Significance of Fracture-Filling Rose-Like Calcite Crystal Clusters in the SE Pyrenees




Abstract
1. Introduction
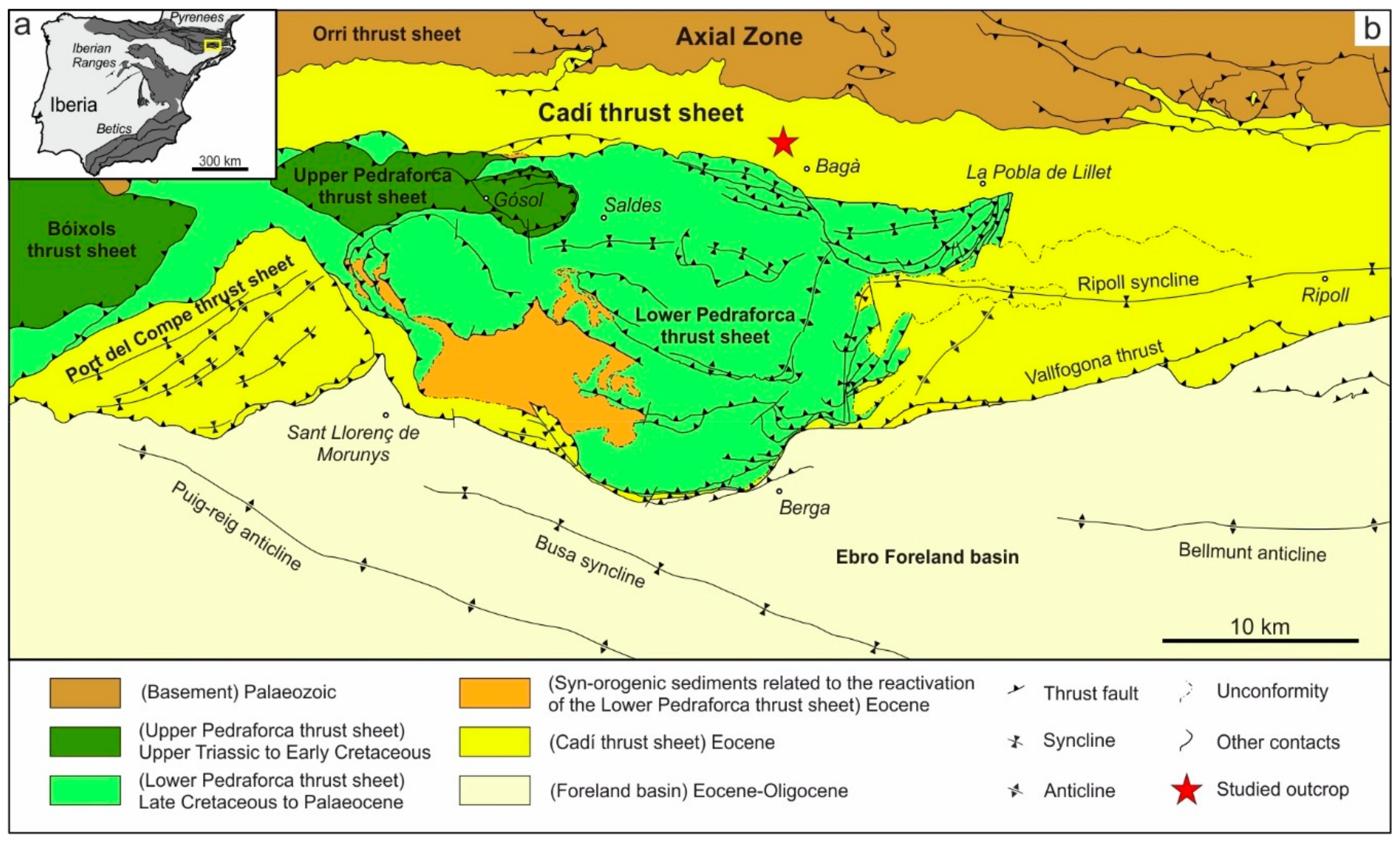
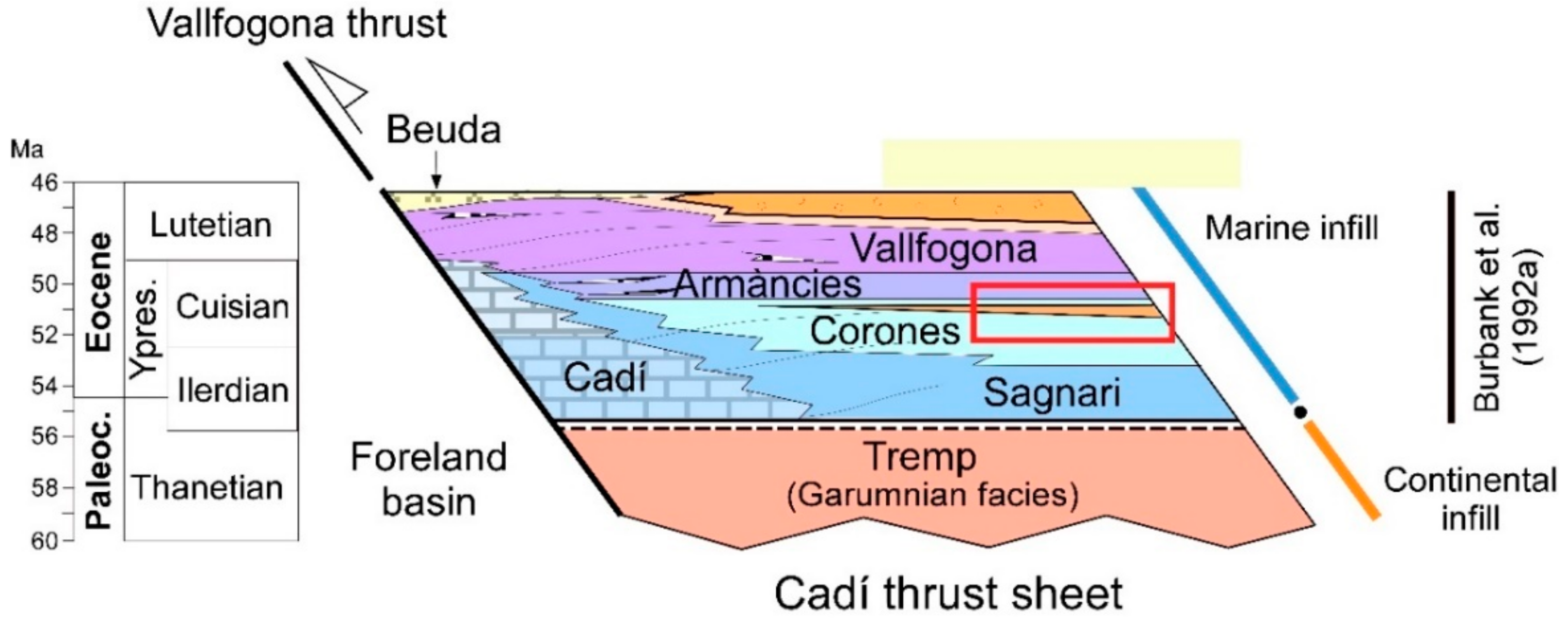
2. Geological Setting
3. Methodology
4. Results
4.1. Description of the Studied Outcrop
4.2. Petrography
4.1.1. The Host Rock
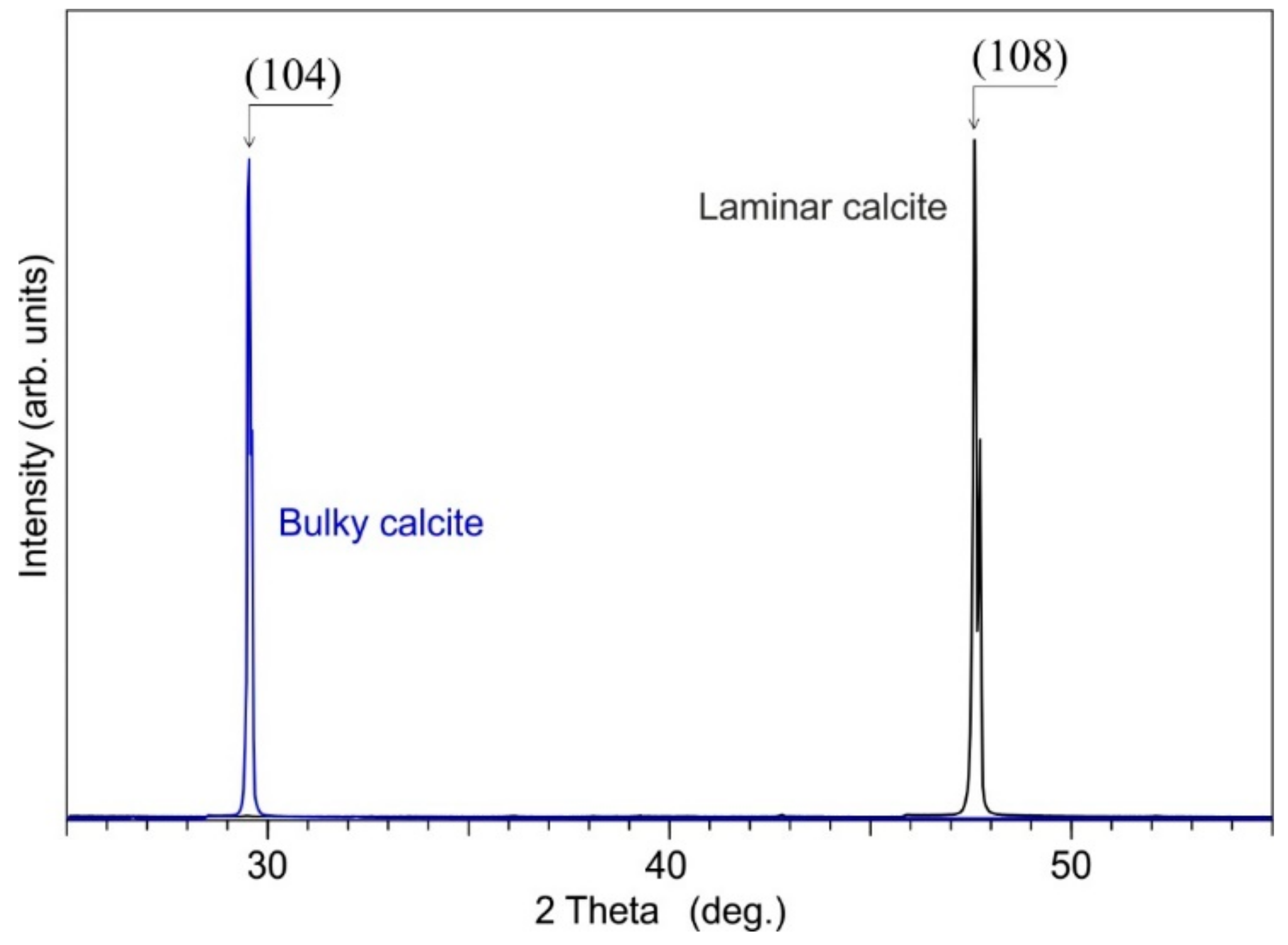
4.1.2. Calcite Cement
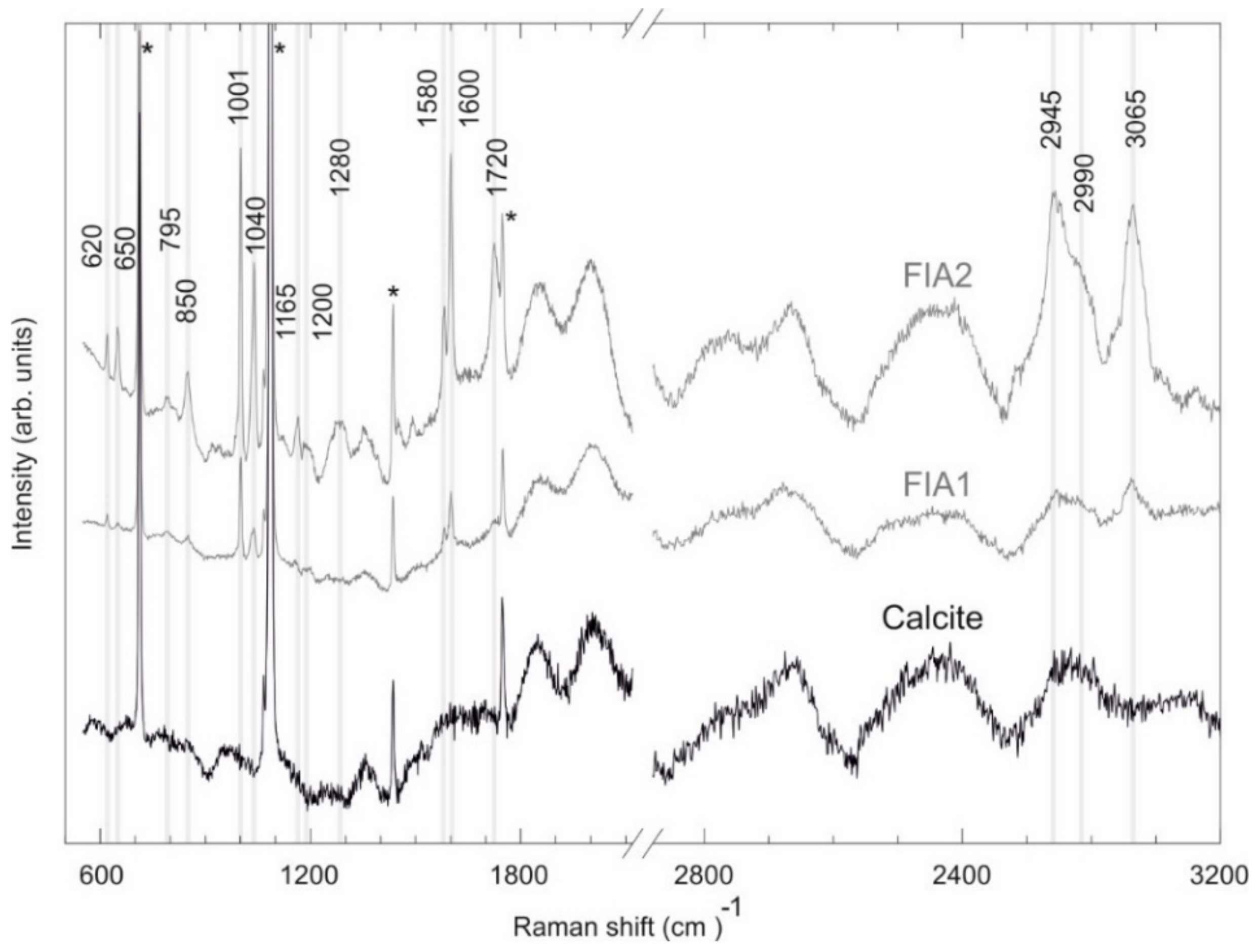
4.3. Fluid Inclusion Studies
4.4. Geochemistry of Bladed Calcite
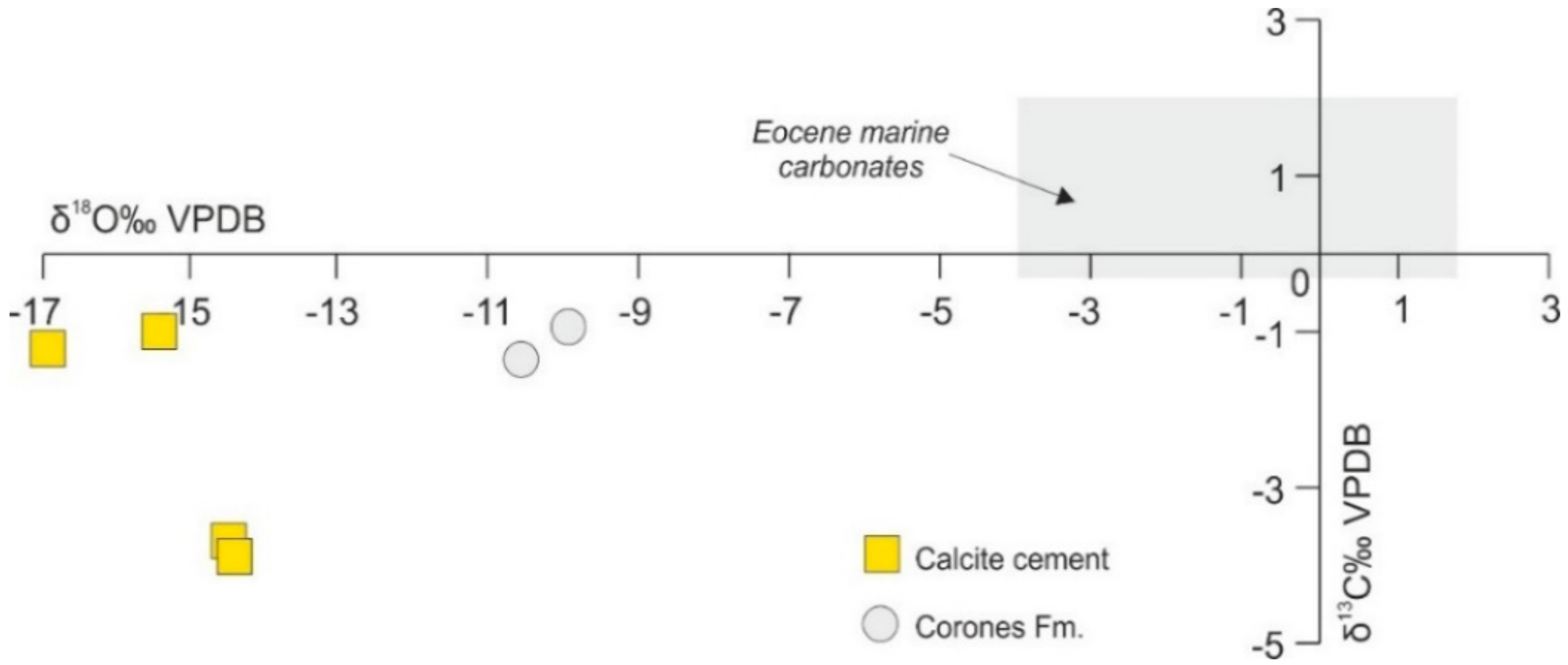
4.4.1. C–O Stable Isotopes
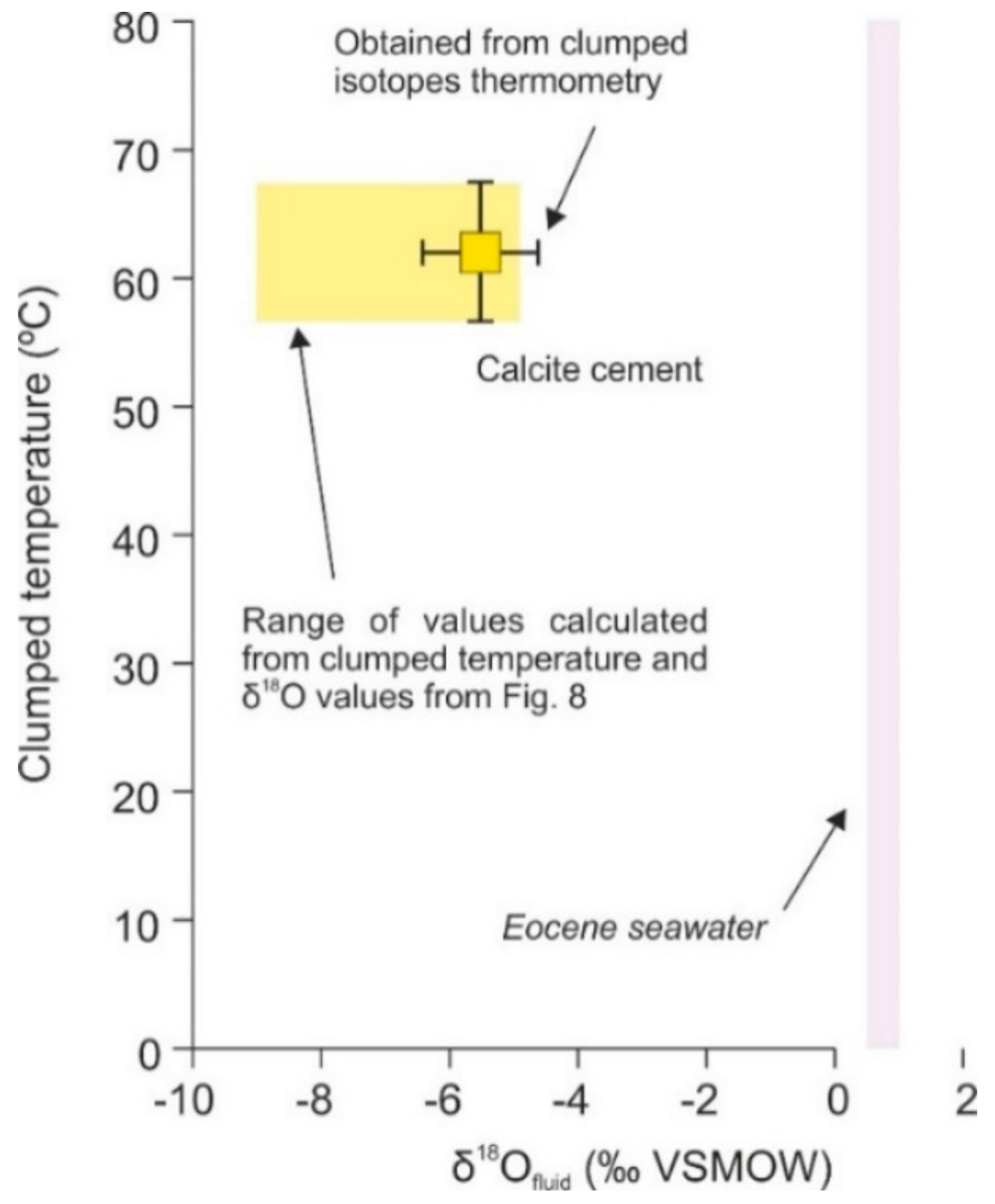
4.4.2. Clumped Isotopes
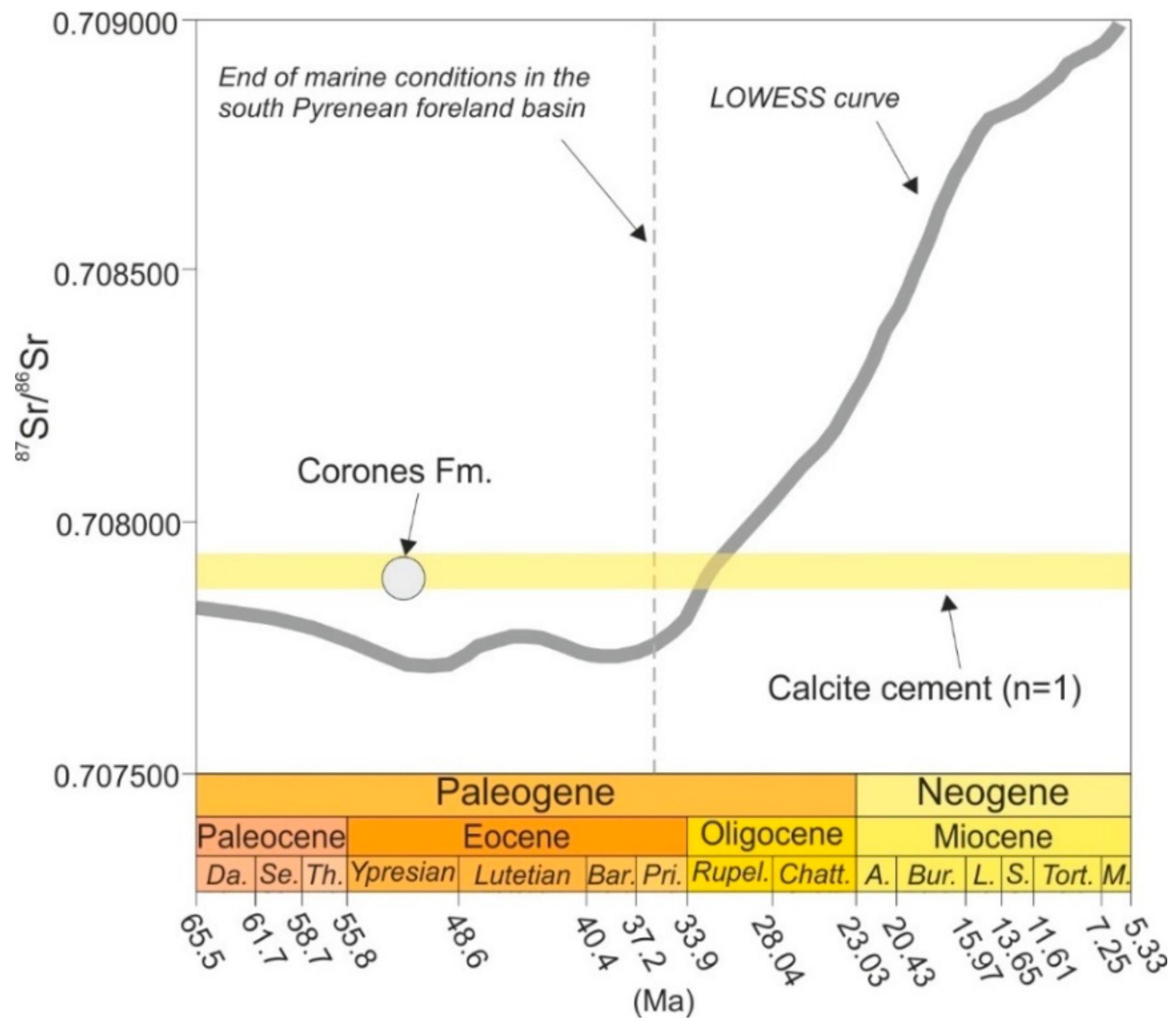
4.4.3. Sr isotopes
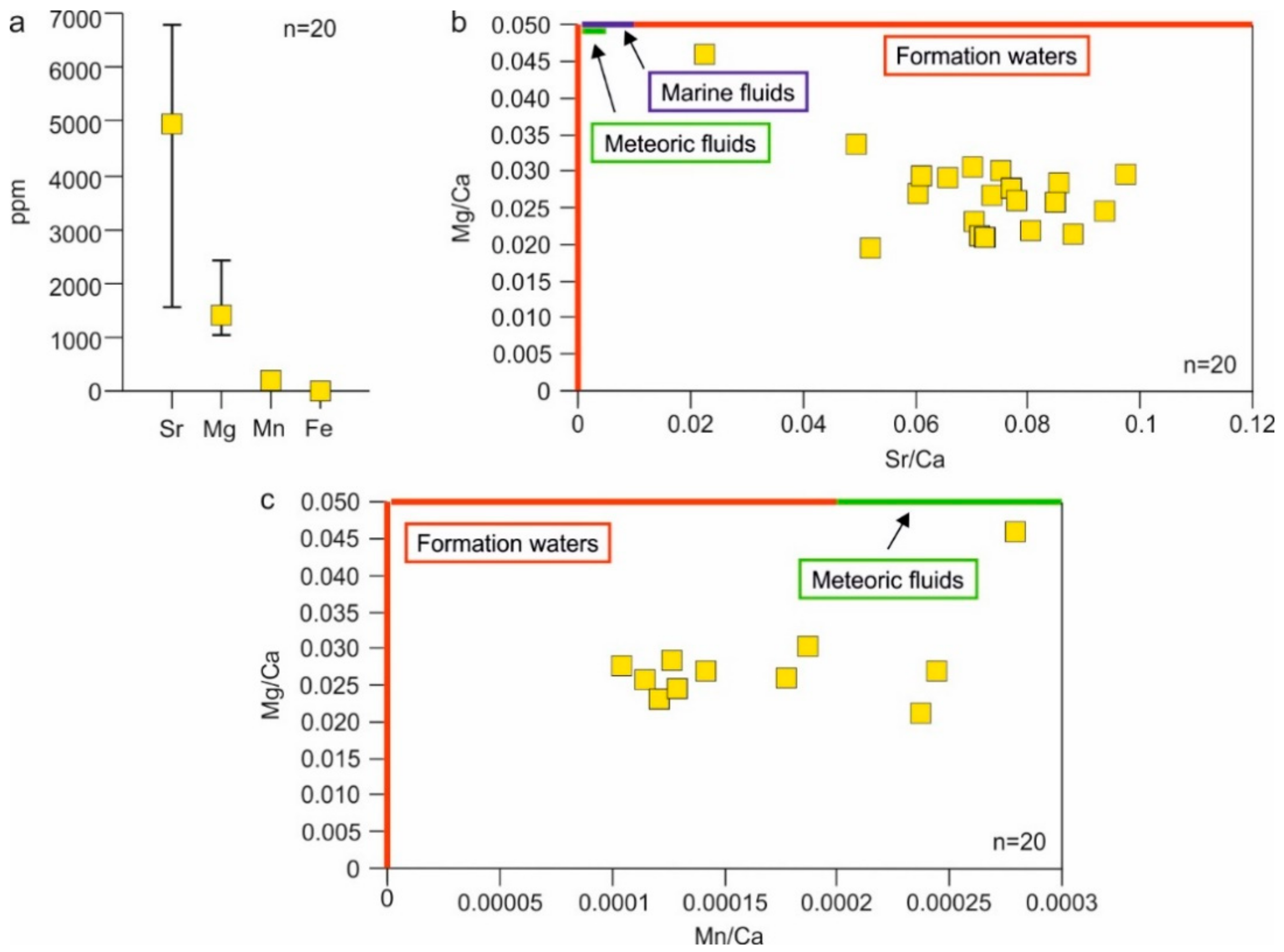
4.4.4. Elemental Composition
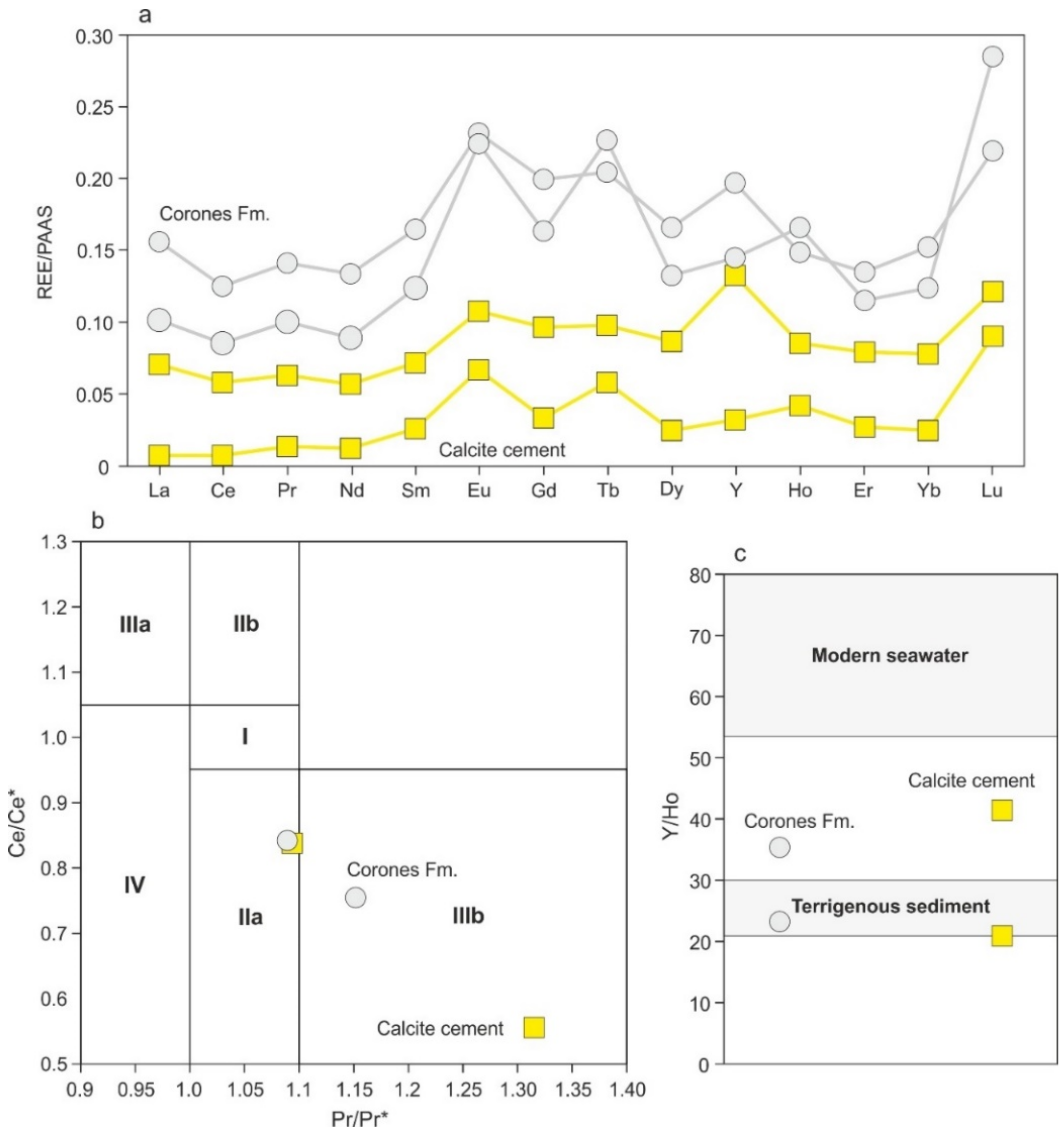
4.4.5. REEs + Y Content
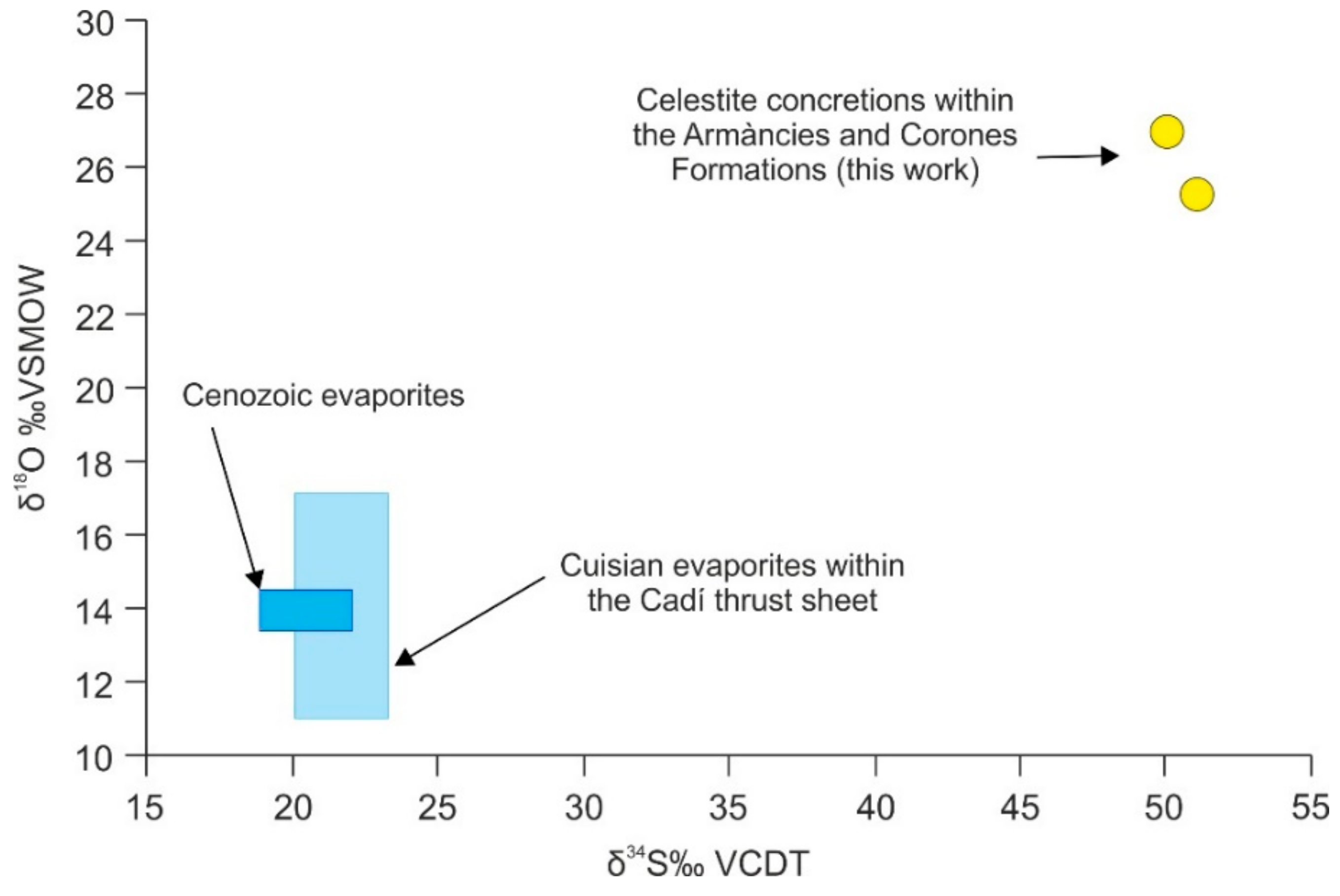
4.4.6. Sulfur and Oxygen Isotopes of Celestite Concretions
5. Discussion
5.1. Host Rock Diagenesis
5.2. Type of Fluids
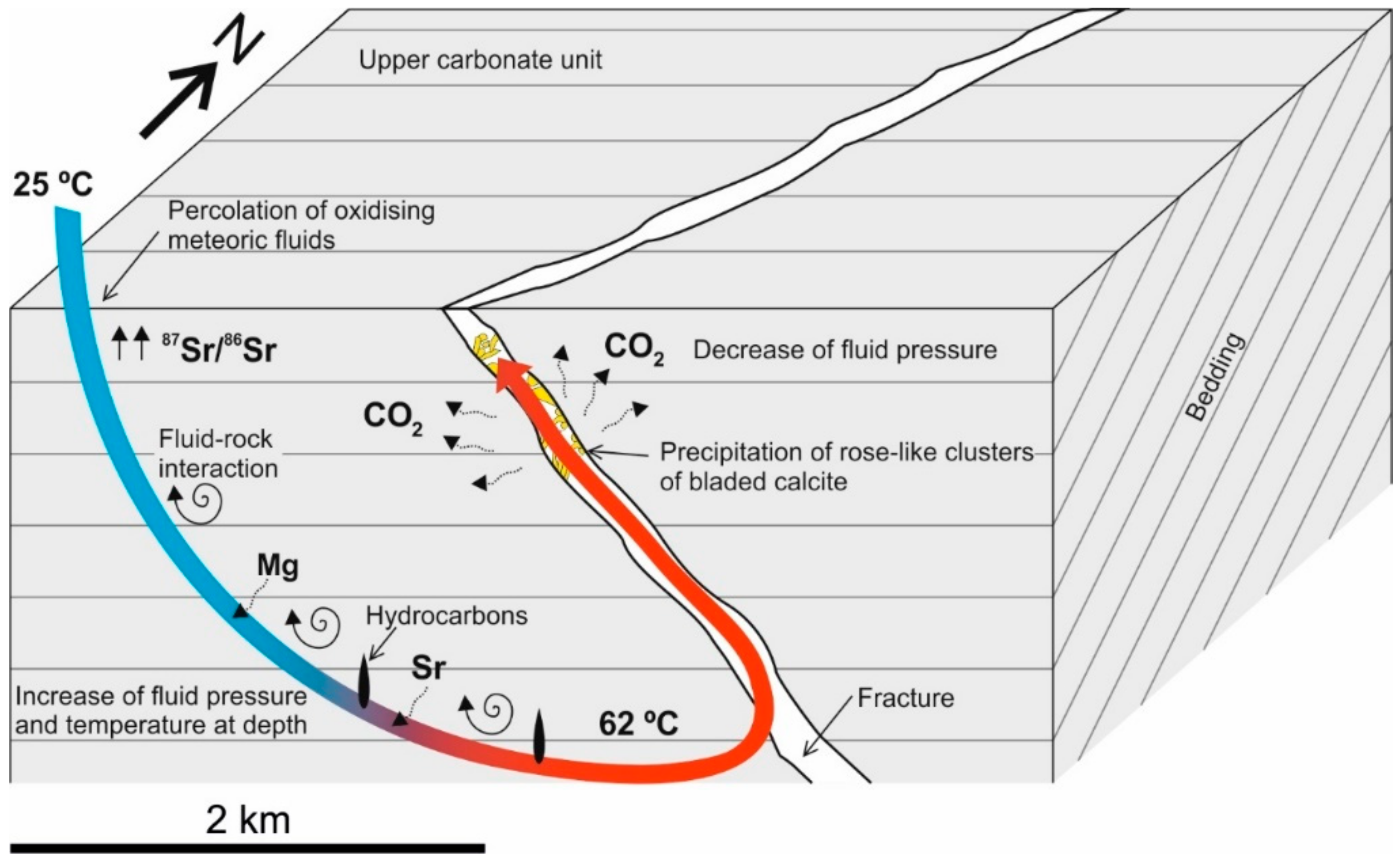
5.3. Precipitation Model for Rose-Like Clusters of Bladed Calcites
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Travé, A.; Calvet, F.; Sans, M.; Vergés, J.; Thirlwall, M. Fluid history related to the Alpine compression at the margin of the south-Pyrenean Foreland basin: The El Guix anticline. Tectonophysics 2000, 321, 73–102. [Google Scholar] [CrossRef]
- Travé, A.; Labaume, P.; Calvet, F.; Soler, A.; Tritlla, J.; Bautier, M.; Potdevin, J.L.; Séguret, M.; Raynaud, S.; Briqueu, L. Fluid migration during Eocene thrust emplacement in the south Pyrenean foreland basin (Spain): An integrated structural, mineralogical and geochemical approach. In Cenozoic Foreland Basins of Western Europe; Mascle, A., Puigdefàbregas, C., LuterBacher, H.P., Fernàndez, M., Eds.; Geological Society, Special Publications: London, UK, 1998; Volume 134, pp. 163–188. [Google Scholar]
- Swennen, R.; Ferket, H.; Benchilla, L.; Roure, F.; Ellam, R.; Team, S. Fluid flow and diagenesis in carbonate dominated Foreland Fold and Thrust Belts: Petrographic inferences from field studies of late-diagenetic fabrics from Albania, Belgium, Canada, Mexico and Pakistan. J. Geochem. Explor. 2003, 78–79, 481–485. [Google Scholar] [CrossRef]
- Hansman, R.J.; Albert, R.; Gerdes, A.; Ring, U. Absolute ages of multiple generations of brittle structures by U-Pb dating of calcite. Geology 2018, 46, 207–210. [Google Scholar] [CrossRef]
- Cantarero, I.; Parcerisa, D.; Plata, M.A.; Gómez-Gras, D.; Gómez-Rivas, E.; Martín-Martín, J.D.; Travé, A. Fracturing and Near-Surface Diagenesis of a Silicified Miocene Deltaic Sequence: The Montjuïc Hill (Barcelona). Minerals 2020, 10, 135. [Google Scholar] [CrossRef]
- Rochelle, C.A.; Czernichowski-Lauriol, I.; Milodowski, A.E. The impact of chemical reactions on CO2 storage in geological formations: A brief review. In Geological Storage of Carbon Dioxide; Baines, S.J., Worden, R.H., Eds.; Geological Society Special Publications: London, UK, 2004; Volume 233, pp. 87–106. [Google Scholar]
- Pham, V.T.H.; Lu, P.; Aagaard, P.; Zhu, C.; Hellevang, H. On the potential of CO2–water–rock interactions for CO2 storage using a modified kinetic model. Int. J. Greenh. Gas Control 2011, 5, 1002–1015. [Google Scholar] [CrossRef]
- Gasparrini, M.; Ruggieri, G.; Brogi, A. Diagenesis versus hydrothermalism and fluid–rock interaction within the Tuscan Nappe of the Monte Amiata CO2-rich geothermal area (Italy). Geofluids 2013, 13, 159–179. [Google Scholar] [CrossRef]
- Banner, J.L.; Hanson, G.N. Calculation of simultaneous isotopic and trace element variations during water-rock interaction with applications to carbonate diagenesis. Geochim. Cosmochim. Acta 1990, 54, 3123–3137. [Google Scholar] [CrossRef]
- Warren, J. Dolomite: Occurrence, evolution and economically important associations. Earth-Sci. Rev. 2000, 52, S0012–S8252. [Google Scholar] [CrossRef]
