1. Introduction
Minimizing the influence of toxic components in acid mine drainage (AMD) and acid rock drainage (ARD) has been a widely discussed topic in the scientific literature since the 1990s [
1,
2,
3,
4,
5,
6,
7,
8,
9]. Along with the use of natural materials as sorbents, including zeolites [
10,
11], clay minerals [
12,
13], plant materials [
14], charcoal ash [
15] and iron and aluminum oxides [
16], to extract various elements from solutions are offered. Other various modified and induced sorbents include aluminosilicates, ferrocyanide sorbents based on hydrated titanium dioxide, resins, organosilicon ion-exchange and complex-forming sorbents and cementitious materials [
17,
18,
19,
20]. Different approaches for the precipitation of metals and metalloids from AMD solutions have been applied, such as the use of zero-valent iron [
21], a pulsed limestone bed treatment system minimizing armor formation [
22], and alkaline industrial wastes [
23]. Currently, the focus is on two directions of the tailings problem: (1) prevention methods; and (2) secondary mineral processing of mine waste [
24].
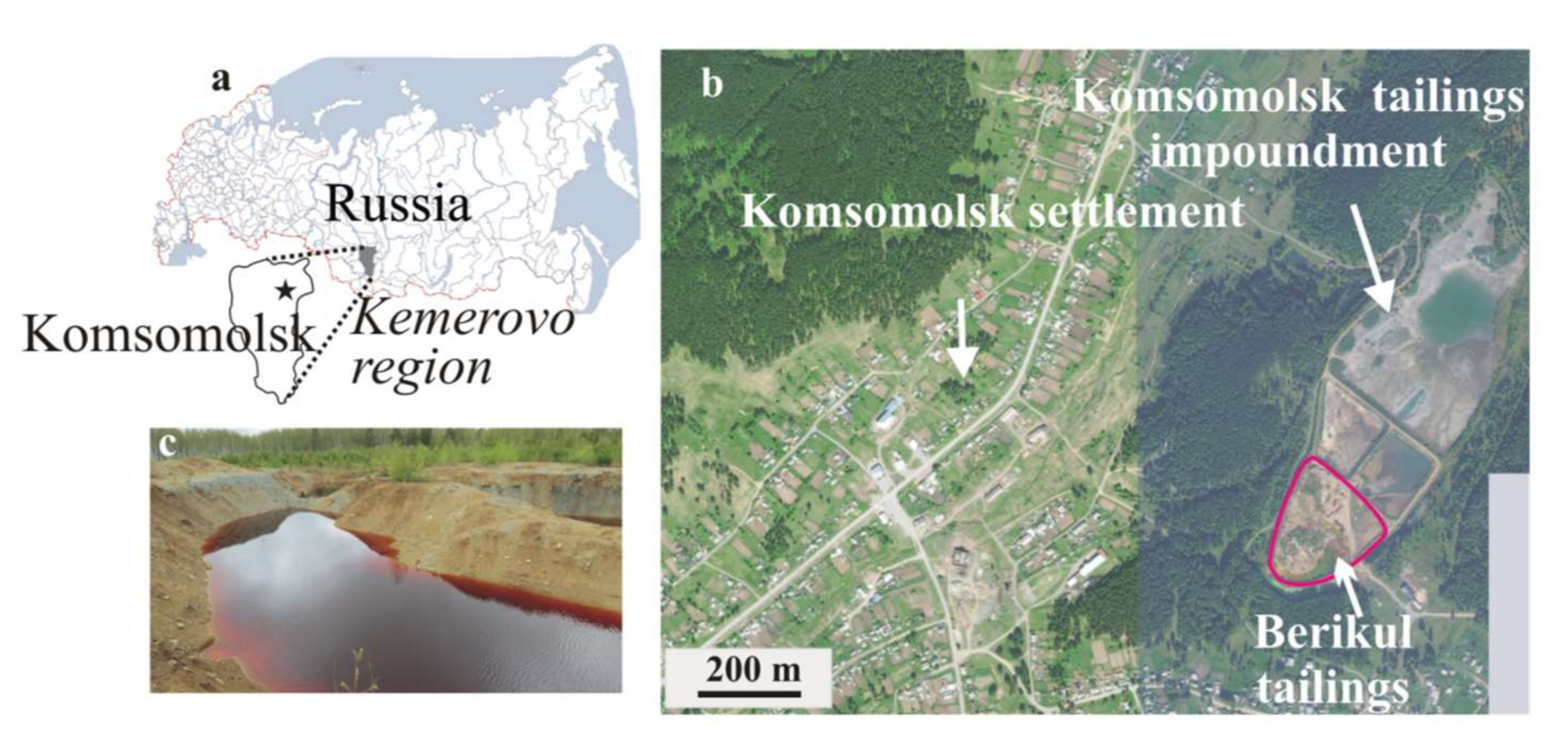
Highly mineralized acid drainage solutions are extremely toxic for the environment; they have to be treated, making a neutral pH and the removal of metal and metalloids necessary to reduce potential hazards [
25]. Nevertheless, the critical environmental situation in the industrial region of southwestern Siberia (Kemerovo region) is the result of the intentional displacement (for reprocessing) and uncontrolled storage for 16 years (due to inexpediency) of the cyanide leaching tailings with high sulfide and arsenic concentrations. Ponds of acidic water with extremely high concentrations of arsenic (up to 4 g/L) and metals are formed on the surface of the solid tailings.
Limestone or portlandite has been used for the neutralization of drainage solutions of different composition and acidity [
25]. These lime neutralizers in case of H
2SO
4 precipitate gypsum (CaSO
4 2H
2O) and passivate the surface. This indicates that alternatives could be needed.
Igarashi et al. [
26] assert that AMD or ARD neutralization is effective but unsustainable in the long term. Estimation of the number of years required for metals in AMD from abandoned tailings dams to decrease below maximum permissible concentrations (MPC) has been done [
27]. The authors of the reactive-transport model have estimated that the formation of AMD and release of Zn will persist for a thousand years. Igarashi et al. [
27] introduced a technique for AMD management, where acid solution with high concentrations of Zn, Cu, and As was treated using a laboratory setup of ferrite flow.
An alternative AMD processing method is the alkaline barium calcium desalination. Both sulfate and metals were reduced below the MPC. The interesting thing about this technology is that low levels of sludge are disposed after useful chemicals are recovered from AMD [
28,
29].
The precipitation of metal sulfides, the solubility and stability of the sulfide ion, and its complexes with metals were reported by Lewis and Hille [
30]. A great attention has been paid to the precipitation of metal sulfides from AMD and saturated leach solutions [
31]. These authors conclude, “notwithstanding and irrespective of the source of sulfide, metallic sulfide precipitation has many challenges. Further research is needed to address these issues”.
A fractional precipitation process was conducted to precipitate metals from the AMD [
32]. With the help of four-step precipitation, AMD was treated to World Health Organization (WHO) requirements. Detoxification of zinc plant leach residues from Kabwe, Zambia has been performed by removing Pb, using a coupled extraction-cementation method in chloride media [
33].
Special attention is devoted to the removal of arsenic from acidic and alkaline drainage, due to its high toxicity and mobility [
34,
35,
36,
37,
38]. The formation of ferric arsenate, or a Fe/Al arsenate phase, as well as strong adsorption of As to Fe-oxyhydroxides/oxides have limited its mobility at low pH conditions [
3]. The initial step in the extraction of As from solution is the oxidation of trivalent arsenic to pentavalent arsenic to remove it in one stable form [
39,
40]. To do this, they use hydrochemical neutralization with lime, resulting in the formation of svabite Ca
5(AsO
4)
3F [
41,
42], sulfide precipitation [
43,
44], coprecipitation with iron and formation of scorodite FeAsO₄·2H
2O [
45,
46], bacterial deposition [
47,
48] and various sorbents [
49,
50,
51]. As(V) in acidic solutions (pH 3–4) can be removed effectively by synthesized schwertmannite [
52], biomineralization [
53] and scorodite precipitation [
54,
55]. Besides, As mobility can be decrease via sorption reactions with carbonates and/or gypsum [
56,
57]. However, the arsenic problem is far from being resolved now.
