Raman Spectroscopy and Single-Crystal High-Temperature Investigations of Bentorite, Ca6Cr2(SO4)3(OH)12·26H2O

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. The Occurrence and Paragenesis of Bentorite
3. Materials and Methods
4. Results
4.1. Chemical Composition
4.2. Raman Spectroscopy
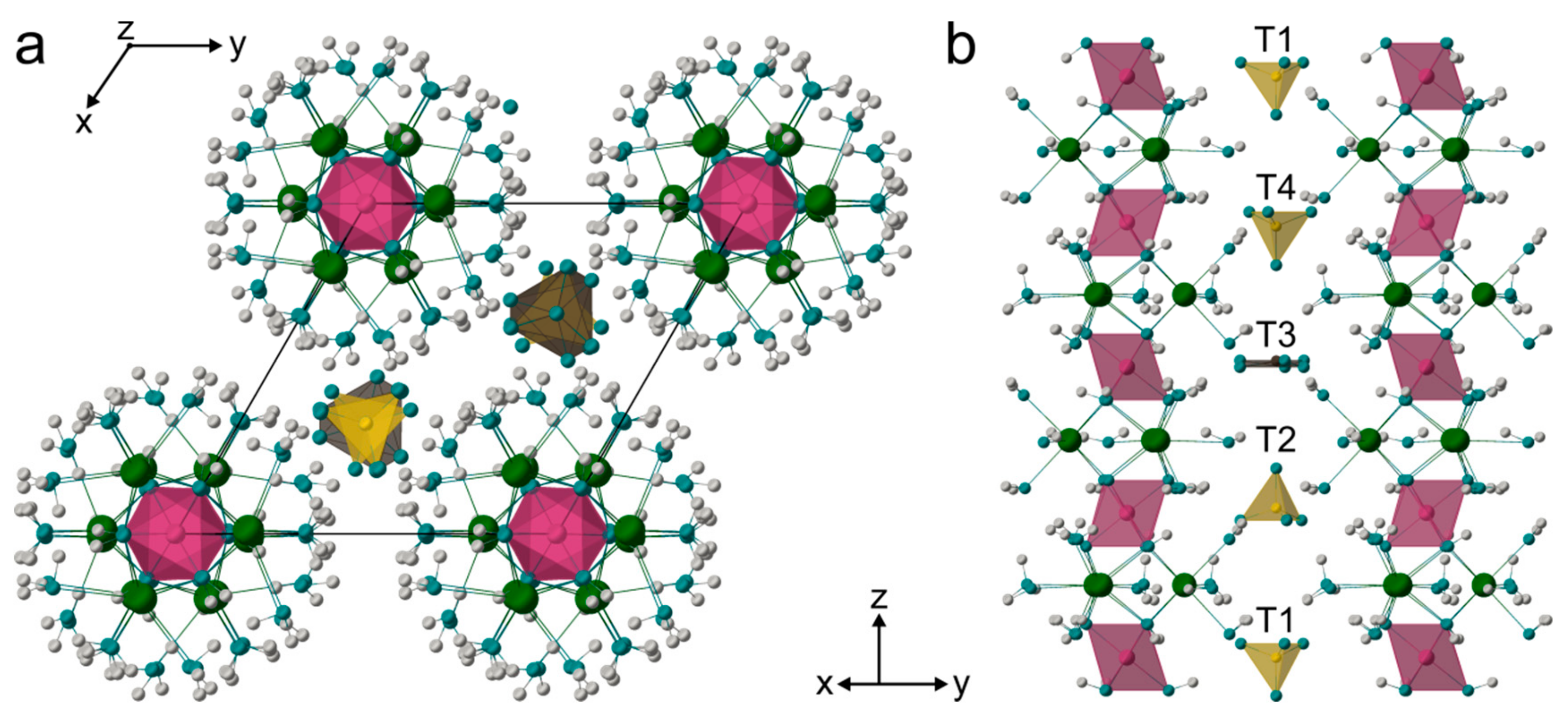
4.3. Crystal Structure
4.4. In Situ HT Single Crystal Study
5. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gross, S. Bentorite-a new mineral from the Hatrurim area, west of the Dead Sea, Israel. Isr. J. Earth Sci. 1980, 29, 81–84. [Google Scholar]
- Wieczorek-Ciurowa, K.; Fela, K.; Kozak, A.J. Chromium(III)-Ettringite Formation and its Thermal Stability. J. Therm. Anal. Calorim. 2001, 65, 655–660. [Google Scholar] [CrossRef]
- Juroszek, R.; Krüger, B.; Galuskina, I.O. Structural reinvestigation of bentorite. In Mitteilungen der Österreichischen Mineralogischen Gesellschaft; Universität Innsbruck: Innsbruck, Austria, 2017; Volume 163, p. 49. [Google Scholar]
- Seryotkin, Y.V.; Sokol, E.V.; Kokh, S.N.; Murashko, M.N. Natural Cr3+-rich ettringite: Occurrence, properties, and crystal structure. Phys. Chem. Miner. 2018, 45, 279–292. [Google Scholar] [CrossRef]
- Seryotkin, Y.V.; Sokol, E.V.; Kokh, S.N.; Sharygin, V.V. Natural bentorite—Cr3+ derivate of ettringite: Determination of crystal structure. Phys. Chem. Miner. 2019, 46, 553–570. [Google Scholar] [CrossRef]
- Drebushchak, V.A.; Seryotkin, Y.V.; Kokh, S.N.; Sokol, E.V. Natural specimen of triple solid solution ettringite–thaumasite–chromate-ettringite. J. Therm. Anal. Calorim. 2013, 114, 777–783. [Google Scholar] [CrossRef]
- Zhou, Q.; Lachowski, E.E.; Glasser, F.P. Metaettringite, a decomposition product of ettringite. Cem. Concr. Res. 2004, 34, 703–710. [Google Scholar] [CrossRef]
- Bentor, Y.K.; Gross, S.; Heller, L. Some unusual minerals from the “Mottled Zone” complex, Israel. Am. Mineral. 1963, 48, 924–930. [Google Scholar]
- Gross, S. The Mineralogy of the Hatrurim Formation, Israel; Geological Survey of Israel: Jerusalem, Israel, 1977. [Google Scholar]
- Burg, A.; Starinsky, A.; Bartov, Y.; Kolodny, Y. Geology of the Hatrurim Formation (“Mottled Zone”) in the Hatrurim basin. Isr. J. Earth Sci. 1991, 40, 107–124. [Google Scholar]