- Aquilano, D.; Otálora, F.; Pastero, L.; García-Ruiz, J.M. Three study cases of growth morphology in minerals: Halite, calcite and gypsum. Prog. Cryst. Growth Charact. Mater. 2016, 62, 227–251. [Google Scholar] [CrossRef]
- Milodowski, A.E.; Bath, A.; Norris, S. Palaeohydrogeology using geochemical, isotopic and mineralogical analyses: Salinity and redox evolution in a deep groundwater system through Quaternary glacial cycles. Chem. Geol. 2018, 97, 40–60. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Astilleros, J.M.; Pina, C.M. The Morphology of calcite crystals grown in a porous medium doped with divalent cations. Chem. Geol. 2006, 225, 314–321. [Google Scholar] [CrossRef]
- Iwatsuki, T.; Satake, H.; Metcalfe, R.; Yoshida, H.; Hama, K. Isotopic and morphological features of fracture calcite from granitic rocks of the Tono area, Japan: A promising palaeohydrogeological tool. Appl. Geochem. 2002, 17, 1241–1257. [Google Scholar] [CrossRef]
- Chen, C.L.; Qi, J.; Tao, J.; Zuckermann, R.N.; Yoreo, J.J.D. Tuning calcite morphology and growth acceleration by a rational design ofhighly stable protein-mimetics. Sci. Rep. 2014, 4, 6266. [Google Scholar] [CrossRef] [PubMed]
- Oral, C.M.; Ercan, B. Influence of pH on morphology, size and polymorph of room temperature synthesized calcium carbonate particles. Powder Technol. 2018, 339, 781–788. [Google Scholar] [CrossRef]
- Harvey, C.C.; Browne, P.R.L. Note on the occurrence and compositions of calcite from the Te Mihi sector of the Wairakei geothermal system, New Zealand. In Proceedings of the 14th New Zealand Geothermal Workshop, Taupo, New Zealand, 14–16 November 2018. [Google Scholar]
- Canet, C.; Franco, S.I.; Prol-Ledesma, R.M.; González-Partida, E.; Villanueva-Estrada, R.E. A model of boiling for fluid inclusion studies: Application to the Bolaños Ag–Au–Pb–Zn epithermal deposit, Western Mexico. J. Geochem. Explor. 2011, 110, 118–125. [Google Scholar] [CrossRef]
- Montcada, D.; Mutchler, S.; Nieto, A.; Reynolds, T.J.; Rimstidt, J.D.; Bodnar, R.J. Mineral textures and fluid inclusion petrography of the epithermal Ag–Au deposits at Guanajuato, Mexico: Application to exploration. J. Geochem. Explor. 2012, 114, 20–35. [Google Scholar] [CrossRef]
- Griffiths, L.; Heap, M.J.; Wang, F.; Daval, D.; Gilg, H.A.; Baud, P.; Schmittbuhl, J.; Genter, A. Geothermal implications for fracture-filling hydrothermalprecipitation. Geothermics 2016, 64, 235–245. [Google Scholar] [CrossRef]
- Lu, Y.C.; Song, S.R.; Taguchi, S.; Wang, P.L.; Yeh, E.C.; Lin, Y.J.; MacDonald, J.; John, C.M. Evolution of hot fluids in the Chingshui geothermal field inferred from crystal morphology and geochemical vein data. Geothermics 2018, 74, 305–318. [Google Scholar] [CrossRef]
- Hutcheon, I.; Shevalier, M.; Abercrombie, H.J. pH buffering by metastable mineral-fluid equilibria and evolution of carbon dioxide fugacity during burial diagenesis. Geochim. Cosmochim. Acta 1993, 57, 1017–1027. [Google Scholar] [CrossRef]
- Simmons, S.F.; Christenson, B.W. Origin of calcite in a boiling geothermal system. Am. J. Sci. 1994, 294, 361–400. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Putnis, C.V.; Rodríguez-Navarro, C.; Putnis, A. Effect of pH on calcite growth at constant aCa2+/aCO32− ratio and supersaturation. Geochim. Cosmochim. Acta 2011, 75, 284–296. [Google Scholar] [CrossRef]
- Randall, M.; Avila, W. The boiling point-composition diagram of insmiscible and partially miscible liquid systems. J. Geochem. Explor. 1940, 17, 536–537. [Google Scholar] [CrossRef]
- Krell, E. (Ed.) Chapter 4. Physical fundamentals of the separation process. In Techniques and Instrumentation in Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 1982; Volume 2, pp. 43–202. [Google Scholar]
- Geankoplis, C.J. Transport Processes and Separation Process Principles, 4th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2003. [Google Scholar]