For example, Yuan et al. [
51] investigated the effect of pH on fast As removal from AMD containing high arsenic. The authors studied Fe/As molar ratio, oxygen flow rate, temperature, initial As concentration and the action of reagents (NaOH vs. Ca(OH)
2). The mechanisms of solid precipitation, including As removal, were deeply discussed. This study provides further evidence on the speciation of As and its distribution in Fe(II/III)-As(III/VI)-S(II/VI) aqueous and solid systems, which affects the fate of arsenic. To date, there is no better summary of arsenic mineralogy than [
58].
In this article, we presented a case study for both acidic solutions neutralization and metal precipitation from a highly mineralized tailings pond water, based on the addition of Ca(OH)2, Na2S, and NaOH in the field. From a geochemical point of view, according to the mechanism, the reagents act as precipitating Ca(OH)2, reducing Na2S, and neutralizing NaOH barriers. Actually, Ca(OH)2 is also a neutralizing reagent, and NaOH may trigger metal precipitation as well; therefore, this difference is conditional for convenience. The above references show the relevance of this case study for a broader international audience. This means that current techniques like chemical neutralization should be improved to facilitate the better and more sustainable management of AMD solutions. In our study, we focus on (1) the optimal S/R ratio for the precipitation of elements during a sequential decrease of its variable; (2) identification of the mineral species of elements formed during neutralization; and (3) description of the chemistry of the water-rock interactions by thermodynamic modeling. Our task was to develop a quick and cheap way to prevent in situ the influence of toxic solutions, within the limits of the population’s habitat.
3. Results
3.1. Precipitation Experiment with Ca(OH)2
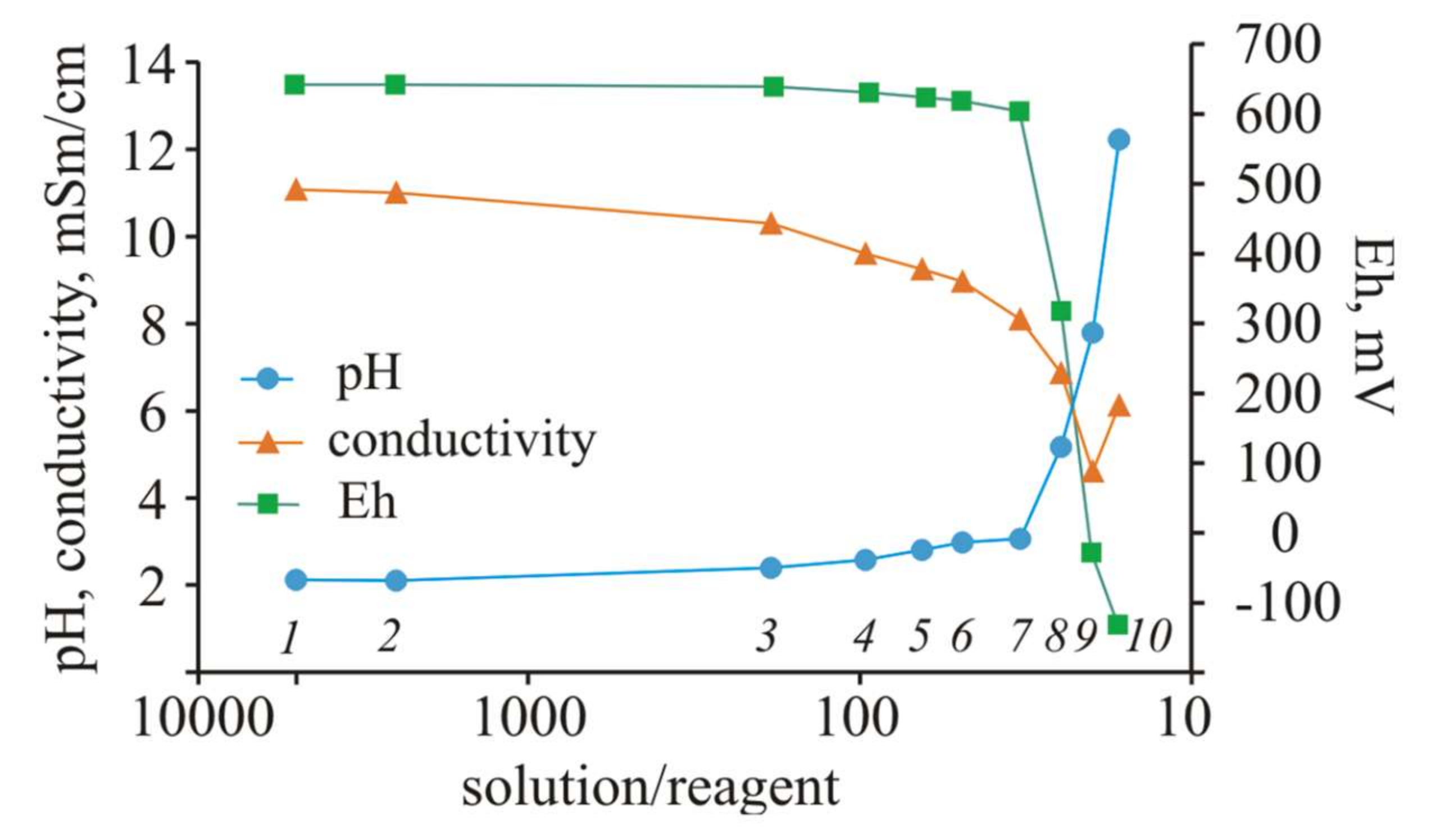
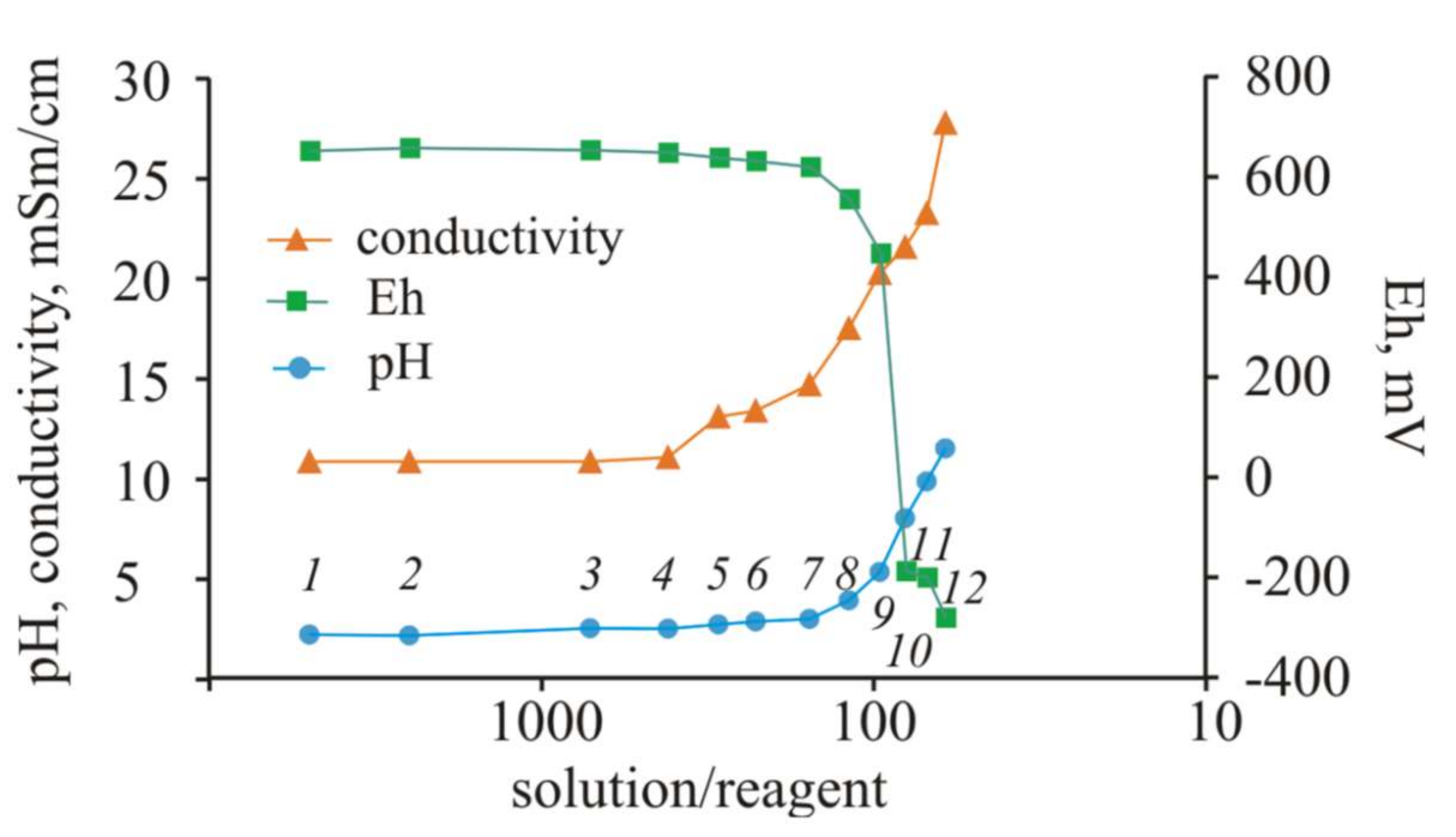
A change in the pH values during the experiment indicates a large buffer capacity of the solution with the initial pH = 2.1. The pH values increased by only 1 unit when S/R = 33 (30 g of Ca(OH)
2 was added,
Figure 2).
