Chlorine Isotope Composition of Apatite from the >3.7 Ga Isua Supracrustal Belt, SW Greenland

 ,
,  , ,
, , 
Abstract
1. Introduction
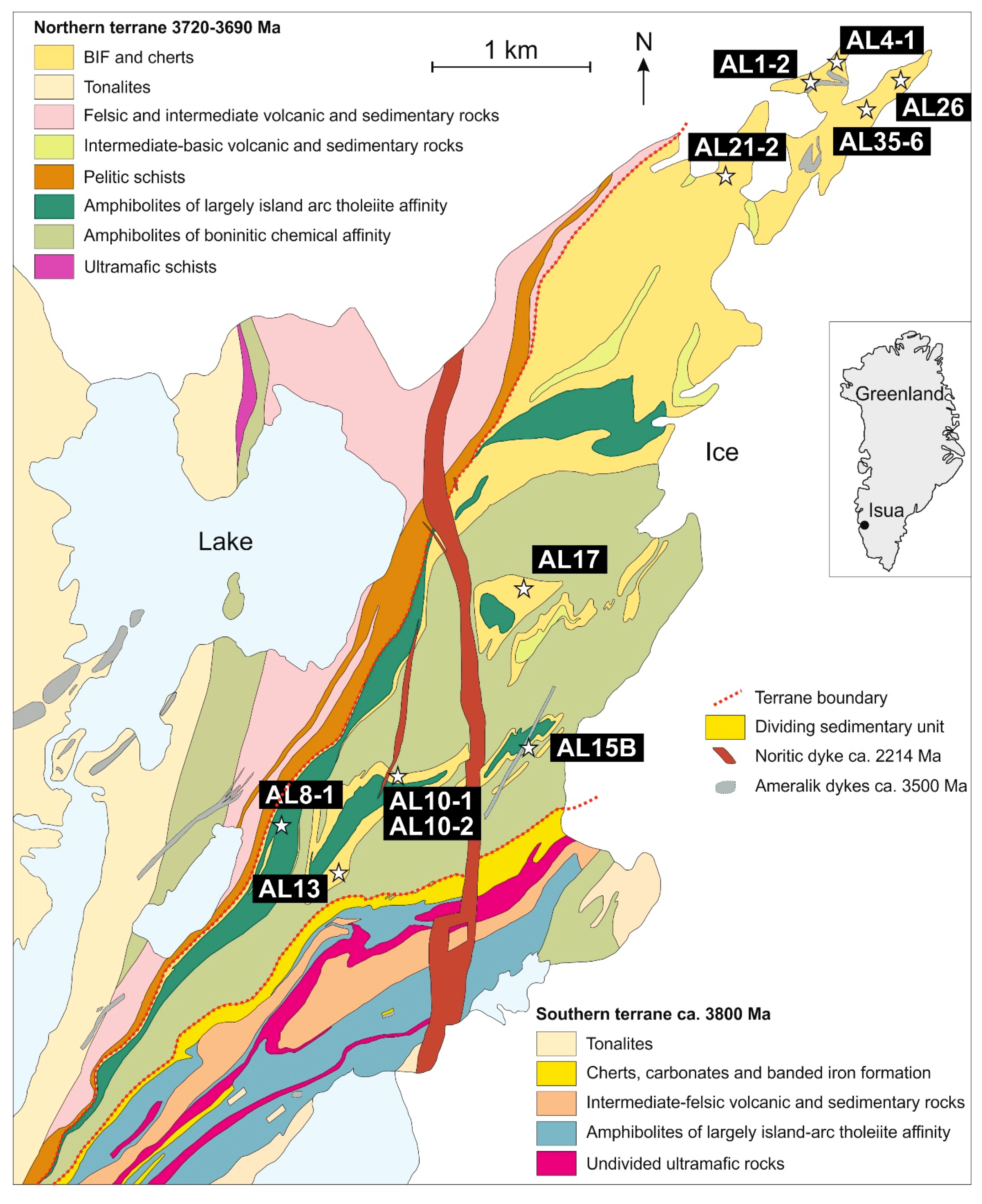
2. Geological Context and Research Material
3. Laboratory Methods
3.1. Transmission Electron Microscopy (TEM)
3.2. Optical-Microscope Cathodoluminescence (CL) Imaging and Spectroscopy
3.3. Secondary Ion Mass Spectrometry (SIMS)
4. Results
4.1. Transmission Electron Microscopy
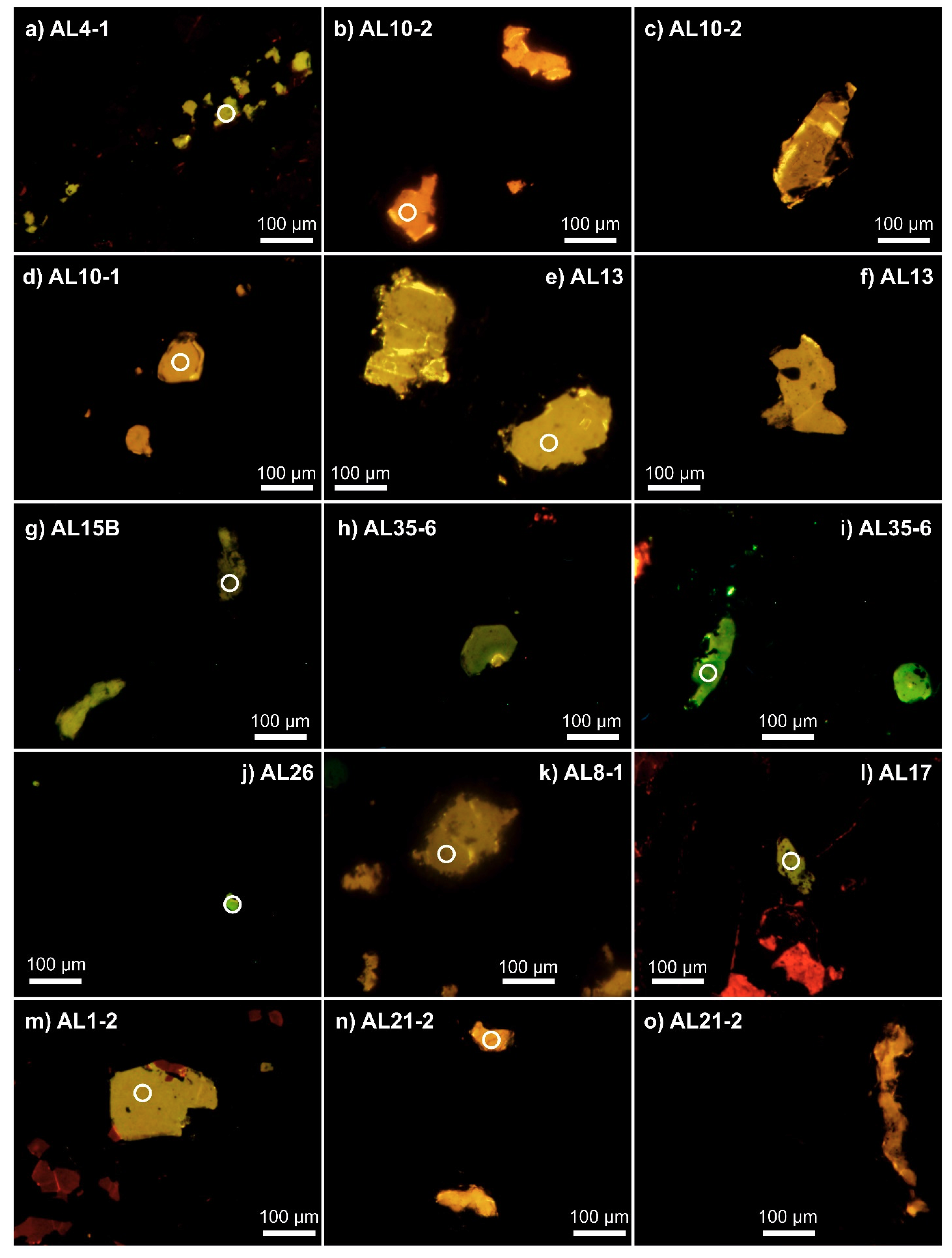
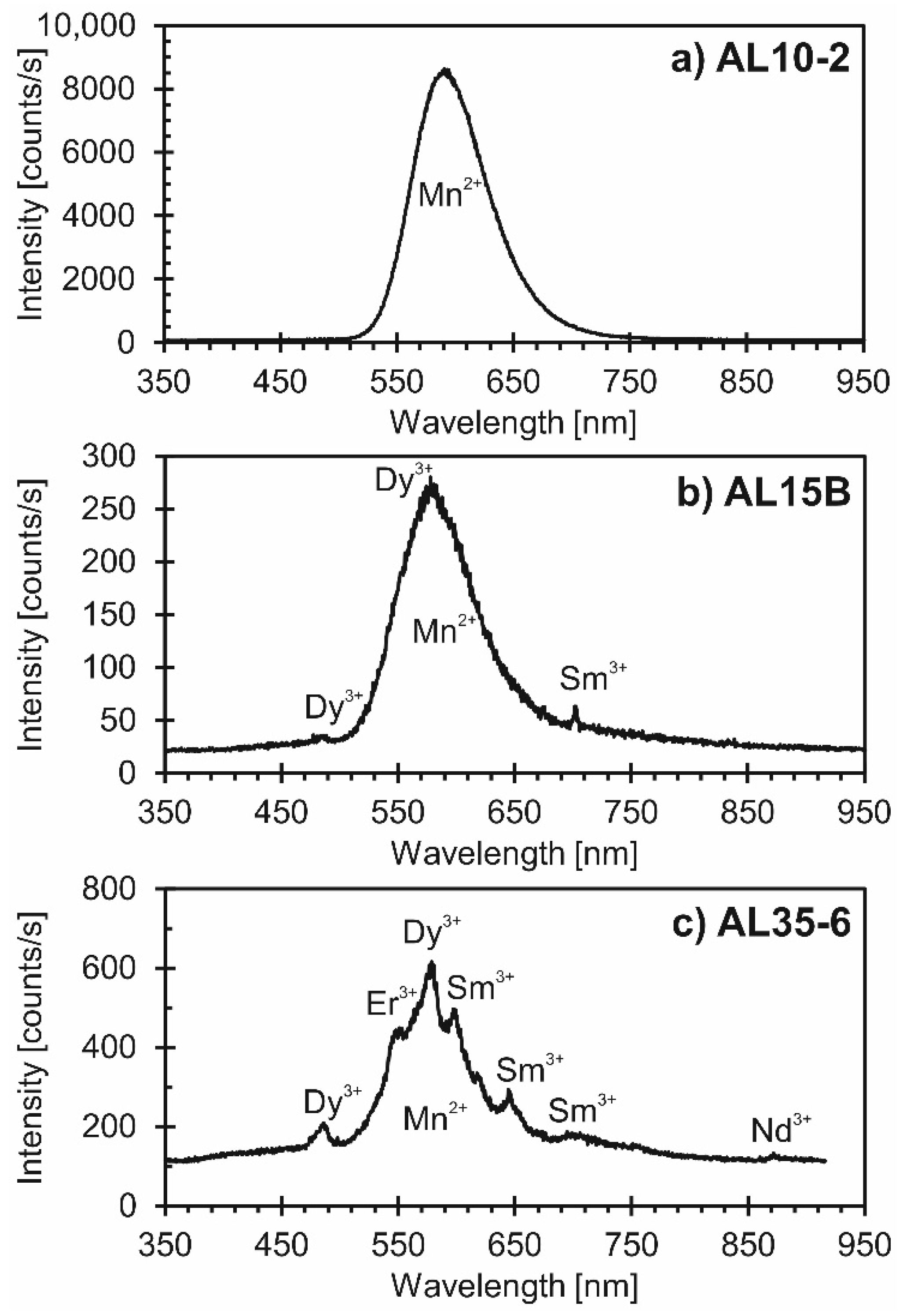
4.2. Cathodoluminescence Imaging and Spectroscopy
4.3. Secondary Ion Mass Spectrometry
5. Discussion
5.1. Micron-Scale Heterogeneity of Isua Apatite Crystals and its Implications for Chlorine Isotope Compositions
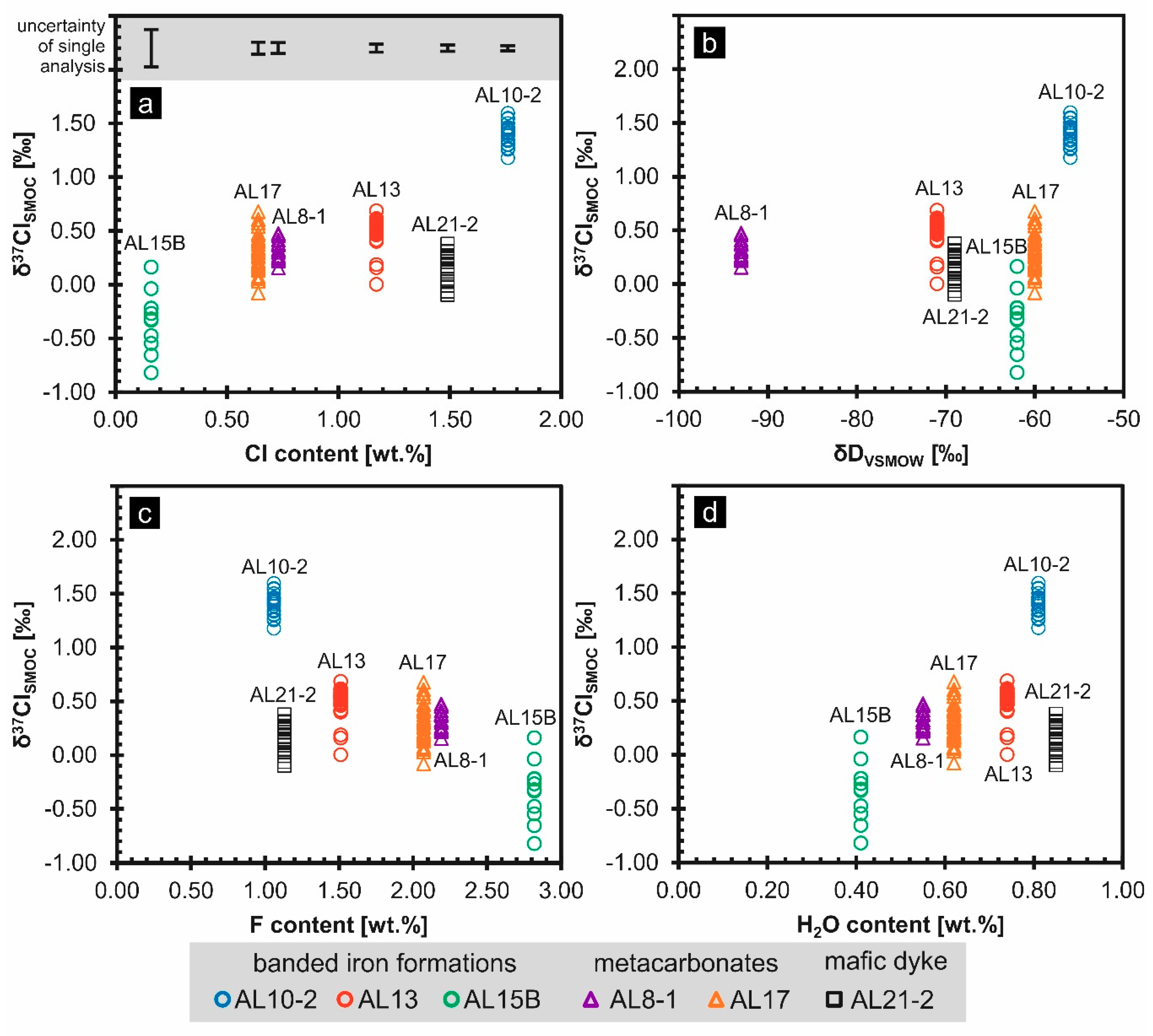
5.2. Chlorine Isotope Ratios
5.3. Evolution Pathways of Volatile Compositions of Isua Apatite
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Black, L.P.; Gale, N.H.; Moorbath, S.; Pankhurst, R.J.; McGregor, V.R. Isotopic dating of very early Precambrian amphibolite facies gneisses from the Godthaab district, West Greenland. Earth Planet. Sci. Lett. 1971, 12, 245–259. [Google Scholar]
- Moorbath, S.; O’nions, R.K.; Pankhurst, R.J. Early Archaean age for the Isua iron formation, West Greenland. Nature 1973, 245, 138–139. [Google Scholar] [CrossRef]
- Frei, R.; Rosing, M.T.; Waight, T.E.; Ulfbeck, D.G. Hydrothermal-metasomatic and tectono-metamorphic processes in the Isua supracrustal belt (West Greenland): A multi-isotopic investigation of their effects on the Earth’s oldest oceanic crustal sequence. Geochim. Cosmochim. Acta 2002, 66, 467–486. [Google Scholar] [CrossRef]
- Schidlowski, M.; Appel, P.W.U.; Eichmann, R.; Junge, C.E. Carbon isotope geochemistry of the 3.7 × 109-yr-old Isua sediments, West Greenland: Implications for the Archaean carbon and oxygen cycles. Geochim. Cosmochim. Acta 1979, 43, 189–199. [Google Scholar] [CrossRef]
- Mojzsis, S.J.; Arrhenius, G.; McKeegan, K.D.; Harrison, T.M.; Nutman, A.P.; Friend, C.R.L. Evidence for life on Earth before 3800 million years ago. Nature 1996, 384, 55. [Google Scholar] [CrossRef] [PubMed]
- Rosing, M.T.; Rose, N.M.; Bridgwater, D.; Thomsen, H.S. Earliest part of Earth’s stratigraphic record: A reappraisal of the >3.7 Ga Isua (Greenland) supracrustal sequence. Geology 1996, 24, 43–46. [Google Scholar] [CrossRef]