- Travé, A.; Labaume, P.; Calvet, F.; Soler, A. Sediment dewatering and pore fluid migration along thrust faults in a foreland basin inferred from isotopic and elemental geochemical analyses (Eocene southern Pyrenees, Spain). Tectonophysics 1997, 282, 375–398. [Google Scholar] [CrossRef]
- Travé, A.; Labaume, P.; Vergés, J. Fluid systems in Foreland Fold and thrust belts: An overview from the Southern Pyrenees. In Thrust Belts and Foreland Basins: From Fold Kinematics to Hydrocarbon Systems; Lacombe, O., Lavé, J., Roure, F., Vergés, J., Eds.; Springer: Amsterdam, The Netherlands, 2007; pp. 93–115. [Google Scholar]
- Caja, M.A.; Permanyer, A.; Marfil, R.; Al-Asm, I.S.; Martín-Crespo, T. Fluid flow record from fracture-fill calcite in the Eocene limestones from the South-Pyrenean Basin (NE Spain) and its relationship to oil shows. J. Geochem. Explor. 2006, 89, 27–32. [Google Scholar] [CrossRef]
- Lacroix, B.; Baumgartner, L.P.; Bouvier, A.S.; Kempton, P.D.; Vennemann, T. Multi fluid-flow record during episodic mode I opening: Amicrostructural and SIMS study (Cotiella Thrust Fault, Pyrenees). Earth Planet. Sci. Lett. 2018, 503, 37–46. [Google Scholar] [CrossRef]
- Lacroix, B.; Buatier, M.; Labaume, P.; Travé, A.; Dubois, M.; Charpentier, D.; Ventalon, S.; Convert-Gaubier, D. Microtectonic and geochemical characterization of thrusting in a foreland basin: Example of the South-Pyrenean orogenic wedge (Spain). J. Struct. Geol. 2011, 33, 1359–1377. [Google Scholar] [CrossRef]
- Lacroix, B.; Travé, A.; Buatier, M.; Labaume, P.; Vennemann, T.; Dubois, M. Syntectonic fluid-flow along thrust faults: Example of the South-Pyrenean fold-and-thrust belt. Mar. Pet. Geol. 2014, 49, 84–98. [Google Scholar] [CrossRef]
- Crognier, N.; Hoareau, G.; Aubourg, C.; Dubois, M.; Lacroix, B.; Branellec, M.; Callot, J.P.; Vennemann, T. Syn-orogenic fluid flow in the Jaca basin (south Pyrenean fold and thrust belt) from fracture and vein analyses. Basin Res. 2017. [Google Scholar] [CrossRef]
- Beaudoin, N.; Huyghe, D.; Bellahsen, N.; Lacombe, O.; Emmanuel, L.; Mouthereau, F.; Ouanhnon, L. Fluid systems and fracture development during syn-depositional fold growth: An example from the Pico del Aguila anticline, Sierras Exteriores, southern Pyrenees, Spain. J. Struct. Geol. 2015, 70, 23–38. [Google Scholar] [CrossRef]
- Cruset, D.; Cantarero, I.; Travé, A.; Vergés, J.; John, C.M. Crestal graben fluid evolution during growth of the Puig-reig anticline (South Pyrenean fold and thrust belt). J. Geodyn. 2016, 101, 30–50. [Google Scholar] [CrossRef]
- Cruset, D.; Cantarero, I.; Vergés, J.; John, C.M.; Muñoz-López, D.; Travé, A. Changes in fluid regime in syn-orogenic sediments during the growth of the south Pyrenean fold and thrust belt. Glob. Planet. Chang. 2018, 171, 207–224. [Google Scholar] [CrossRef]
- Nardini, N.; Muñoz-López, D.; Cruset, D.; Cantarero, I.; Martín-Martín, J.D.; Benedicto, A.; Gómez-Rivas, E.; John, C.M.; Travé, A. From early contraction to post-folding fluid evolution in the frontal part of the Bóixols thrust sheet (southern Pyrenees) as revealed by the texture and geochemistry of calcite cements. Minerals 2019, 9, 117. [Google Scholar] [CrossRef]
- Choukroune, P.; Team ECORS. The ECORS Pyrenean deep seismic profile reflection data and the overall structure of an orogenic belt. Tectonics 1989, 8, 23–39. [Google Scholar] [CrossRef]
- Roure, F.; Choukroune, P.; Berastegui, J.; Muñoz, J.A.; Villien, A.; Matheron, P.; Bareyt, M.; Seguret, M.; Camara, P.; Deramond, J. Ecors deep seismic data and balanced cross sections: Geometric constraints on the evolution of the Pyrenees. Tectonics 1989, 8, 41–50. [Google Scholar] [CrossRef]
- Muñoz, J.A. Evolution of a continental collision belt: ECORS–Pyrenees crustal balanced section. In Thrust Tectonics; McClay, K.R., Ed.; Chapman & Hall: London, UK, 1992; pp. 235–246. [Google Scholar]