A noticeable increase in pH to 5.17 occurred when 40 g of reagent was added to the initial solution with a volume of 1 L (S/R = 25). In the next steps of the experiment, the acidity of the solution began to decrease sharply. At S/R = 20, the pH increased to 7.79, and at the end of the experiment, when 60 g of Ca(OH)2 was added to the solution (S/R = 17), the pH increased sharply to 12.22.
The pH value increased during the experiment, whereas the EC, reflecting the total mineralization of the solution, gradually decreased to the ratio S/R = 20 (pH = 7.79), due to the precipitation of the Ca-containing solid phases. This phenomenon is evidenced by the stable Ca concentrations in the solution throughout the experiment, despite the gradual addition of Ca(OH)2. However, at the last step of the experiment (pH = 12.22), the electrical conductivity sharply increased by 1.5 mSm/cm, which indicates the leaching of elements into the solution. This result is not unexpected, since alkaline solutions are very aggressive. The Eh in the first seven steps of the experiment changed from 657 to 619 mV; in step No. 8, the Eh decreased to 333 mV and became negative in the last two subsequent steps (−13 and −116 mV).
The pH value increased during the experiment, whereas the EC, reflecting the total mineralization of the solution, gradually decreased to the ratio S/R = 20 (Ph = 7.79), due to the precipitation of the Ca-containing solid phases. This phenomenon is evidenced by the stable Ca concentrations in the solution throughout the experiment, despite the gradual addition of Ca(OH)2. However, at the last step of the experiment (pH = 12.22), the electrical conductivity sharply increased by 1.5 mSm/cm, which indicates the leaching of elements into the solution. This result is not unexpected, since alkaline solutions are very aggressive. The Eh in the first seven steps of the experiment changed from 657 to 619 mV; in step No. 8, the Eh decreased to 333 mV and became negative in the last two subsequent steps (−13 and −116 mV).
According to the Nernst equation [
62], the dependence of Eh-pH suggests such a coherent change. However, we note that, in ponds exposed to the atmosphere, the Eh value is only 657 mV, which indicates that the potential-determining system is most likely the pair Fe
2+/Fe(OH)
3(s), since the initial concentration of iron is 8600 mg/L (
Table 1). Thus, the process can be divided into two main stages: S/R = 33 (a slight increase in pH) and S/R = 25-20-17 (a total of 60.4 g of Ca(OH)
2 was added), Equations (1)–(3):
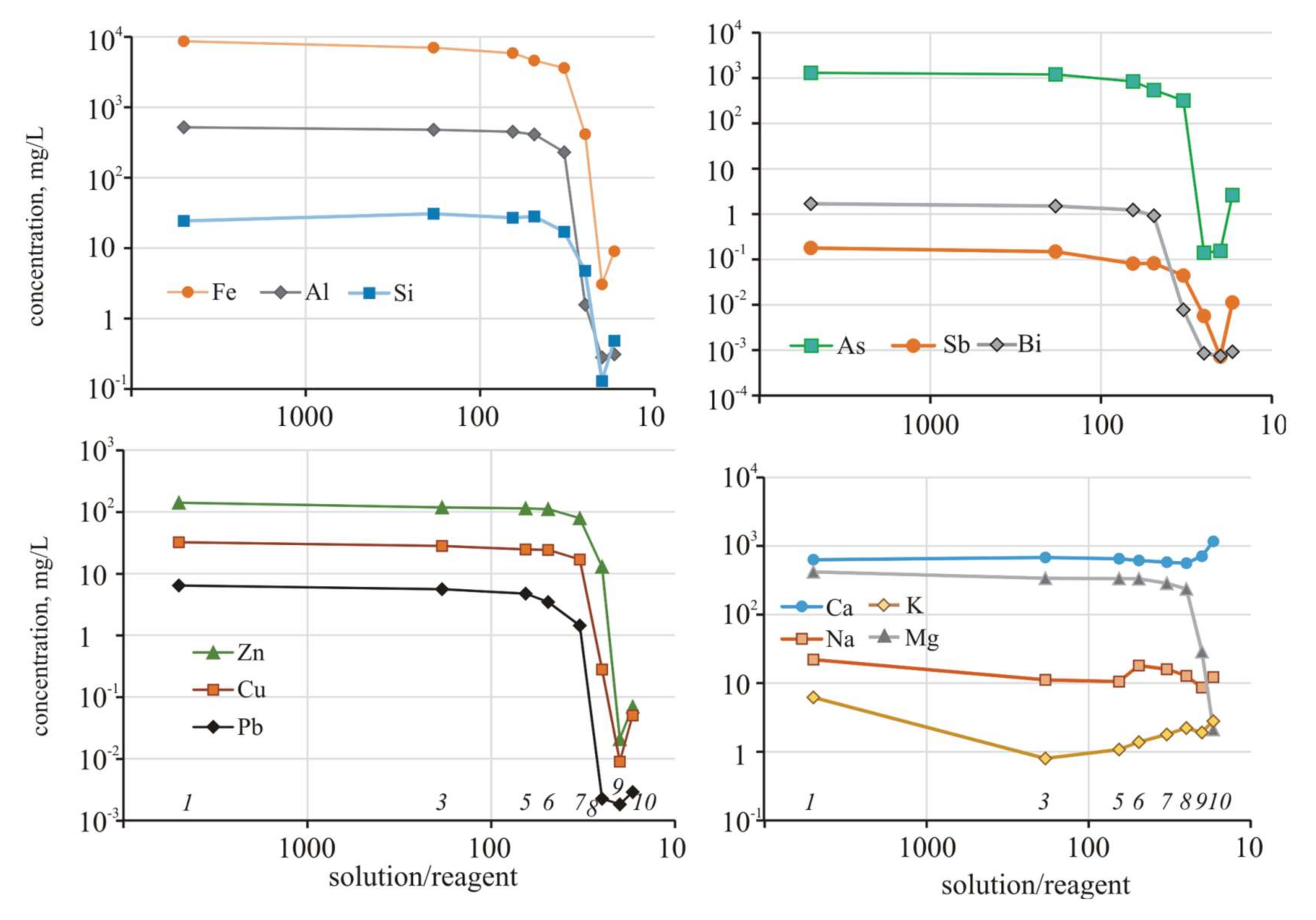
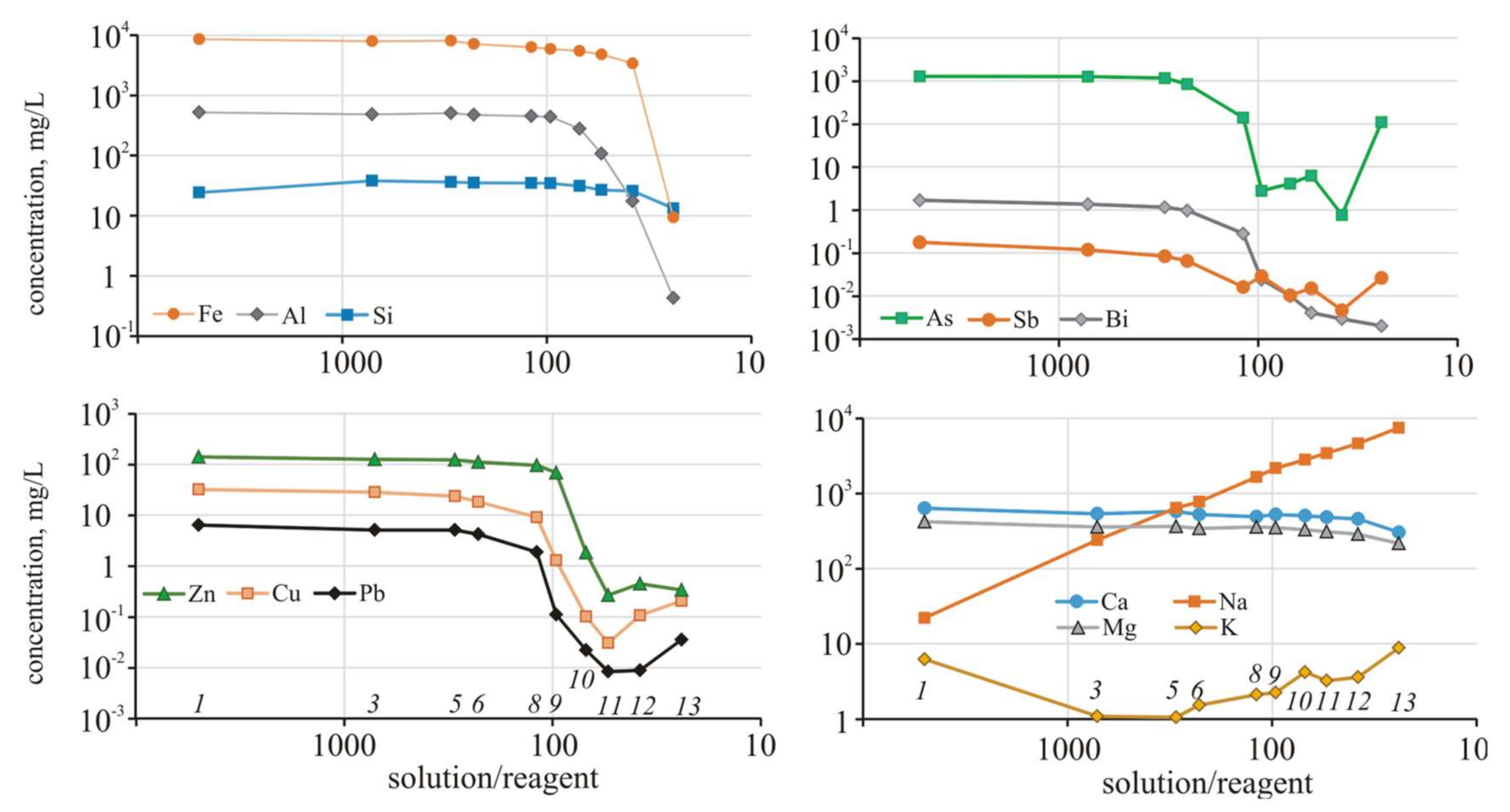
The buffer capacity of the solution was high, and allowed for the stability of pH values in a narrow range of one unit. After reaching S/R = 33, other reactions involving arsenic and metals take place in the system. The content of Fe, Cu, Zn, Pb, and other metals, as well as metalloids (As, Sb, Bi, Sn), slightly decreased while the solution was acidic (to pH < 3.06), which corresponded to S/R = 33 (
Figure 3).