- Ueno, Y.; Yurimoto, H.; Yoshioka, H.; Komiya, T.; Maruyama, S. Ion microprobe analysis of graphite from ca. 3.8 Ga metasediments, Isua supracrustal belt, West Greenland: Relationship between metamorphism and carbon isotopic composition. Geochim. Cosmochim. Acta 2002, 66, 1257–1268. [Google Scholar] [CrossRef]
- van Zuilen, M.A.; Lepland, A.; Teranes, J.; Finarelli, J.; Wahlen, M.; Arrhenius, G. Graphite and carbonates in the 3.8 Ga old Isua supracrustal belt, southern West Greenland. Precambrian Res. 2003, 126, 331–348. [Google Scholar] [CrossRef]
- Lepland, A.; van Zuilen, M.A.; Arrhenius, G.; Whitehouse, M.J.; Fedo, C.M. Questioning the evidence for Earth’s earliest life—Akilia revisited. Geology 2005, 33, 77–79. [Google Scholar] [CrossRef]
- Craddock, P.R.; Dauphas, N. Iron and carbon isotope evidence for microbial iron respiration throughout the Archean. Earth Planet. Sci. Lett. 2011, 303, 121–132. [Google Scholar] [CrossRef]
- Ohtomo, Y.; Kakegawa, T.; Ishida, A.; Nagase, T.; Rosing, M.T. Evidence for biogenic graphite in early Archaean Isua metasedimentary rocks. Nat. Geosci. 2014, 7, 25. [Google Scholar] [CrossRef]
- Yoshiya, K.; Sawaki, Y.; Hirata, T.; Maruyama, S.; Komiya, T. In-situ iron isotope analysis of pyrites in ~3.7 Ga sedimentary protoliths from the Isua supracrustal belt, southern West Greenland. Chem. Geol. 2015, 401, 126–139. [Google Scholar] [CrossRef]
- Nutman, A.P.; Bennett, V.C.; Friend, C.R.L.; Van Kranendonk, M.J.; Chivas, A.R. Rapid emergence of life shown by discovery of 3700-million-year-old microbial structures. Nature 2016, 537, 535. [Google Scholar] [CrossRef] [PubMed]
- Hassenkam, T.; Andersson, M.P.; Dalby, K.N.; Mackenzie, D.M.A.; Rosing, M.T. Elements of Eoarchean life trapped in mineral inclusions. Nature 2017, 548, 78. [Google Scholar] [CrossRef] [PubMed]
- Allwood, A.C.; Rosing, M.T.; Flannery, D.T.; Hurowitz, J.A.; Heirwegh, C.M. Reassessing evidence of life in 3700-million-year-old rocks of Greenland. Nature 2018, 563, 241. [Google Scholar] [CrossRef]
- Nutman, A.P.; Bennett, V.C.; Friend, C.R.L.; Van Kranendonk, M.J.; Rothacker, L.; Chivas, A.R. Cross-examining Earth’s oldest stromatolites: Seeing through the effects of heterogeneous deformation, metamorphism and metasomatism affecting Isua (Greenland) similar to 3700 Ma sedimentary rocks. Precambrian Res. 2019, 331, 105347. [Google Scholar] [CrossRef]