- Muñoz, J.A. The Pyrenees. In The Geology of Spain; Gibbons, W., Moreno, T., Eds.; Geological Society: London, UK, 2002; pp. 370–385. [Google Scholar]
- Vergés, J.; Fernàndez, M.; Martínez, A. The Pyrenean orogen: Pre-, syn-, and post-collisional evolution. In Reconstruction of the Evolution of the Alpine-Himalayan Orogen, Journal of the Virtual Explorer; Rosenbaum, G., Lister, G., Eds.; Research School of Earth Sciences, Australian National University: Canberra, Australia, 2002; Volume 8, pp. 55–74. [Google Scholar]
- Vergés, J.; Millán, H.; Roca, E.; Muñoz, J.A.; Marzo, M.; Cirés, J.; Bezemer, T.D.; Zoetemeijer, R.; Cloetigh, S. Eastern Pyrenees and related foreland basins: Pre-, syn- and post-collisional cristal-scale cross-sections. Mar. Pet. Geol. 1995, 12, 893–915. [Google Scholar] [CrossRef]
- Teixell, A.; Labaume, P.; Ayarza, P.; Espurt, N.; Blanquat, M.d.S.; Lagabrielle, Y. Crustal structure and evolution of the Pyrenean-Cantabrian belt: A review and new interpretations from recent concepts and data. Tectonophysics 2018, 724–725, 146–170. [Google Scholar] [CrossRef]
- Mató, E.; Saula, E.; Martínez-Rius, A.; Muñoz, J.A.; Escuer, J. Memoria de la Hoja n° 293 (Berga). In Mapa Geológico de España E. 1:50.000 (MAGNA), Segunda Serie, Primera Edición; IGME: Madrid, Spain, 1994; p. 66. [Google Scholar]
- Pi, M.E.; Samsó-Escola, J.M.; Vilella, L.; Arbués, P.; Casanovas, J.; Berastegui, X. Mapa geológico de la Hoja n° 291 (Oliana). In Mapa Geológico de España E. 1:50.000. Segunda Serie (MAGNA), Primera Edición; IGME: Madrid, Spain, 2001. [Google Scholar]
- Muñoz, J.A.; Vergés, J.; Martínez, A.; Fleta, J.; Cirés, J.; Casas, J.M.; Sàbat, F. Mapa geológico de la Hoja n° 256 (Ripoll). In Mapa Geológico de España E. 1:50.000. Segunda Serie (MAGNA), Primera Edición; IGME: Madrid, Spain, 1994. [Google Scholar]
- Vergés, J.; Martínez, A.; Domingo, F.; Muñoz, J.A.; Losantos, M.; Fleta, J.; Gisbert, J. Mapa geológico de la Hoja n° 255 (La Pobla de Lillet). In Mapa Geológico de España E. 1:50.000. Segunda Serie (MAGNA), Primera Edición; IGME: Madrid, Spain, 1994. [Google Scholar]
- Puigdefàbregas, C.; Muñoz, J.A.; Marzo, M. Thrust Belt Development in the Eastern Pyrenees and Related Depositional Sequences in the Southern Foreland Basin. In Foreland Basins; Allen, P.A., Homewood, P., Eds.; Blackwell Publishing Ltd.: Oxford, UK, 1986; Volume 8, pp. 229–246. [Google Scholar]
- Burbank, D.W.; Vergés, J.; Muñoz, J.A.; Bentham, P. Coeval hinward- and forward-imbricating thrusting in the south-central Pyrenees, Spain: Timing and rates of shortening and deposition. Geol. Soc. Am. Bull. 1992, 104, 3–17. [Google Scholar] [CrossRef]
- Burbank, D.W.; Puigdefàbregas, C.; Muñoz, J.A. The chronology of the Eocene tectonic and stratigraphic development of the Eastern Pyrenean Foreland Basin. NE Spain. Geol. Soc. Am. Bull. 1992, 104, 1101–1120. [Google Scholar] [CrossRef]
- Vergés, J.; Burbank, D.W. Eocene-Oligocene thrusting and basin configuration in the eastern and central Pyrenees (Spain). In Tertiary Basins of Spain; Friend, P., Dabrio, C., Eds.; World and Regional Geology E11; Cambridge University Press: Cambridge, UK, 1996; pp. 120–133. [Google Scholar]
- Vergés, J. Estudi Geològic del Vessant sud del Pirineu Oriental i Central, Evolució cinemàtica en 3D. Ph.D. Thesis, Universitat de Barcelona, Barcelona, Spain, 1993. [Google Scholar]
- Gich, M. Estudio geológico del Eoceno prepirenaico del Ripollés oriental. Acta Geol. Hisp. 1973, 8, 120–124. [Google Scholar]
- Barnolas, A. Evolución sedimentaria de la Cuenca Surpirenaica Oriental durante el Eoceno. Acta Geol. Hisp. 1992, 27, 15–31. [Google Scholar]
- Giménez-Monsant, J. Análisis de Cuenca del Eoceno Inferior de la Unidad Cadí (Pirineo oriental). El Sistema Deltaico y de Plataforma Carbonática de la Formación Corones. Ph.D. Thesis, Universitat de Barcelona, Barcelona, Spain, 1993. [Google Scholar]