Then, with a noticeable increase in pH to 5.17 at S/R = 25, the concentrations of almost all the elements decreased sharply and continued to decrease in the next step when the pH reached slightly alkaline values (pH = 7.79, S/R = 20). Adsorption-coprecipitation with hydrous ferric oxides and sorption reactions with kaolinite and montmorillonite play a significant role in mobility of Zn and Pb [
63]. However, with a further increase in alkalinity (pH = 12.2, S/R = 17), the concentrations of the elements began to increase. The contents of the alkali metals (Na, K) fluctuated insignificantly throughout the experiment.
The precipitate formed as a result of the interaction of Ca(OH)
2 with the solution was a semiamorphous dark gray substance; after drying, it changed to a fine-grained, earthy mass. The XRD analysis data showed the presence of mainly gypsum and the amorphous phase of iron and aluminum hydroxides (plohmite) in the products from the final phase after the experiment. Ettringite, bassanite, alunite, and carbonates of Ca and Fe were formed in smaller amounts. In addition, traces of pickeringite (precipitated Mg) and arsenosiderite Ca
3Fe
4(AsO
4)(OH)
6·3H
2O were found in the sample (
Table 3). Thus, we have evidence of the formation of aqueous sulfates and complex compositions, hydroxides, and carbonates. The mineral phases of Cu and Zn were not identified.
However, as a result of the electron microscopy studies of the precipitate, Cu and Zn impurities were detected in the Fe-Ca minerals. In addition, ultrafine inclusions of the As-containing phases, presumably arsenosiderite, which were identified by XRD analysis (
Figure 4), were found in some grains of gypsum and Fe-Ca minerals.
Numerical Simulation of the Ca(OH)2 Experiment
A theoretical model with one liter of the Fe-As-sulfate solution (
Table 1) by milk of lime has shown that, when 2.1 g of Ca(OH)
2 is added, the solution becomes supersaturated with, Fe(OH)
3(am), gypsum, and anglesite (
Table 4).
Due to the formation of suspensions, 2800 mg of iron and 4.6 mg of lead can precipitate from the solution. The Ca content in the solution remains stable because it is controlled by the solubility of gypsum. The amounts of the formed suspension obtained in the numerical simulation are slightly higher than the decrease in the concentration of elements in the experimental solutions at the corresponding stages; however, taking the analysis error (10%) into account at such high concentrations of elements in the solution, the agreement is satisfactory. At pH ~3 (S/R = 49 and 33), the situation changes, such that gibbsite, alunite (aluminum minerals), apatite, and traces of shultenite and conichalcite (arsenates containing heavy metals) appear. Even though only 40 mg of arsenic precipitates from the solution, the precipitation process begins. With optimal S/R ratios of 25 and 20, the precipitation of arsenic occurs in the form of aqueous calcium arsenate, and carbonates may be present in the solid phase (
Table 4). The formation of Ca
3(AsO
4)
2∙4H
2O; occurs by many technological schemes in mining plants, although it is a toxic compound for humans [
64,
65].