- Dodd, M.S.; Papineau, D.; She, Z.-B.; Manikyamba, C.; Wan, Y.-S.; O’Neil, J.; Karhu, J.A.; Rizo, H.; Pirajno, F. Widespread occurrences of variably crystalline 13C-depleted graphitic carbon in banded iron formations. Earth Planet. Sci. Lett. 2019, 512, 163–174. [Google Scholar] [CrossRef]
- Whitehouse, M.J.; Dunkley, D.J.; Kusiak, M.A.; Wilde, S.A. On the true antiquity of Eoarchean chemofossils—Assessing the claim for Earth’s oldest biogenic graphite in the Saglek Block of Labrador. Precambrian Res. 2019, 323, 70–81. [Google Scholar] [CrossRef]
- Tashiro, T.; Ishida, A.; Hori, M.; Igisu, M.; Koike, M.; Méjean, P.; Takahata, N.; Sano, Y.; Komiya, T. Early trace of life from 3.95 Ga sedimentary rocks in Labrador, Canada. Nature 2017, 549, 516. [Google Scholar] [CrossRef]
- Appel, P.W.U.; Fedo, C.M.; Moorbath, S.; Myers, J.S. Recognizable primary volcanic and sedimentary features in a low-strain domain of the highly deformed, oldest known (~3.7–3.8 Gyr) Greenstone Belt, Isua, West Greenland. Terra Nova 1998, 10, 57–62. [Google Scholar] [CrossRef]
- Appel, P.W.U.; Rollinson, H.R.; Touret, J.L.R. Remnants of an early Archaean (>3.75 Ga) sea-floor, hydrothermal system in the Isua Greenstone Belt. Precambrian Res. 2001, 112, 27–49. [Google Scholar] [CrossRef]
- Lepland, A.; Arrhenius, G.; Cornell, D. Apatite in early Archean Isua supracrustal rocks, southern West Greenland: Its origin, association with graphite and potential as a biomarker. Precambrian Res. 2002, 118, 221–241. [Google Scholar] [CrossRef]
- Klein, C. Some Precambrian banded iron-formations (BIFs) from around the world: Their age, geologic setting, mineralogy, metamorphism, geochemistry, and origins. Am. Mineral. 2005, 90, 1473–1499. [Google Scholar] [CrossRef]
- Frei, R.; Polat, A. Source heterogeneity for the major components of ~3.7 Ga Banded Iron Formations (Isua Greenstone Belt, Western Greenland): Tracing the nature of interacting water masses in BIF formation. Earth Planet. Sci. Lett. 2007, 253, 266–281. [Google Scholar] [CrossRef]
- Pope, E.C.; Bird, D.K.; Rosing, M.T. Isotope composition and volume of Earth’s early oceans. Proc. Natl. Acad. Sci. USA 2012, 109, 4371–4376. [Google Scholar] [CrossRef]