- Gimènez-Montsant, J.; Calvet, F. Ciclos y ciclicidad en depósitos de ostrácodos del Cuisiense inferior del sur de los Pirineos orientales. In Proceedings of the I Congreso del Grupo Español del Terciario, Vic, Spain, 12–17 April 1998. [Google Scholar]
- Calvet, F.; Playà, E.; Giménez-Montsant, J.; Permanyer, A. Fifth-order cyclicity and organic matter contents relationship (Lower Eocene, Pyrenees). Geol. Acta 2007, 5, 59–75. [Google Scholar]
- Cruset, D.; Cantarero, I.; Vergés, J.; Benedicto, A.; John, C.M.; Gerdes, A.; Albert, R.; Travé, A. Tectonically-induced oil migration in the south-eastern Pyrenean foreland basin. In Proceedings of the 34th International Meeting of Sedimentology (IAS), Rome, Italy, 10–13 September 2019. [Google Scholar]
- Permanyer, A.; Vallés, D.; Dorronsoro, C. Source rocks potential of an Eocene carbonate ramps: The Armàncies Formation of the southern Pyrenean Basin, northeast Spain. AAPG Bull. 1988, 72, 1019. [Google Scholar]
- Permanyer, A.; Gallego, J.L.R.; Caja, M.A.; Dessort, D. Crude oil biodegradation and environmental factors at the Riutort oil shale mine, SE Pyrenees. J. Pet. Geol. 2010, 33, 123–140. [Google Scholar] [CrossRef]
- Jones, B.; Renaut, R.W. Origin of Platy calcite crystals in hot-spring deposits in the Kenya Rift Valley. J. Sediment. Res. 1998, 68, 913–927. [Google Scholar] [CrossRef]
- McCrea, J.M. On the Isotopic Chemistry of Carbonates and a Paleotemperature Scale. J. Chem. Phys. 1950, 18, 849–957. [Google Scholar] [CrossRef]
- Craig, H.; Gordon, I.-I. Deuterium and oxygen-18 variations in the ocean and the marine atmosphere. In A Conference on Stable Isotopes in Oceanographic Studies and Paleotemperatures; Tongiorgi, E., Ed.; Consiglio Nazionale delle Richerche, Laboratorio di Geologia Nucleare: Pisa, Italy, 1965; pp. 9–130. [Google Scholar]
- Claypool, G.E.; Kaplan, W.T.; Kaplan, I.R.; Sakai, H.; Zak, I. The age curves of sulfur and oxygen isotopes in marine sulfate and their mutual interpretations. Chem. Geol. 1980, 28, 199–260. [Google Scholar] [CrossRef]
- John, C.M.; Bowen, D. Community software for challenging isotope analysis: First applications of “Easotope” to clumped isotopes. Rapid Commun. Mass Spectrom. 2016, 30, 2285–2300. [Google Scholar] [CrossRef]
- Guo, W.; Mosenfelder, J.L.; Goddard, W.A.; Eiler, J.M. Isotopic fractionations associated with phosphoric acid digestion of carbonate minerals: Insights from first-principles theoretical modeling and clumped isotope measurements. Geochim. Cosmochim. Acta 2009, 73, 7203–7225. [Google Scholar] [CrossRef]
- Huntington, K.W.; Eiler, J.M.; Affeck, H.P.; Guo, W.; Bonifacie, M.; Yeung, L.Y.; Thiagarajan, N.; Passey, B.; Tripati, A.; Daëron, M.; et al. Methods and limitations of ‘clumped’ CO2 isotope (Δ47) analysis by gas-source isotope ratio mass spectrometry. J. Mass Spectrom. 2009, 44, 1318–1329. [Google Scholar] [CrossRef]
- Bernasconi, S.M.; Hu, B.; Wacker, U.; Fiebig, J.; Breitenbach, S.F.M.; Rutz, T. Background effects on Faraday collectors in gas-source mass spectrometry and implications for clumped isotope measurements. Rapid Commun. Mass Spectrom. 2013, 27, 603–612. [Google Scholar] [CrossRef]
- Dennis, K.J.; Affeck, H.P.; Passey, B.H.; Schrag, D.P.; Eiler, J.M. Defining an absolute reference frame for ‘clumped’ isotope studies of CO2. Geochim. Cosmochim. Acta 2011, 75, 7117–7131. [Google Scholar] [CrossRef]
- Kim, S.T.; O’Neil, J.R. Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates. Geochim. Cosmochim. Acta 1997, 61, 3461–3475. [Google Scholar] [CrossRef]
- Davies, A.J.; John, C.M. The clumped (13C18O) isotope composition of echinoid calcite: Further evidence for “vital effects” in the clumped isotope proxy. Geochim. Cosmochim. Acta 2019, 245, 172–189. [Google Scholar] [CrossRef]