However, due to the precipitation of Ca
3(AsO
4)
2∙4H
2O, the arsenic concentration equals 0.14 g/L. At the last step of the experiment (pH 12.2, Eh −116 mV), ettringite (an indicator of alkaline solutions and a product of concrete corrosion) and (hydr)oxides (tenorite CuO, hausmannite Mn
2+Mn
3+2O
4, zincite ZnO) appear. Indeed, the model solution becomes richer in Fe, Cu, and Ca, but does not become richer in arsenic. The increase in the arsenic concentrations at the end of the experiment is because the stability of iron hydroxocomplexes increases at pH 12.2; here, its solid phases dissolve, which releases trace elements into the solution. In the air, calcium arsenate is converted to carbonate, with the release of arsenic into the solution, according to the following reaction (Equation (4)):
Portlandite and Brusite were not detected by the microscopy. Not all phases can be taken into account in the model, due to the lack of thermodynamic data. That is why, instead of complex sulfate-arsenate phases, simple hydroxides such as Ca(OH)2 could appear under alkaline conditions. Some supersaturation of the solution in respect to portlandite cannot be ruled out upon its final addition.
Arsenic species in solutions change with increasing pH and mineralization (EC). In the first stages of the experiment, the majority of arsenic in the solution was in the form of positively charged complexes with iron FeH
2AsO
4+, FeHAsO
4+ and FeH
2AsO
42+ and arsenic acid H
3AsO
4 (
Figure S1). With a decrease in the S/R ratio to 49, due to the binding of iron in Fe(OH)
3(am), H
2AsO
4− becomes a major species, with a meaningful proportion of arsenate complexes with the metals MgH
2AsO
4+, CaH
2AsO
4+ and AlHAsO
4+. At the next steps of the numerical simulation, at subalkaline and alkaline pH values, arsenic is in the form of a neutral complex of CaHAsO
4 and anionic CaAsO
4−, which remains the only compound of arsenic in solution, at a concentration ~0.1 mg/L. Iron also has a variety of species. In acidic solutions, Fe
2+ (0.01 mole/L), sulfate and arsenite complexes (FeH
2AsO
4+) are the predominant species, with a meaningful proportion of arsenate complexes. At pH 7.79 and 12.22, hydroxocomplexes (Fe(OH)
4−) are the predominant species. The study of the elemental species is important from a scientific (cycle of elements in the biosphere) and practical (technology) perspective. This phenomenon can be traced back to the example of As in the S/R ratio, changing from 25 through 20 to 17. In the first case (S/R = 25), the proportion of the As complexes is low, solutions are supersaturated with respect to minerals and 3310 mg of precipitate are formed (supersaturation is calculated through the product of the uncomplexed ions activity). In the second case (S/R = 20), the proportion of As complexes is almost 50%, and only 150 mg of precipitate are formed; in addition, at S/R = 17, despite the fact that AsO
43− < 1%, 620 mg of Ca
3(AsO
4)
2∙4H
2O is precipitated because it is no longer gypsum, and ettringite controls a higher concentration of Ca in the solution (
Figure 3).
3.2. Redox Experiment with Na2S
It is known that sulfur, in the form of sulfide, forms low soluble compounds with heavy metals under reducing conditions [
66]. Therefore, in nature, there are massive deposits of hypogenic sulfides and sulfides of the secondary enrichment zone. The highest activity, due to the significant content of sulfur in the form of sulfide, has sodium sulfide (Na
2S), which yields strongly alkaline solutions. In the experiment with Na
2S, the acidity of the solutions decreased, due to a slightly different scheme (
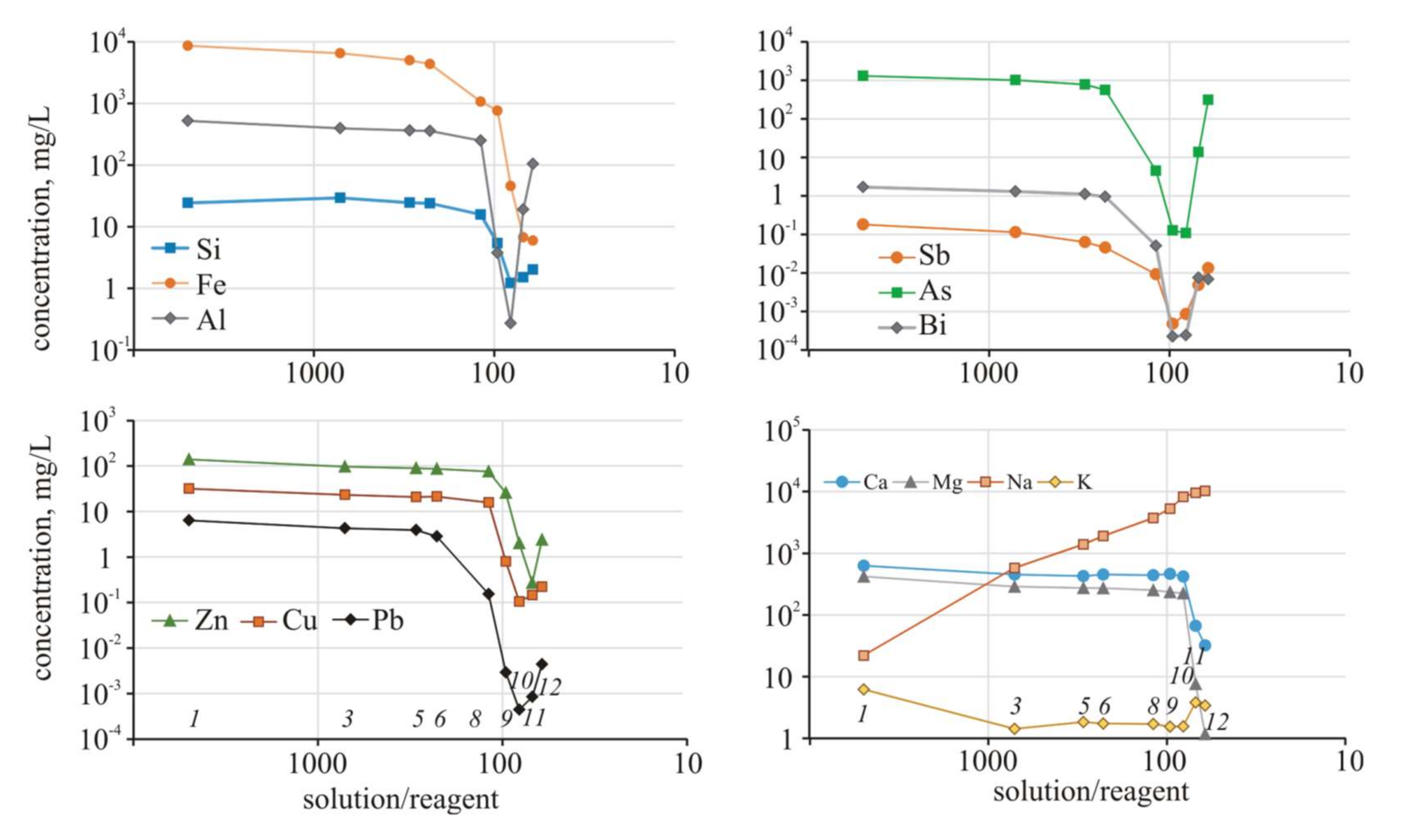
Figure 5).
Initially, at high S/R ratios, the increase in pH was insignificant (0.5 units), which was similar in the experiment with Ca(OH)2. Moreover, for an identical decrease in acidity, three times less reagent was required than in the experiment with Ca(OH)2 (pH 2.9 requires 6.4 g of Na2S and 20.4 g of Ca(OH)2). With S/R = 24–25, the pH value in the experiment with Na2S was 7.29, and in the experiment with Ca(OH)2, it was only 5.17.