- Aoki, S.; Kabashima, C.; Kato, Y.; Hirata, T.; Komiya, T. Influence of contamination on banded iron formations in the Isua supracrustal belt, West Greenland: Reevaluation of the Eoarchean seawater compositions. Geosci. Front. 2018, 9, 1049–1072. [Google Scholar] [CrossRef]
- Wudarska, A.; Wiedenbeck, M.; Słaby, E.; Lepland, A.; Birski, Ł.; Simon, K. Halogen chemistry and hydrogen isotopes of apatite from the >3.7 Ga Isua supracrustal belt, SW Greenland. Precambrian Res. 2018, 310, 153–164. [Google Scholar] [CrossRef]
- D’Andres, J.; Kendrick, M.A.; Bennett, V.C.; Nutman, A.P. Halogens in serpentinites from the Isua supracrustal belt, Greenland: An Eoarchean seawater signature and biomass proxy? Geochim. Cosmochim. Acta 2019, 262, 31–59. [Google Scholar] [CrossRef]
- Nishizawa, M.; Takahata, N.; Terada, K.; Komiya, T.; Ueno, Y.; Sano, Y. Rare-earth element, lead, carbon, and nitrogen geochemistry of apatite-bearing metasediments from the ~3.8 Ga Isua Supracrustal Belt, West Greenland. Int. Geol. Rev. 2005, 47, 952–970. [Google Scholar] [CrossRef]
- Higashi, Y.; Itoh, S.; Hashiguchi, M.; Sakata, S.; Hirata, T.; Watanabe, K.; Sakaguchi, I. Hydrogen diffusion in the apatite-water system: Fluorapatite parallel to the c-axis. Geochem. J. 2017, 51, 115–122. [Google Scholar] [CrossRef]
- Brenan, J. Kinetics of fluorine, chlorine and hydroxyl exchange in fluorapatite. Chem. Geol. 1993, 110, 195–210. [Google Scholar] [CrossRef]
- Sharp, Z.D.; Shearer, C.K.; McKeegan, K.D.; Barnes, J.D.; Wang, Y.Q. The chlorine isotope composition of the moon and implications for an anhydrous mantle. Science 2010, 329, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- Tartèse, R.; Anand, M.; Joy, K.H.; Franchi, I.A. H and Cl isotope systematics of apatite in brecciated lunar meteorites Northwest Africa 4472, Northwest Africa 773, Sayh al Uhaymir 169, and Kalahari 009. Meteorit. Planet. Sci. 2014, 49, 2266–2289. [Google Scholar] [CrossRef]
- Treiman, A.H.; Boyce, J.W.; Gross, J.; Guan, Y.; Eiler, J.M.; Stolper, E.M. Phosphate-halogen metasomatism of lunar granulite 79215: Impact-induced fractionation of volatiles and incompatible elements. Am. Mineral. 2014, 99, 1860–1870. [Google Scholar] [CrossRef]