- Techer, I.; Khoury, H.N.; Salameh, E.; Rassineux, F.; Claude, C.; Clauer, N.; Pagel, M.; Lancelot, J.; Hamelin, B.; Jacquot, E. Propagation of high-alkaline fluids in an argillaceous formation: Case study of the Khushaym Matruk natural analogue (Central Jordan). J. Geochem. Explor. 2006, 90, 53–67. [Google Scholar] [CrossRef]
- Sokol, E.; Novikov, I.; Zateeva, S.; Vapnik, Y.; Shagam, R.; Kozmenko, O. Combustion metamorphism in the Nabi Musa dome: New implications for a mud volcanic origin of the Mottled Zone, Dead Sea area. Basin Res. 2010, 22, 414–438. [Google Scholar] [CrossRef]
- Geller, Y.I.; Burg, A.; Halicz, L.; Kolodny, Y. System closure during the combustion metamorphic “Mottled Zone” event, Israel. Chem. Geol. 2012, 334, 25–36. [Google Scholar] [CrossRef]
- Novikov, I.; Vapnik, Y.; Safonova, I. Mud volcano origin of the Mottled Zone, South Levant. Geosci. Front. 2013, 4, 597–619. [Google Scholar] [CrossRef]
- Matthews, A.; Gross, S. Petrological Evolution of the “Mottled Zone” (Hatrurim) Metamorphic Complex of Israel. Isr. J. Earth Sci. 1980, 29, 93–106. [Google Scholar]
- Grapes, R. Pyrometamorphism, 2nd ed.; Springer: Heidelberg/Berlin, Germany, 2010; ISBN 978-3-642-15587-1. [Google Scholar]
- Khoury, H.N.; Sokol, E.V.; Clark, I.D. Calcium uranium oxide minerals from Central Jordan: Assemblages, chemistry, and alteration products. Can. Mineral. 2015, 53, 61–82. [Google Scholar] [CrossRef]
- Kolodny, Y.; Burg, A.; Geller, Y.I.; Halicz, L.; Zakon, Y. Veins in the combusted metamorphic rocks, Israel; Weathering or a retrograde event? Chem. Geol. 2014, 385, 140–155. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. Sect. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.38.43; Rigaku Corporation: Oxford, UK, 2015. [Google Scholar]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General features. Z. Krist. Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Krüger, H.; Breil, L. Computer-controlled high-temperature single-crystal X-ray diffraction experiments and temperature calibration. J. Appl. Cryst. 2009, 42, 140–142. [Google Scholar] [CrossRef]
- Stoe. Cie X-AREA 1.84; Stoe & Cie GmbH: Darmstadt, Germany, 2018. [Google Scholar]