It is reasonable that the redox conditions of the solutions changed more significantly, and at the end of the experiment (S/R = 24), Eh was −244 mV, which corresponds to the reducing environment at pH 7.29. The fundamental difference from the experiment with Ca(OH)
2 is a change in EC. Due to an increase in the Na concentrations (up to 7.4 g/L), the EC increased. Unlike calcium, Na was deposited in minimal amounts at the last steps of the experiment. The Na concentration, starting from S/R = 700, exceeds the MPC; additionally, at the end of the experiment, the excess concentration was 60 times higher, which is certainly an unfavorable result. The iron concentration during the experiment began to decrease under acidic conditions (S/R = 227, pH 2.69) and then gradually decreased as the conditions changed to S/R = 38 and pH 5.32 (
Figure 5).
In the next step of the experiment (S/R = 24, pH 7.29), the Fe content decreased sharply (from 3410 to 9 mg/L), accompanied by a decrease in Eh to 244 mV. The behavior of Al
(III), affected only by pH, was similar. A significant decrease in the Al concentrations (to 278 mg/L, almost two-fold) began with the ratio S/R = 69 and pH 4.41. It is noteworthy that arsenic precipitates from the solution, even at higher ratios (S/R = 119, pH 3.25) and under oxidizing conditions (Eh = 587 mV). Thus, the behavior of As is affected by the Eh value, pH value and sulfide sulfur concentration. In addition, at the subsequent steps of the experiment, As concentrations fluctuated and reached a minimum (0.78 mg/L) at S/R = 38 and pH 5.32. The experiment was terminated because it had already reached pH 7.22, and arsenic was released back to the solution at concentrations up to 113 mg/L. We assume that, due to reducing conditions, bound As
(V) began to release into the solution in the form of HAsO
32−, which is the highly toxic acid of As
(III). Antimonous acid is its analog (green and red lines,
Figure 6).
A decrease in the concentrations of Pb and Cu also began under acidic conditions S/R = 119 and pH 3.25, but the Zn concentration decreased in the next step of the experiment at pH 3.61. The minimum metal concentrations were achieved under slightly acidic conditions (pH 4.87), and then the concentrations began to increase.
The precipitate formed in the experiment was a dark gray, almost black substance; after drying, it was a fine-grained mass. According to XRD analysis, sulfides, sulfates and arsenates formed as a result of the solution reduction (
Table 5).
Pyrite, gypsum, native sulfur, and amorphous Fe-phases are the main phases. The following phases were determined in minor amounts: thenardite, scorodite, tennantite, chalcophyllite; in addition, the following phases were determined in trace amounts: uzonite, basaluminite, mirabilite, As-phases, and Fe, Cu and Zn sulfate (solid solution). We do not exclude the possibility of the formation of soluble Na salts thenardite and mirabilite from residual solutions during the drying of the precipitates.
Using electron microscopy, crystals of native sulfur, gypsum (
Figure 7a), Fe and Na sulfates with impurities of As and Zn (
Figure 7a–c), thenardite (
Figure 7b), pyrite veins and borders with an admixture of As were found.
Numerical Simulation for the Na2S Experiment
Thermodynamic modeling was carried out according to the same scheme as for the Ca(OH)
2 experiment: titration of the sulfate Fe-As solution with Na
2S. After adding 0.2 g of Na
2S, the solution was supersaturated with respect to silica, Fe(OH)
3(am), gypsum, and anglesite, and this condition determined the beginning of a decrease in the concentrations of elements, such as Si, Fe, Ca, and Pb. Here, at pH 2.3 (10.4 g Na
2S, S/R = 96), symplesite Fe
(II)3(AsO
4)
2·8H
2O begins to precipitate (
Table 6).
The binding of arsenic to symplesite explains the sharp decrease in the arsenic concentrations in the numerical simulation from 1300 to 35.8 mg/L, and the decrease in iron concentrations to 668 mg/L. The process can be described by the reaction as follows (Equation (5)):
We did not see any symplesite Fe
2+3(AsO
4)
2·8H2O in our experimental solids. Instead, a mixture of Fe(OH)
3 + Scorodite FeAsO4∙2H2O + Chalcophyllite Cu
18Al
2(AsO
4)
4(SO
4)
3(OH)
24∙36H
2O was discovered (
Table 5). Nevertheless, the precipitation of symplesite at S/R ~100, and then its gradual dissolution, correctly described the variation of As and Fe in according experimental solutions.
Because of the active binding and precipitation of a large amount of As, in the next steps of the modeling, the amount of formed simplesite sharply decreases due to the formation of pyrite, which can be seen in
Table 6. The negative result is high sulfate ion concentrations in solution, which is clearly indicated by the calculations. To precipitate the sulfate ion at this stage, Ca(OH)
2 should probably be added (using a combined scheme), which would lead to the formation of gypsum. Of the metal sulfides, chalcocite (Cu
2S) first appeared, which in nature marks the zone of secondary sulfide enrichment, and this explains the decrease in the concentration of Cu from 32 to 1.3 mg/L (at 25 times). A subsequent decrease in the Cu concentrations is also determined by the formation of chalcocite to pH ~5.0. In parallel with Cu sulfide, sulfates, such as gypsum, alunite, gibbsite, and kaolinite, are formed. Then, during the transition from weakly oxidizing to reducing conditions, sulfides, such as pyrite, chalcopyrite, sphalerite, galena, and argentite, begin to form in neutral and subalkaline conditions. Arsenopyrite was not recorded in the calculations.
The change in the As species in an acidic solution is similar to the experiment with Ca(OH)
2. At high S/R ratios, iron arsenate cationic complexes, FeH
2AsO
4+, FeHAsO
4+, FeH
2AsO
42+, and arsenic acid H
3AsO
4, are the predominant species of As (
Figure S2). Arsenate complexes with Al (AlHAsO
4+) and Mg (MgH
2AsO
4+) were formed in a significantly smaller amount. Starting with the S/R = 69, the differences in the experiment with Ca(OH)
2 are dramatic, since there is no Ca in the solution; i.e., Ca completely precipitated (gypsum and then apatite). Arsenic in solution is present in the form of highly toxic arsenic acid H
3As
(III)O
3. In this case, in the model solutions, a clear increase in the As content is observed, which agrees with the experimental results.