- Boyce, J.W.; Treiman, A.H.; Guan, Y.; Ma, C.; Eiler, J.M.; Gross, J.; Greenwood, J.P.; Stolper, E.M. The chlorine isotope fingerprint of the lunar magma ocean. Sci. Adv. 2015, 1, e1500380. [Google Scholar] [CrossRef]
- Potts, N.J.; Barnes, J.J.; Tartèse, R.; Franchi, I.A.; Anand, M. Chlorine isotopic compositions of apatite in Apollo 14 rocks: Evidence for widespread vapor-phase metasomatism on the lunar nearside ~4 billion years ago. Geochim. Cosmochim. Acta 2018, 230, 46–59. [Google Scholar] [CrossRef]
- Wang, Y.; Hsu, W.; Guan, Y. An extremely heavy chlorine reservoir in the Moon: Insights from the apatite in lunar meteorites. Sci. Rep. 2019, 9, 5727. [Google Scholar] [CrossRef]
- Sharp, Z.; Williams, J.; Shearer, C.; Agee, C.; McKeegan, K. The chlorine isotope composition of Martian meteorites 2. Implications for the early solar system and the formation of Mars. Meteorit. Planet. Sci. 2016, 51, 2111–2126. [Google Scholar] [CrossRef]
- Bellucci, J.J.; Whitehouse, M.J.; John, T.; Nemchin, A.A.; Snape, J.F.; Bland, P.A.; Benedix, G.K. Halogen and Cl isotopic systematics in Martian phosphates: Implications for the Cl cycle and surface halogen reservoirs on Mars. Earth Planet. Sci. Lett. 2017, 458, 192–202. [Google Scholar] [CrossRef]
- John, T.; Layne, G.D.; Haase, K.M.; Barnes, J.D. Chlorine isotope evidence for crustal recycling into the Earth’s mantle. Earth Planet. Sci. Lett. 2010, 298, 175–182. [Google Scholar] [CrossRef]
- Barnes, J.D.; Sharp, Z.D. Chlorine isotope geochemistry. Rev. Mineral. Geochem. 2017, 82, 345–378. [Google Scholar] [CrossRef]
- Morton, R.D.; Catanzaro, E.J. Stable chlorine isotope abundances in apatites from Ødegårdens verk, Norway. Nor. Geol. Tidsskr. 1964, 44, 307–313. [Google Scholar]
- Kusebauch, C.; John, T.; Whitehouse, M.J.; Engvik, A.K. Apatite as probe for the halogen composition of metamorphic fluids (Bamble Sector, SE Norway). Contrib. Mineral. Petrol. 2015, 170, 34. [Google Scholar] [CrossRef]
- Eggenkamp, H. The Geochemistry of Stable Chlorine and Bromine Isotopes; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–172. [Google Scholar]
- Schauble, E.A.; Rossman, G.R.; Taylor, H.P., Jr. Theoretical estimates of equilibrium chlorine-isotope fractionations. Geochim. Cosmochim. Acta 2003, 67, 3267–3281. [Google Scholar] [CrossRef]
