholar] [CrossRef]
- Kalnicky, D.J.; Singhvi, R. Field portable XRF analysis of environmental samples. J. Hazard. Mater. 2001, 83, 93–122. [Google Scholar] [CrossRef]
- Berger, M.; Zou, L.; Schleicher, R. Analysis of Sulfur in the Copper Basin and Muddy River Sites. Int. J. Soil Sediment Water 2009, 2, 3. [Google Scholar]
- Hürkamp, K.; Raab, T.; Völkel, J. Two and three-dimensional quantification of lead contamination in alluvial soils of a historic mining area using field portable X-ray fluorescence (pXRF) analysis. Geomorphology 2009, 110, 28–36. [Google Scholar] [CrossRef]
- Ge, L.; Lai, W.; Lin, Y.; Zhou, S. In Situ Applications of FPXRF Techniques in Mineral Exploration. In In Situ Applications of X-Ray Fluorescence Techniques; IAEA-TECDOC-1456: 61. 2005. Available online: http://www-pub.iaea.org/books/IAEABooks/7212/In-Situ-Applications-of-X-Ray-Fluorescence-Techniques (accessed on 15 September 2014).
- Ge, L.; Lai, W.; Lin, Y. Influence of and correction for moisture in rocks, soils and sediments on in situ XRF analysis. X-Ray Spectrom. 2004, 34, 28–34. [Google Scholar] [CrossRef]
- Padilla, J.T.; Hormes, J.; Selim, H.M. Use of portable XRF: Effect of thickness and antecedent moisture of soils on measured concentration of trace elements. Geoderma 2019, 337, 143–149. [Google Scholar] [CrossRef]
- Ramsey, M. Contaminated Land: Cost-effective Investigation within Sampling Constraints. In IAEA, In Situ Applications of X Ray Fluorescence Techniques; IAEA-TECDOC-1456 (ISBN: 92-0-107105-1). 2005. Available online: http://www-pub.iaea.org/books/IAEABooks/7212/In-Situ-Applications-of-X-Ray-Fluorescence-Techniques (accessed on 15 September 2014).
- Ho, H.H.; Swennen, R.; Cappuyns, V.; Vassilieva, E.; Tran, T.V. Necessity of normalization to aluminium to assess the contamination by heavy metals and arsenic in sediments near Haiphong Harbor, Vietnam. J. Asian Earth Sci. 2012, 56, 229–239. [Google Scholar] [CrossRef]
- Haffert, L.; Craw, D. Field quantification and characterisation of extreme arsenic concentrations at a historic mine processing site, Waiuta, New Zealand. N. Zeal. J. Geol. Geophys. 2009, 52, 261–272. [Google Scholar] [CrossRef]
- Goff, K.; Schaetzl, R.J.; Chakraborty, S.; Weindorf, D.C.; Kasmerchak, C.; Bettis, E.A. Impact of sample preparation methods for characterizing the geochemistry of soils and sediments by portable X-ray fluorescence. Soil Sci. Soc. Am. J. 2020, 84, 131–143. [Google Scholar] [CrossRef]
- Newlander, K.; Goodale, N.; Jones, G.T.; Bailey, D.G. Empirical study of the effect of count time on the precision and accuracy of pXRF data. J. Archaeol. Sci. Rep. 2015, 3, 534–548. [Google Scholar] [CrossRef]
- Scholze, F.; Longoni, A.; Fiorini, C.; Strüder, L.; Meidinger, N.; Hartmann, R.; Kawahara, N.; Shoji, T. X-Ray Detectors and XRF Detection Channels. In Handbook of Practical X-Ray Fluorescence Analysis; Springer: Berlin/Heidelberg, Germany, 2007; pp. 199–308. [Google Scholar]
- Brand, N.W.; Brand, C.J. Performance comparison of portable XRF instruments. Geochem. Explor. Environ. Anal. 2014, 14, 125–138. [Google Scholar] [CrossRef]
- Simandl, G.J.; Fajber, R.; Paradis, S. Portable X-ray fluorescence in the assessment of rare earth element-enriched sedimentary phosphate deposits. Geochem. Explor. Environ. Anal. 2014, 14, 161–169. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Fundamental Parameters (FP) | Compton | Empirical | Positive Material Identification (PMI) |
|---|---|---|---|---|
| Names | Mining, Geochemistry, Alloys, Precious metals, Plastics | Soil | Consumer Safety, RoHS, WEEE, Fast Identification | |