3.3. Neutralization Experiment with NaOH
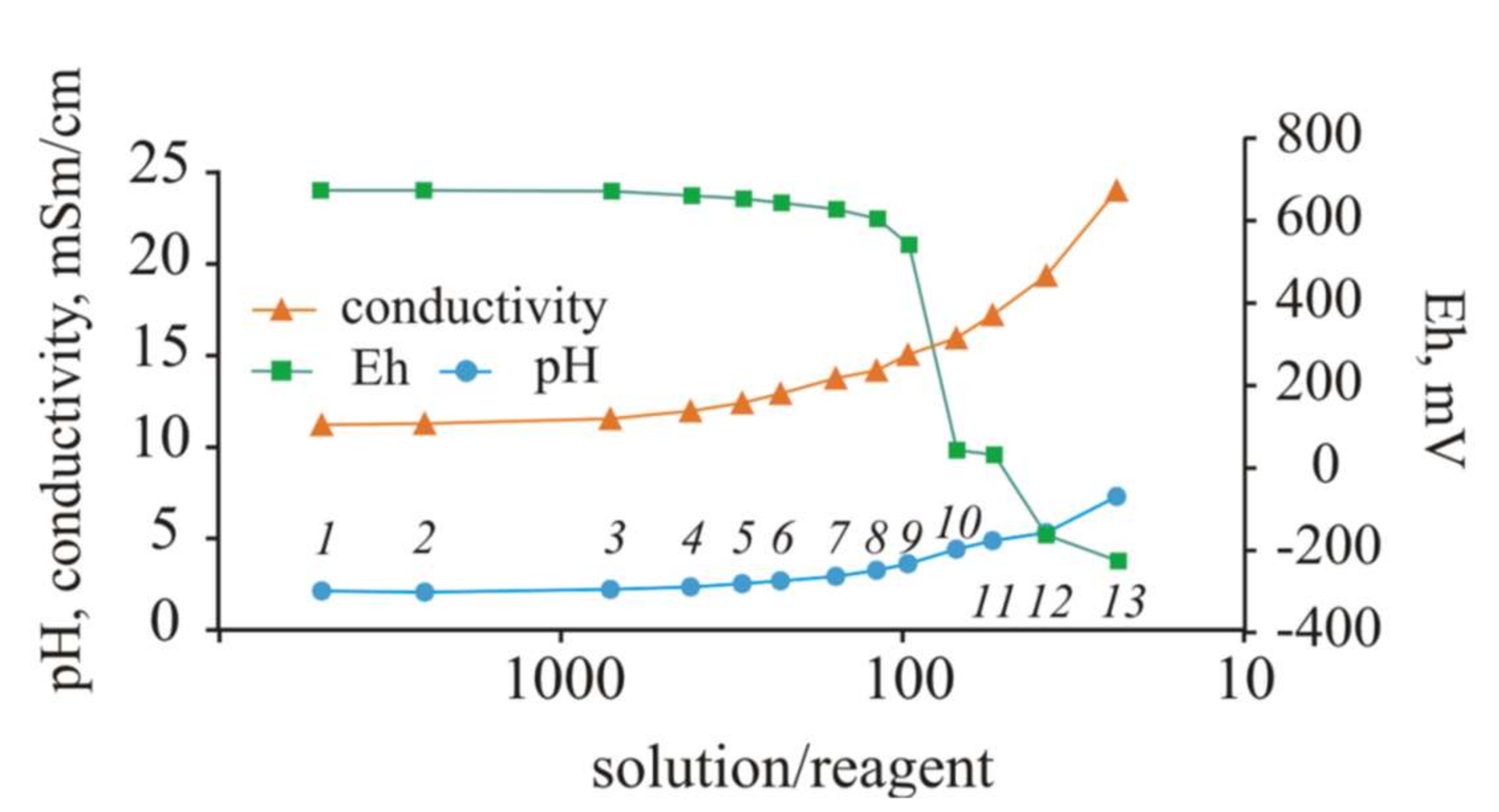
During the experiment with NaOH, the pH values gradually increased to pH = 2.94 (S/R = 156). Subsequently, at S/R = 119, the pH value increased by almost 1 unit (pH 3.86), and then the pH value further increased at each step by 1–1.5 units (
Figure 8).
The EC due to the release of sodium into the solution remained stable only in the first four steps. When the S/R ratio was decreased, the EC sharply increased. The redox potential began to change markedly when the medium became near-neutral, and then, in the alkaline region, the conditions changed to reducing conditions.
The concentration of Fe decreased by four times with the increase in pH from 2.94 to 3.86, and then continued to decrease until alkaline conditions were established (
Figure 9).
The Al concentration began to decrease one step later, with a change in pH from 3.86 to 5.29; however, the decrease was sharper than for the iron. In addition, the pH range of its minimum values was even narrower: in an alkaline environment, aluminum actively began releasing into the solution. The behavior of As, Sb and Bi was similar. The minimum As concentration (0.13–0.11 mg/L) was reached in the neutral-slightly alkaline conditions, and As increased sharply (up to 310 mg/L) at pH = 11.45.
Metals (Zn, Cu, and Pb) began to precipitate in neutral conditions; however, as expected under alkaline conditions, they were released into solution. Similar to the experiment with Na
2S, the Na concentration increased linearly during the experiment, and at S/R = 700, the Na concentration was 4.5 times higher than that of the MPC. By the end of the experiment, Na was almost 2 orders of magnitude higher than that of the MPC. The precipitate mainly consists of thenardite, gypsum, and iron hydroxides (
Table 7,
Figure 10a–c). Calcite, chalcophyllite, scorodite, kaatialaite, and untitled phases (Na and Al sulfate and Mg hydroxo arsenate) were identified as impure minerals.
In the electron microscopy study, As was revealed in the amorphous Fe-phase together with an admixture of Cu (
Figure 10a). Impurities of Cu, Zn, Al, and As were revealed in small inclusions of Fe-sulfate in thenardite (
Figure 10b). Perhaps these inclusions are the smallest grains of As minerals identified by XRD, and the impurities in them are due to matrix capture. The grains of calcite are surrounded by the druse of tenardite (
Figure 10c).
Numerical Simulation for the NaOH Experiment
The acidic solutions are supersaturated with respect to silica, Fe(OH)
3(am), gypsum, anglesite and simplesite Fe
3(AsO
4)
2·8H
2O (
Table 8). The neutralization processes are faster than in the experiment with Ca(OH)
2. It was possible to achieve a pH = 5.29 with the addition of only 10.4 g of NaOH. At this moment (Eh 0.44), gibbsite, alunite, and apatite were precipitated, and conichalcite CaCuAsO
4(OH) (
Table 8) were formed instead of symplesite and shultenite. At pH 9.8 and 11.6, the removal of As was associated with the formation of calcium arsenate, which was mixed with illite, apatite, and goethite. Therefore, the formation of native copper and austenite CaZnAsO
4(OH) is possible.
At the beginning of the experiment, the As species in solution are the same, namely, iron arsenate complexes with a small proportion of magnesium and aluminum complexes, but arsenic acid predominates (H
3AsO
4 and H
2AsO
4−,
Figure S3). When the S/R = 96, the As concentration sharply decreases, and the proportion of arsenic complexes with metals also decreased. In general, for the experiment with NaOH, the As complexes with metals are not characteristic in the solution, due to the sharp decrease in the iron concentration in the solution at high S/R ratio, which will be discussed subsequently. HAsO
42− and AsO
43− are the predominant species, with a small proportion of calcium arsenate.
So, in experimental settings, abundant Cu, Al, Fe, Mg arsenates precipitation can occur compared to model mineral phases via pH increase Fe arsenate symplesite → Ca, Cu, Zn, Pb arsenates (Conichalcite, Shultenite Austinite) → Ca arsenate Ca3(AsO4)2∙4H2O. HCh modeling, together with experimental results, indicate that Ca in these solutions is very low due to the low solubility of its arsenates. Most likely, that native copper, quartz, as well as illite require higher reaction time in order to precipitate in the experiment.
4. Discussion
For a comparative assessment of the effectiveness of the reagents used, the changes in the different parameters are summarized in the corresponding diagrams (
Figures S4 and S5). A decrease in the acidity of the solution (increasing pH values) most rapidly occurred in the experiment with NaOH, and the slightly acidic conditions (pH 5.29) were recorded at S/R = 96 (10.4 g of reagent was added). The same pH value (5.32) in the experiment with Na
2S was reached at S/R = 38 (26.4 g was added); in addition, in the experiment with Ca(OH)
2, a pH of 5.17 was reached only after adding 40.4 g (S/R = 25).
In experiments with NaOH and Na2S, the mineralization of the solution increased with the decrease in the S/R ratio, due to an increase in the Na concentration. Sodium precipitation in the form of thenardite and mirabilite did not have a significant effect on its concentration in the solution, due to the high solubility of thenardite and mirabilite. However, in the experiment with Ca(OH)2, the mineralization decreased with increasing pH, due to the formation of a large amount of gypsum in the first stages, and then due to the formation of calcite. The element deposition also occurred differently. The most effective deposition of Fe, As, and Zn occurred in the experiment with NaOH and began at earlier stages. However, in the experiment with NaOH, the release of arsenic to 310 mg/L into the solution was observed at earlier stages compared with the other experiments, and this release negated the effectiveness of this reagent. An increase in the As concentrations occurred in the other two experiments, up to 2.6 mg/L in the experiments with Ca(OH)2, and up to 113 mg/L in experiments with Na2S. Note that Cu began to precipitate earlier in the experiment with Na2S, which was facilitated by the formation of a chalcocite suspension.