- Myneni, S.C.B.; Traina, S.J.; Waychunas, G.A.; Logan, T.J. Vibrational spectroscopy of functional group chemistry and arsenate coordination in ettringite. Geochim. Cosmochim. Acta 1998, 62, 3499–3514. [Google Scholar] [CrossRef]
- Renaudin, G.; Segni, R.; Mentel, D.; Nedelec, J.-M.; Leroux, F.; Taviot-Gueho, C. A Raman Study of the Sulfated Cement Hydrates: Ettringite and Monosulfoaluminate. ACT 2007, 5, 299–312. [Google Scholar] [CrossRef]
- Frost, R.L.; López, A.; Xi, Y.; Scholz, R.; da Costa, G.M.; Lima, R.M.F.; Granja, A. The spectroscopic characterization of the sulphate mineral ettringite from Kuruman manganese deposits, South Africa. Vib. Spectrosc. 2013, 68, 266–271. [Google Scholar] [CrossRef][Green Version]
- Scholtzová, E.; Kucková, L.; Kožíšek, J.; Tunega, D. Structural and spectroscopic characterization of ettringite mineral–combined DFT and experimental study. J. Mol. Struct. 2015, 1100, 215–224. [Google Scholar] [CrossRef]
- You, K.S.; Ahn, J.W.; Cho, H.C.; Han, G.C.; Han, D.Y.; Cho, K.H. Competing Ion Effect of Stabilization by Cr(III) & Cr(VI) in Ettringite Crystal Structure. Solid State Phenom. 2007, 124–126, 1629–1632. [Google Scholar]
- Kloprogge, J.T.; Hickey, L.; Frost, R.L. The effect of varying synthesis conditions on zinc chromium hydrotalcite: A spectroscopic study. Mater. Chem. Phys. 2005, 89, 99–109. [Google Scholar] [CrossRef]
- Barkley, M.C.; Yang, H.; Evans, S.H.; Downs, R.T.; Origlieri, M.J. Redetermination of despujolsite, Ca3Mn4+(SO4)2(OH)6·3H2O. Acta Crystallogr. Sect. E 2011, 67, i47–i48. [Google Scholar] [CrossRef] [PubMed]
- Antao, S.M.; Hassan, I. Temperature dependence of the structural parameters in the transformation of aragonite to calcite, as determined from in situ synchrotron powder X-ray-diffraction data. Can. Mineral. 2010, 48, 1225–1236. [Google Scholar] [CrossRef]
- Galuskin, E.V.; Krüger, B.; Galuskina, I.O.; Krüger, H.; Vapnik, Y.; Pauluhn, A.; Olieric, V. Stracherite, BaCa6(SiO4)2[(PO4)(CO3)]F, the first CO3-bearing intercalated hexagonal antiperovskite from Negev Desert, Israel. Am. Mineral. 2018, 103, 1699–1706. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | |
|---|---|
| Refined chemical composition | C0.499H60Al0.387Ca6Cr1.613O49.725S2.75 |