- McLennan, S. Rare earth elements in sedimentary rocks; influence of provenance and sedimentary processes. Rev. Mineral. Geochem. 1989, 21, 277–290. [Google Scholar]
- Halas, S.; Szaran, J. Improved thermal decomposition of sulfates to SO2 and mass spectrometric determination of δ34S of IAEA SO-5, IAEA SO-6 and NBS-127 sulfate standards. Rapid Commun. Mass Spectrom. 2001, 15, 1618–1620. [Google Scholar] [CrossRef]
- Checa, A.G.; Esteban-Delgado, F.J.; Rodríguez-Navarro, A.B. Crystallographic structure of the foliated calcite of bivalves. J. Struct. Biol. 2007, 157, 393–402. [Google Scholar] [CrossRef]
- Orange, D.; Knittle, E.; Farber, D.; Williams, Q. Raman spectroscopy of crude oils and hydrocarbon fluid inclusions: A feasibility study. In Mineral Spectroscopy: A Tribute to Roger G. Burns; Dyar, M.D., McCammon, C., Schaefer, M.W., Eds.; The Geochemical Society; Special Publication: Washington, DC, USA, 1996; Volume 5, pp. 65–81. [Google Scholar]
- Hurai, V.; Huraiová, M.; Slobondník, M.; Thomas, R. (Eds.) Chapter 7-Raman and Infrared Spectroscopic Analysis. In Geofluids: Developments in Microthermometry, Spectroscopy, Thermodynamics, and Stable Isotopes; Elsevier: Amsterdam, The Netherlands, 2015; pp. 231–279. [Google Scholar] [CrossRef]
- Veizer, J.; Ala, D.; Azmy, K.; Bruckschen, P.; Buhl, D.; Bruhn, F.; Carden, G.A.F.; Diener, A.; Ebneth, S.; Godderis, Y.; et al. 87Sr/86Sr, d13C and d18O evolution of Phanerozoic seawater. Chem. Geol. 1999, 161, 59–88. [Google Scholar] [CrossRef]
- Friedman, I.; O’Neil, J.R. Compilation of stable isotope fractionation factors of geochemical interest. In Data of Geochemistry; Fleischer, M., Ed.; U.S. Government Printing Office: Washington, DC, USA, 1977; pp. 1–12. [Google Scholar]
- Tindall, J.; Flecker, R.; Valdes, P.; Schmidt, D.N.; Markwick, P.; Harris, J. Modelling the oxygen isotope distribution of ancient seawater using a coupled ocean–atmosphere GCM: Implications for reconstructing early Eocene climate. Earth Planet. Sci. Lett. 2010, 292, 265–273. [Google Scholar] [CrossRef]
- McArthur, J.M.; Howarth, R.J.; Bailey, T.R. Strontium Isotope Stratigraphy: LOWESS Version 3: Best Fit to the Marine Sr-Isotope Curve for 0–509 Ma and Accompanying Look-up Table for Deriving Numerical Age. J. Geol. 2001, 109, 155–170. [Google Scholar] [CrossRef]
- McIntire, W.L. Trace element partition coefficients, a review of theory and applications to geology. Geochim. Cosmochim. Acta 1963, 27, 1209–1264. [Google Scholar] [CrossRef]
- Tucker, M.E.; Wright, P.V. Carbonate Sedimentology; Blackwell: Oxford, UK, 1990; p. 482. [Google Scholar]
- Howson, M.R.; Pethybridge, A.D.; House, W.A. Synthesis and distribution coefficients of low-magnesium calcites. Chem. Geol. 1987, 64, 79–87. [Google Scholar] [CrossRef]
- Kolker, A.; Chou, C.L. Cleat-Filling Calcite in Illinois Basin Coals: Trace-Element Evidence for Meteoric Fluid Migration in a Coal Basin. J. Geol. 1994, 102, 111–116. [Google Scholar] [CrossRef]
- Steuber, T.; Rauch, M. Evolution of the Mg/Ca ratio of Cretaceous seawater: Implications from the composition of biological low-Mg calcite. Mar. Geol. 2005, 217, 199–213. [Google Scholar] [CrossRef]
- Ligi, M.; Bonatti, E.; Cuffaro, M.; Brunelli, D. Post-Mesozoic Rapid Increase of Seawater Mg/Ca due to Enhanced Mantle-Seawater Interaction. Sci. Rep. 2013, 3, 2752. [Google Scholar] [CrossRef] [PubMed]
- Katz, A. The interaction of magnesium with calcite during crystal growth at 25–90 °C and one atmosphere. Geochim. Cosmochim. Acta 1973, 39, 486–508. [Google Scholar] [CrossRef]
- Lorens, R. Sr, Cd, Mn and Co distribution coefficients in calcite as a function of calcite precipitation rate. Geochim. Cosmochim. Acta 1981, 45, 553–561. [Google Scholar] [CrossRef]
- Kinsman, D.J.J. Interpretation of Sr2+ concentrations in carbonate minerals and rocks. J. Sediment. Petrol. 1969, 39, 486–508. [Google Scholar]