These processes can be explained by the thermodynamic calculation of the amounts and mineral compositions of the suspension formed during the experiments at different S/R ratios. As a result of the interaction of the solution with Ca(OH)
2, gypsum and Fe(OH)
3(am) are already formed, even at high S/R ratios. Only at S/R = 49 do small amounts of schultenite and conichalcite begin to form, which leads to the removal of 40 mg of arsenic from the solution in the form of As phases. At the same stage (S/R = 49), gibbsite and alunite are formed. In a field experiment with S/R = 49, 760 mg of arsenic was removed from the solution, which we associated with sorption on the surface of amorphous Fe-hydroxides (
Figure S6). The effective purification of As-containing solutions using iron sulfate (the formation of amorphous iron hydroxides) has been known for a long time and is widely used [
67].
The most significant binding and As removal occurred when the solution was at saturation with respect to the Ca3(AsO4)2·4H2O. When the S/R ratio was 25, 990 mg of As precipitated according to the numerical simulation results. In the field experiment, it was at this stage (S/R = 25) that a sharp decrease in As concentrations from 320 to 0.14 mg/L was also recorded.
Unlike Ca(OH)
2, the Na
2S at much earlier stages caused the binding of arsenic in the form of symplesite, which began to form under acidic conditions at S/R = 96 (
Figure S7). At this stage, the formation of its highest amount took place; in the subsequent steps of the experiment, symplesite remained the only As mineral. However, if the As-containing phases were formed up to the very end in the experiment with Ca(OH)
2, then the formation of symplesite in the experiment with Na
2S was reduced up to the end of the experiment. Perhaps this formation of As-containing phases, in addition to desorption from iron hydroxides, explains the sharp increase in the As concentrations at the end of the experiment with Na
2S.
The formation of As-containing pyrite in the last stages of the experiment with Na
2S under alkaline conditions led to a decrease in the Fe concentration (
Figure 6).
From the first steps of the experiment with NaOH, due to the neutralization process, a large suspension quantity was formed, mainly Fe(OH)
3(am) and symplesite, which immediately removed 240 mg of arsenic (
Figure S8). In the subsequent stages, As minerals were formed in greater or lesser amounts. Moreover, if there was only symplesite at the first two steps of the experiments with NaOH, then in the future steps, conichalcite and schultenite were formed, and then again, the symplesite was in a mixture with austenite; at the final stages, the Ca
3(AsO
4)
2·4H
2O phase appeared, which led to a sharp decrease in the As concentration. However, although the As-containing minerals in this experiment could be formed until the end of the process, As leaching in the alkaline solutions was also observed, due to desorption and dissolution.
NaOH is the most effective compound for the deposition of As and Fe: the pH value increased to 11.45 at S/R = 60. To achieve the neutral pH value of the solution, it was necessary to add approximately 10 g of NaOH, 30 g of Na2S, and 50 g of Ca(OH)2. It is advisable to use NaOH in the first stage of the technological treatment. The removal of 99.9% arsenic and other elements from the solution was achieved in a narrow pH range for experiments with all three reagents. When using Ca(OH)2, the best result is obtained at S/R ~20; in the experiment with Na2S, this S/R ratio can be ~55–50; and with NaOH, the best result is obtained at S/R ~100–80. In the experiments with Ca(OH)2 and NaOH, the minimum As concentrations were 0.14–0.15 mg/L and 0.13–0.11 mg/L, respectively. This result required 40–50 g Ca(OH)2 and 10–12 g NaOH. In the experiment with Na2S, the minimum As concentration was 0.78 mg/L, which is a high value even for technological effluents. In this case, the presence of Fe has a negative effect. Since Fe is the first element to be reduced in the solution, a competing reaction of iron sulfide formation occurs. For the precipitation of Zn and Cu, Ca(OH)2 was the most effective reagent; the minimum concentrations of Zn and Cu were 0.021 mg/L and 0.009 mg/L, respectively. Lead was best removed in the experiment with NaOH. A serious drawback of the use of Na2S and NaOH is the high Na mobility. At the end of the experiment with Na2S, the excess concentration of Na was 60 times higher than the MPC for Na; additionally, in the experiment with NaOH, it was almost two orders of magnitude higher than the MPC for Na.
The correlation analysis of As bonds with other elements in the solution after the interaction with Ca(OH)
2, Na
2S, and NaOH and deposition of solid phases was performed (
Table 9). High positive correlation coefficients were revealed between arsenic and metals (Cu, Zn, Pb, Cd, Cr, Ni, Ag, V, Mo, Sn, In) and metalloids (Sb, Bi, P) when interacting with all reagents. This means similar behavior of these components in all three experiments and deposition of solid phases of complex composition: 1) arsenates, sulfo- and hydroxo-arsenates, oxides of Ca, Mg, Fe, Zn, Cu, and Pb; 2) colloidal hydroxides of Fe, Mn, and Al with metals and As sorbed on their surface; 3) compounds of unknown composition that do not occur in nature.
Positive correlation between As and Ca, Mg, Al, Fe, Zn, Cu, and Pb in solutions after interaction with Ca(OH)
2 is explained by the formation of arsenates, sulfo- and hydroxo-arsenates and other compounds confirmed by thermodynamic modeling and found in sediments using XRD analysis: Ca
3(AsO
4)
2∙4aq, Fe-Ca minerals containing As, arsenosiderite Ca
3Fe
4(AsO
4)(OH)
6·3H
2O austinite, conichalcite, shultenite (
Table 3 and
Table 4,
Figure 8). The positive correlations of As with Fe, Zn, Cu in the experiment with Na
2S are due to the formation of symplesite, scorodite, Fe-Na sulfates with As and Zn, pyrite with As, chalcophyllite, tennantite, and Zn
2As
2O
7 (
Table 5 and
Table 6,
Figure 9). Positive correlations of As and Ca, Mg, Pb, Zn, Cu, and Fe, in the experiment with NaOH, are associated with the formation of Ca
3(AsO
4)
2∙4aq, Mg(H
2AsO
4)
2, shultenite PbHAsO
4, austinite CaZnAsO
4(OH), conichalcite CaCuAsO
4, symplesite Fe
2+3(AsO
4)
2·8H
2O, scorodite FeAsO
4·2H
2O, chalcophyllite Cu
18Al
2(AsO
4)
4(SO
4)
3(OH)
24·36H
2O, and kaatialaite FeAs
3O
9·8H
2O (
Table 7 and
Table 8,
Figure 10).
The deposition of arsenic due to sorption on colloidal hydroxide iron (III), manganese (IV) and aluminum (III) compounds when the pH shifts to the alkaline values in experiments with Ca(OH)
2 and NaOH is indicated by positive significant correlations of the contents of As and Al, Fe, and Mn (r = 0.8/0.9). Above, we showed the formation of colloforms of Fe minerals containing As (up to 3.2%) and Cu (0.34%) after the interaction with NaOH (
Figure 10) and colloform mixture of gypsum, goethite, and hydroxosulfates of Al, Mg, and Fe, with ultrafine inclusions of As-containing grains after the interaction with Ca(OH)
2 using scanning electron microscopy (SEM analysis) (
Figure 4). When forming a suspension, not only those phases that were found in precipitation and determined by the thermodynamic modelling, but also multicomponent compounds, which, possibly, have no analogues in nature, including Cr, Ni, Bi, In, V, Mo, Sn, and Ag. The co-precipitation of many elements must be taken into account when developing practical recommendations for the subsequent use of the precipitate.
On the whole, the use of NaOH is advisable in the first steps of the solution neutralization, and the further precipitation of metals and metalloids should be carried out using Ca(OH)2.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}