- Sharp, Z.D.; Barnes, J.D.; Fischer, T.P.; Halick, M. An experimental determination of chlorine isotope fractionation in acid systems and applications to volcanic fumaroles. Geochim. Cosmochim. Acta 2010, 74, 264–273. [Google Scholar] [CrossRef]
- Kusebauch, C.; John, T.; Barnes, J.D.; Klügel, A.; Austrheim, H.O. Halogen element and stable chlorine isotope fractionation caused by fluid—Rock interaction (Bamble Sector, SE Norway). J. Petrol. 2015, 56, 299–324. [Google Scholar] [CrossRef]
- Barnes, J.D.; Selverstone, J.; Sharp, Z.D. Chlorine isotope chemistry of serpentinites from Elba, Italy, as an indicator of fluid source and subsequent tectonic history. Geochem. Geophys. Geosyst. 2006, 7. [Google Scholar] [CrossRef]
- Bonifacie, M.; Jendrzejewski, N.; Agrinier, P.; Humler, E.; Coleman, M.; Javoy, M. The chlorine isotope composition of Earth’s mantle. Science 2008, 319, 1518–1520. [Google Scholar] [CrossRef]
- Sarafian, A.R.; John, T.; Roszjar, J.; Whitehouse, M.J. Chlorine and hydrogen degassing in Vesta’s magma ocean. Earth Planet. Sci. Lett. 2017, 459, 311–319. [Google Scholar] [CrossRef]
- Sharp, Z.D.; Mercer, J.A.; Jones, R.H.; Brearley, A.J.; Selverstone, J.; Bekker, A.; Stachel, T. The chlorine isotope composition of chondrites and Earth. Geochim. Cosmochim. Acta 2013, 107, 189–204. [Google Scholar] [CrossRef]
- Knauth, L.P. Salinity history of the Earth’s early ocean. Nature 1998, 395, 554. [Google Scholar] [CrossRef] [PubMed]
- De Ronde, C.E.; Channer, D.M.; Faure, K.; Bray, C.J.; Spooner, E.T. Fluid chemistry of Archean seafloor hydrothermal vents: Implications for the composition of circa 3.2 Ga seawater. Geochim. Cosmochim. Acta 1997, 61, 4025–4042. [Google Scholar] [CrossRef]
- Pinti, D.L. The origin and evolution of the oceans. In Lectures in Astrobiology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 83–112. [Google Scholar]
- Nutman, A.P. The early Archaean to Proterozoic history of the Isukasia area, southern West Greenland. Bull. Gronl. Geol. Unders. 1986, 154, 55–68. [Google Scholar]
- Komiya, T.; Maruyama, S.; Masuda, T.; Nohda, S.; Hayashi, M.; Okamoto, K. Plate tectonics at 3.8–3.7 Ga: Field evidence from the Isua accretionary complex, southern West Greenland. J. Geol. 1999, 107, 515–554. [Google Scholar] [CrossRef]
- Myers, J.S. Protoliths of the 3.8–3.7 Ga Isua greenstone belt, west Greenland. Precambrian Res. 2001, 105, 129–141. [Google Scholar] [CrossRef]