- Bau, M.; Dulski, P. Distribution of yttrium and rare-earth elements in the Penge and Kuruman iron-formations, Transvaal Supergroup, South Africa. Precambr. Res. 1996, 79, 37–55. [Google Scholar] [CrossRef]
- Webb, G.E.; Kamber, B.S. Rare earth elements in Holocene reefal microbialites: A new shallow seawater proxy. Geochim. Cosmochim. Acta 2000, 64, 1557–1565. [Google Scholar] [CrossRef]
- Bau, M.; Dulski, P. Evolution of the yttrium-holmium systematics of seawater through time. Mineral. Mag. 1994, 58, 61–62. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Z.Q.; Algeo, T.J.; Chen, J.; Chen, Y.; Tong, J.; Gao, S.; Zhou, L.; Hu, Z.; Liu, Y. Rare-earth element patterns in conodont albid crowns: Evidence for massive inputs of volcanic ash during the latest Permian biocrisis? Glob. Planet. Chang. 2013, 105, 135–151. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Creig, A.; Collerson, K.D.; Kamber, B.S. Rare earth element and yttrium variability in south east Queensland Waterways. Aquat. Geochem. 2006, 12, 39–72. [Google Scholar] [CrossRef]
- Carrillo, E. The Evaporites of the Southeastern Pyrenean Basin (Late Cuisian–Lutetian): Sedimentology and Structure. Ph.D. Thesis, University of Barcelona, Barcelona, Spain, 2012. [Google Scholar]
- Meyer, E.E.; Quicksall, A.N.; Landis, J.D.; Link, P.K.; Bostick, B.C. Trace and rare earth elemental investigation of a Sturtian cap carbonate, Pocatello, Idaho: Evidence for ocean redox conditions before and during carbonate deposition. Precambr. Res. 2012, 192–195, 89–106. [Google Scholar] [CrossRef]
- Tostevin, R.; Shields, G.A.; Tarbuck, G.M.; He, T.; Clarckson, M.O.; Wood, R.A. Effective use of cerium anomalies as a redox proxy in carbonate-dominated marine settings. Chem. Geol. 2016, 438, 146–162. [Google Scholar] [CrossRef]
- Clark, I.D.; Fritz, P. Environmental Isotopes in Hydrogeology; CRC Press/Lewis Publishers: Boca Raton, FL, USA, 1997. [Google Scholar]
- Aharon, P.; Fu, B. Sulfur and oxygen isotopes of coeval sulfate–sulfide in pore fluids of cold seep sediments with sharp redox gradients. Chem. Geol. 2003, 195, 201–218. [Google Scholar] [CrossRef]
- Giménez-Montsant, J.; Calvet, F.; Tucker, M.E. Silica diagenesis in Eocene shallow-water platform carbonates, southern Pyrenees. Sedimentology 1999, 46, 969–984. [Google Scholar] [CrossRef]
- Esteban, M.; Taberner, C. Secondary porosity development during late burial in carbonate reservoirs as a result of mixing and/or cooling of brines. J. Geochem. Explor. 2003, 78–79, 355–359. [Google Scholar] [CrossRef]
- Taylor, B.E. Stable isotope geochemistry of ore-forming fluids. In Short Course in Stable Isotope Geochemistry of Low Temperature Fluids; Kyser, T.K., Ed.; Mineral Association of Canada: Saskatoon, SK, Canada, 1987; Volume 13, pp. 337–418. [Google Scholar]
- Froelich, P.N.; Klinkhammer, G.P.; Bender, M.L.; Luedtke, N.A.; Heath, G.R.; Cullen, D.; Dauphin, P.; Hammond, D.; Hartman, B.; Maynard, V. Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: Suboxic diagenesis. Geochim. Cosmochim. Acta 1979, 43, 1075–1090. [Google Scholar] [CrossRef]
- Barnaby, R.J.; Rimstidt, J.D. Redox conditions of calcite cementation interpreted from Mn and Fe contents of authigenic calcites. Geol. Soc. Am. Bull. 1989, 101, 795–804. [Google Scholar] [CrossRef]
- Travé, A.; Roca, E.; Playà, E.; Parcerisa, D.; Gómez-Gras, D.; Martín-Martín, J.D. Migration of Mn-rich fluids through normal faults and fine-grained terrigenous sediments during early development of the Neogene Vallès-Penedès half-graben (NE Spain). Geofluids 2009, 9, 303–320. [Google Scholar] [CrossRef]
- Lécuyer, C.; Reynard, B.; Grandjean, P. Rare earth element evolution of Phanerozoic seawater recorded in biogenic apatites. Chem. Geol. 2004, 204, 63–102. [Google Scholar] [CrossRef]
- Camprubí, A.; Albinson, T. Epithermal deposits in mexico update of current knowledge, and an empirical reclassification. Geol. Soc. Am. Spec. Pap. 2007, 422, 377–415. [Google Scholar]