| Materials analysed | Rocks, ores, soils, waste, plastics, and alloys | Rocks, soils, and sediments data | All matrices, paint, fine deposit, dust | Minerals, waste, alloys |
| Lab XRF | pXRF | No Film | KaptonTM Film | Paper | PVC |
|---|---|---|---|---|---|
| Ni | Ni | Y = 1.26x | Y = 1.74x | Y = 1.62x | Y = 1.68x |
| Fe2O3 | Fe | Y = 1.46x − 6.35 | Y = 1.91x − 9.65 | Y = 1.80x − 8.49 | Y = 1.85x − 9.84 |
| Co | Co | Y = 1.20x | Y = 0.74x | Y = 0.97x | Y = 1.19x |
| Cr2O3 | Cr | Y = 2.37x | Y = 2.37x | Y = 2.39x | Y = 2.13x |
| Mn | Mn | Y = 1.07x | Y = 1.32x | Y = 1.31x | Y = 1.27x |
| SiO2 | Si | Y = 2.06x | N/A | N/A | N/A |
| Al2O3 | Al | Y = 1.5x + 1 | N/A | N/A | N/A |
| Z | Symbol | Element | Kα | Kβ | Lα | Lβ | Lγ |
|---|---|---|---|---|---|---|---|
| 12 | Mg | Magnesium | 1.25 | 1.30 | - | - | - |
| 13 | Al | Aluminium | 1.49 | 1.55 | - | - | - |
| 14 | Si | Silicon | 1.74 | 1.83 | - | - | - |
| 15 | P | Phosphorus | 2.02 | 2.14 | - | - | - |
| 16 | S | Sulfur | 2.31 | 2.46 | - | - | - |
| 17 | Cl | Chlorine | 2.62 | 2.82 | - | - | - |
| 18 | Ar | Argon | 2.96 | 3.19 | - | - | - |
| 19 | K | Potassium | 3.31 | 3.59 | - | - | - |
| 20 | Ca | Calcium | 3.69 | 4.01 | 0.34 | - | - |
| 21 | Sc | Scandium | 4.09 | 4.46 | 0.40 | - | - |
| 31 | Ga | Gallium | 9.24 | 10.26 | 1.10 | 1.12 | |
| 32 | Ge | Germanium | 9.88 | 10.98 | 1.19 | 1.21 | |
| 33 | As | Arsenic | 10.53 | 11.73 | 1.28 | 1.32 | |
| 34 | Se | Selenium | 11.21 | 12.50 | 1.38 | 1.42 | |
| 35 | Br | Bromine | 11.91 | 13.30 | 1.48 | 1.53 | |
| 36 | Kr | Krypton | 12.64 | 14.11 | 1.59 | 1.64 | |
| 37 | Rb | Rubidium | 13.38 | 14.97 | 1.69 | 1.75 | |
| 38 | Sr | Strontium | 14.15 | 15.85 | 1.81 | 1.87 | |
| 39 | Y | Yttrium | 14.95 | 16.74 | 1.92 | 2.00 | |
| 40 | Zr | Zirconium | 15.77 | 17.68 | 2.04 | 2.12 | 2.30 |
| 41 | Nb | Niobium | 16.61 | 18.64 | 2.17 | 2.26 | 2.46 |
| 42 | Mo | Molybdenum | 17.48 | 19.63 | 2.29 | 2.40 | 2.62 |
| 43 | Tc | Technetium | 18.41 | 20.66 | 2.42 | 2.54 | 2.79 |
| 44 | Ru | Ruthenium | 19.28 | 21.66 | 2.56 | 2.68 | 2.96 |
| 45 | Rh | Rhodium | 20.21 | 22.75 | 2.70 | 2.83 | 3.14 |
| 46 | Pd | Palladium | 21.18 | 23.85 | 2.84 | 2.99 | 3.33 |
| 47 | Ag | Silver | 22.16 | 24.96 | 2.98 | 3.16 | 3.52 |
| 48 | Cd | Cadmium | 23.17 | 26.12 | 3.13 | 3.32 | 3.72 |
| 49 | In | Indium | 24.21 | 27.30 | 3.29 | 3.49 | 3.92 |
| eV | Line | Interferent | Distance (eV) | Reference |
|---|---|---|---|---|
| 4090.6 | 21 Sc Kα1 | 20 Ca Kβ1 | 78 | |
| 5414.72 | 24 Cr Kα1 | 23 V Kβ1 | 13 | |
| 5898.75 | 25 Mn Kα1 | 24 Cr Kβ1 | 48 | |
| 6490.45 | 25 Mn Kβ1 | 26 Fe Kα1 | 5 | [40] |
| 6930.32 | 27 Co Kα1 | 26 Fe Kβ1 | 128 | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laperche, V.; Lemière, B. Possible Pitfalls in the Analysis of Minerals and Loose Materials by Portable XRF, and How to Overcome Them. Minerals 2021, 11, 33. https://doi.org/10.3390/min11010033
Laperche V, Lemière B. Possible Pitfalls in the Analysis of Minerals and Loose Materials by Portable XRF, and How to Overcome Them. Minerals. 2021; 11(1):33. https://doi.org/10.3390/min11010033
Chicago/Turabian StyleLaperche, Valérie, and Bruno Lemière. 2021. "Possible Pitfalls in the Analysis of Minerals and Loose Materials by Portable XRF, and How to Overcome Them" Minerals 11, no. 1: 33. https://doi.org/10.3390/min11010033
APA StyleLaperche, V., & Lemière, B. (2021). Possible Pitfalls in the Analysis of Minerals and Loose Materials by Portable XRF, and How to Overcome Them. Minerals, 11(1), 33. https://doi.org/10.3390/min11010033