- Nutman, A.P.; Friend, C.R.L. New 1: 20,000 scale geological maps, synthesis and history of investigation of the Isua supracrustal belt and adjacent orthogneisses, southern West Greenland: A glimpse of Eoarchaean crust formation and orogeny. Precambrian Res. 2009, 172, 189–211. [Google Scholar] [CrossRef]
- Rollinson, H. The metamorphic history of the Isua greenstone belt, West Greenland. Geol. Soc. Lond. Spec. Publ. 2002, 199, 329–350. [Google Scholar] [CrossRef]
- Rollinson, H. Metamorphic history suggested by garnet-growth chronologies in the Isua Greenstone Belt, West Greenland. Precambrian Res. 2003, 126, 181–196. [Google Scholar] [CrossRef]
- Hayashi, M.; Komiya, T.; Nakamura, Y.; Maruyama, S. Archean regional metamorphism of the Isua supracrustal belt, southern West Greenland: Implications for a driving force for Archean plate tectonics. Int. Geol. Rev. 2000, 42, 1055–1115. [Google Scholar] [CrossRef]
- Komiya, T.; Hayashi, M.; Maruyama, S.; Yurimoto, H. Intermediate-P/T type Archean metamorphism of the Isua supracrustal belt: Implications for secular change of geothermal gradients at subduction zones and for Archean plate tectonics. Am. J. Sci. 2002, 302, 806–826. [Google Scholar] [CrossRef]
- Arai, T.; Omori, S.; Komiya, T.; Maruyama, S. Intermediate P/T-type regional metamorphism of the Isua Supracrustal Belt, southern west Greenland: The oldest Pacific-type orogenic belt? Tectonophysics 2015, 662, 22–39. [Google Scholar] [CrossRef]
- Nutman, A.P.; Hagiya, H.; Maruyama, S. SHRIMP U-Pb single zircon geochronology of a Proterozoic mafic dyke, Isukasia, southern West Greenland. Bull. Geol. Soc. Den. 1995, 42, 17–22. [Google Scholar]
- White, R.V.; Crowley, J.L.; Myers, J.S. Earth’s oldest well-preserved mafic dyke swarms in the vicinity of the Isua greenstone belt, southern West Greenland. Geol. Greenl. Surv. Bull. 2000, 186, 65–72. [Google Scholar]
- Komiya, T. Geochemistry of the oldest MORB and OIB of the World, Isua (3.8 Ga), Greenland. EOS Trans. 1995, 76, 700. [Google Scholar]
- Birski, Ł.; Wirth, R.; Słaby, E.; Wudarska, A.; Lepland, A.; Hofmann, A.; Schreiber, A. (Ca-Y)-phosphate inclusions in apatite crystals from Archean rocks from the Barberton Greenstone Belt and Pilbara Craton: First report of natural occurrence. Am. Mineral. 2018, 103, 307–313. [Google Scholar] [CrossRef]