| Crystal system | trigonal |
| Space group | P31c |
| Unit cell dimensions | a = 11.1927 (5) Å |
| b = 11.1927 (5) Å | |
| c = 21.7121 (10) Å | |
| Unit cell volume | 2355.60 (18) Å3 |
| Formula weight | 1285 |
| Density (calculated) | 1.8115 g/cm3 |
| Z Crystal size | 2 0.19 × 0.16 × 0.12 mm |
| Data Collection | |
| Diffractometer | four-circle diffractometer, Rigaku Oxford Diffraction Gemini R Ultra |
| Radiation wavelength | 0.71073 Å |
| min. and max. theta (°) | 3.53, 29.6 |
| Time of exposure | 130 s |
| Reflection ranges | −14 ≤ h ≤ 15; −13 ≤ k ≤ 13; −29 ≤ l ≤ 26 |
| Refinement of Structure | |
| Reflection measured | 38,295 |
| No. of unique reflections | 3998 |
| No. of observed unique reflections [I > 3σ(I)] | 2714 |
| Refined parameters | 248 |
| Rint | 0.046 |
| R1 | 0.0388 |
| Goof | 1.91 |
| Δρmin (e Å−3) | −0.81 |
| Δρmax (e Å−3) | 1.34 |
| wt % | Arad Stone Quarry | ||
|---|---|---|---|
| Mean | S.D. | Range | |
| n = 10 | |||
| SO3 | 18.48 | 0.32 | 17.79–18.93 |
| SiO2 | 0.29 | 0.12 | 0.13–0.50 |
| Al2O3 | 0.90 | 0.32 | 0.42–1.42 |
| Cr2O3 | 10.00 | 0.56 | 9.31–11.20 |
| CaO | 25.89 | 0.83 | 24.59–27.02 |
| H2O a | 44.44 | ||
| Total | 100.00 | ||
| Calculated on 8 framework cations | |||
| S6+ | 3.00 | ||
| Si4+ | 0.06 | ||
| Al3+ | 0.23 | ||
| Cr3+ | 1.71 | ||
| Ca2+ | 6.00 | ||
| H2O | 52.00 | ||
| OH | 12.00 | ||
| Ca1 | Ca2 | ||
|---|---|---|---|
| Ca1-O1 | 2.421(7) | Ca2-O2 | 2.443(7) |
| Ca1-O1 | 2.462(5) | Ca2-O2 | 2.373(5) |
| Ca1-O3 | 2.416(5) | Ca2-O4 | 2.413(7) |
| Ca1-O3 | 2.447(7) | Ca2-O4 | 2.390(5) |
| Ca1-O6 | 2.400(8) | Ca2-O5 | 2.493(8) |
| Ca1-O8 | 2.448(8) | Ca2-O7 | 2.403(8) |
| Ca1-O10 | 2.743(9) | Ca2-O9 | 2.597(7) |
| Ca1-O12 | 2.675(5) | Ca2-O11 | 2.626(4) |
| average | 2.502 | average | 2.467 |
| M1 = 0.799 Cr + 0.201 Al | M2 = 0.814 Cr + 0.186 Al | ||
| M1-O1 | 1.985(6) | M2-O3 | 1.971(6) |
| M1-O1 | 1.985(5) | M2-O3 | 1.971(5) |
| M1-O1 | 1.985(7) | M2-O3 | 1.971(8) |
| M1-O2 | 1.951(6) | M2-O4 | 1.977(6) |
| M1-O2 | 1.951(5) | M2-O4 | 1.977(5) |
| M1-O2 | 1.951(7) | M2-O4 | 1.977(7) |
| average | 1.968 | average | 1.974 |
| T1 = 0.940 S + 0.060 C | |||
| T1-O13 | 1.428(8) | ||
| T1-O16 | 1.473(4) | ||
| T1-O16 | 1.473(6) | ||
| T1-O16 | 1.473(4) | ||