- Bons, P.D.; Elburg, M.A.; Gómez-Rivas, E. A review of the formation of tectonic veins and their microstructures. J. Struct. Geol. 2012, 43, 33–62. [Google Scholar] [CrossRef]
- Beaudoin, N.; Bellahsen, N.; Lacombe, O.; Emmanuel, L.; Pironon, J. Crustal-scale fluid flow during the tectonic evolution of the Bighorn Basin (Wyoming, USA). Basin Res. 2014, 26, 403–435. [Google Scholar] [CrossRef]
- Kerkhof, A.M.V.D.; Hein, U.F. Fluid inclusion petrography. Lithos 2001, 55, 27–47. [Google Scholar] [CrossRef]
- Ke, C.W.; Xu, Y.H.; Chang, X.C.; Lin, W.B. Composition and distribution of NSO compounds in two different shales at the early maturity stage characterized by negative ion electrospray ionization coupled with Fourier transform ion cyclotron resonance mass spectrometry. Pet. Sci. 2018, 15, 289–296. [Google Scholar] [CrossRef]
- Beamud, E.; Muñoz, J.A.; Fitzgerald, P.G.; Baldwin, S.L.; Garcés, M.; Cabrera, L.; Metcalf, J.R. Magnetostratigraphy and detrital apatite fission track thermochronology in syntectonic conglomerates: Constraints on the exhumation of the South-Central Pyrenees. Basin Res. 2010, 23, 309–331. [Google Scholar] [CrossRef]
- Rushlow, C.R.; Barnes, J.B.; Ehlers, T.A.; Vergés, J. Exhumation of the southern Pyrenean fold-thrust-belt (Spain from orogenic growth to decay. Tectonics 2013, 32, 843–860. [Google Scholar] [CrossRef]
- Taillefer, A.; Soliva, R.; Guillou-Frottier, L.; Goff, E.L.; Martin, G.; Saranne, M. Fault-Related Controls on Upward Hydrothermal Flow: An Integrated Geological Study of the Têt Fault System, Eastern Pyrénées (France). Geofluids 2017, 8190109. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Observed Peak (cm−1) | Functional Group | Mode Assignment | Compound | Measured Peaks by [78] (cm−1) | Measured Peaks by [77] (cm−1) |
|---|---|---|---|---|---|
| 620 | C–C | Bending | Aromatic hydrocarbons | 620–625 | - |
| 650 | S–C | Stretching | Aromatic hydrocarbons | 620–715 | - |
| 795 | C–C | Ring breathing/stretching | Cycloalkanes | 700–1000 | - |
| 850 | C–C | Stretching | n-Alkanes | 837–905 | - |
| 1001 | Ring | Ring deformation | Benzene and derivates | 990–1060 | 999 |
| 1040 | Ring | Ring deformation | Benzene and derivates | 990–1060 | - |
| 1165 | C–C | Stretching/Bending | Aromatic hydrocarbons | 1020–1200 | - |
| 1200 | C–C | Stretching/Bending | Aromatic hydrocarbons | 1020–1200 | - |
| 1280 | C–C | Stretching | Aromatic hydrocarbons | 1284–1295 | - |
| 1580 | Ring | Ring Scissors | Benzene and derivates | - | 1580 |
| 1600 | C–C | Stretching | Aromatic hydrocarbons | 1600–1605 | - |
| 1720 | C–H | Stretching | Single aromatic compounds | 1700–2000 | - |
| 2945 | C–H | Stretching | Aromatic hydrocarbons | 2927–3085 | - |
| 2990 | C–H | Stretching | Aromatic hydrocarbons | 2927–3085 | - |
| 3065 | C–H | Stretching | Benzene and derivates | 3057–3060 | 3064 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruset, D.; Ibáñez-Insa, J.; Cantarero, I.; John, C.M.; Travé, A. Significance of Fracture-Filling Rose-Like Calcite Crystal Clusters in the SE Pyrenees. Minerals 2020, 10, 522. https://doi.org/10.3390/min10060522
Cruset D, Ibáñez-Insa J, Cantarero I, John CM, Travé A. Significance of Fracture-Filling Rose-Like Calcite Crystal Clusters in the SE Pyrenees. Minerals. 2020; 10(6):522. https://doi.org/10.3390/min10060522
Chicago/Turabian StyleCruset, David, Jordi Ibáñez-Insa, Irene Cantarero, Cédric M. John, and Anna Travé. 2020. "Significance of Fracture-Filling Rose-Like Calcite Crystal Clusters in the SE Pyrenees" Minerals 10, no. 6: 522. https://doi.org/10.3390/min10060522
APA StyleCruset, D., Ibáñez-Insa, J., Cantarero, I., John, C. M., & Travé, A. (2020). Significance of Fracture-Filling Rose-Like Calcite Crystal Clusters in the SE Pyrenees. Minerals, 10(6), 522. https://doi.org/10.3390/min10060522