| average | 1.462 | ||
| T2 = S | |||
| T2-O14 | 1.482(10) | ||
| T2-O17 | 1.452(7) | ||
| T2-O17 | 1.452(11) | ||
| T2-O17 | 1.452(6) | ||
| average | 1.460 | ||
| T3 = 0.25 C | |||
| T3-O18a | 1.43(3) | T3-O18b | 1.413(15) |
| T3-O18a | 1.43(3) | T3-O18b | 1.41(2) |
| T3-O18a | 1.43(2) | T3-O18b | 1.413(12) |
| average | 1.43 | average | 1.412 |
| T4 = 0.809 S + 0.191 C | |||
| T4-O15 | 1.453(10) | ||
| T4-O19 | 1.474(5) | ||
| T4-O19 | 1.474(4) | ||
| T4-O19 | 1.474(8) | ||
| average | 1.469 | ||
| Bentorite from the Arad Stone Quarry | Bentorite from Ma’ale Adummim | Band Assignment |
|---|---|---|
| 157, 193 | 160, 197 | lattice vibrations |
| 442 | 442 | ν2 (SO4)2− |
| 537 | 538 | Al/Cr–OH stretching vibrations of Al/Cr(OH)63− unit |
| 601 | 598, 641 | ν4 (SO4)2− |
| 757 | 756 | hydroxyl deformation vibration of Cr–OH units |
| - | 853 | Al–OH bending vibrations |
| 983 | 983 | ν1 (SO4)2− |
| 1109 | 1111 | ν3 (SO4)2− |
| 1682 | 1685 | (OH)− bending vibration of H2O molecules |
| 3314, 3400, 3469 | 3295, 3384, 3475 | (OH)− stretching vibration of H2O molecules |
| 3618 | 3616 | (OH)− stretching vibration of the OH units |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juroszek, R.; Krüger, B.; Galuskina, I.; Krüger, H.; Tribus, M.; Kürsten, C. Raman Spectroscopy and Single-Crystal High-Temperature Investigations of Bentorite, Ca6Cr2(SO4)3(OH)12·26H2O. Minerals 2020, 10, 38. https://doi.org/10.3390/min10010038
Juroszek R, Krüger B, Galuskina I, Krüger H, Tribus M, Kürsten C. Raman Spectroscopy and Single-Crystal High-Temperature Investigations of Bentorite, Ca6Cr2(SO4)3(OH)12·26H2O. Minerals. 2020; 10(1):38. https://doi.org/10.3390/min10010038
Chicago/Turabian StyleJuroszek, Rafał, Biljana Krüger, Irina Galuskina, Hannes Krüger, Martina Tribus, and Christian Kürsten. 2020. "Raman Spectroscopy and Single-Crystal High-Temperature Investigations of Bentorite, Ca6Cr2(SO4)3(OH)12·26H2O" Minerals 10, no. 1: 38. https://doi.org/10.3390/min10010038
APA StyleJuroszek, R., Krüger, B., Galuskina, I., Krüger, H., Tribus, M., & Kürsten, C. (2020). Raman Spectroscopy and Single-Crystal High-Temperature Investigations of Bentorite, Ca6Cr2(SO4)3(OH)12·26H2O. Minerals, 10(1), 38. https://doi.org/10.3390/min10010038