- Birski, Ł.; Słaby, E.; Wirth, R.; Koch-Müller, M.; Simon, K.; Wudarska, A.; Götze, J.; Lepland, A.; Hofmann, A.; Kuras, A. Archaean phosphates: A case study of transformation processes in apatite from the Barberton greenstone belt. Contrib. Mineral. Petrol. 2019, 174, 25. [Google Scholar] [CrossRef]
- Harlov, D.E. Apatite: A fingerprint for metasomatic processes. Elements 2015, 11, 171–176. [Google Scholar] [CrossRef]
- Bouzari, F.; Hart, C.J.R.; Bissig, T.; Barker, S. Hydrothermal alteration revealed by apatite luminescence and chemistry: A potential indicator mineral for exploring covered porphyry copper deposits. Econ. Geol. 2016, 111, 1397–1410. [Google Scholar] [CrossRef]
- Słaby, E.; Gros, K.; Förster, H.-J.; Wudarska, A.; Birski, Ł.; Hamada, M.; Götze, J.; Martin, H.; Jayananda, M.; Moyen, J.-F.; et al. Mineral–fluid interactions in the late Archean Closepet granite batholith, Dharwar Craton, southern India. Geol. Soc. Lond. Spec. Publ. 2019, 489, SP489–SP2019. [Google Scholar] [CrossRef]
- Wirth, R. Focused Ion Beam (FIB) A novel technology for advanced application of micro-and nanoanalysis in geosciences and applied mineralogy. Eur. J. Mineral. 2004, 16, 863–876. [Google Scholar] [CrossRef]
- Wirth, R. Focused Ion Beam (FIB) combined with SEM and TEM: Advanced analytical tools for studies of chemical composition, microstructure and crystal structure in geomaterials on a nanometre scale. Chem. Geol. 2009, 261, 217–229. [Google Scholar] [CrossRef]
- Neuser, R.D.; Bruhn, F.; Götze, J.; Habermann, D.; Richter, D.K. Kathodolumineszenz: Methodik und anwendung. Z. Geol. Paläontol. Teil I H 1995, 1, 287–306. [Google Scholar]
- McCubbin, F.M.; Hauri, E.H.; Elardo, S.M.; Vander Kaaden, K.E.; Wang, J.; Shearer, C.K., Jr. Hydrous melting of the martian mantle produced both depleted and enriched shergottites. Geology 2012, 40, 683–686. [Google Scholar] [CrossRef]
- Sarafian, A.R. Personal Communication; Woods Hole Oceanographic Institution: Woods Hole, MA, USA, 2016. [Google Scholar]
- Wudarska, A.; Słaby, E.; Wiedenbeck, M.; Barnes, J.D.; Bonifacie, M.; Sturchio, N.C.; Couffignal, F.; Glodny, J.; John, T.; Kusebauch, C.; et al. Inter-Laboratory Characterization of Apatite Reference Materials for Chlorine Isotope Analysis. Manuscript in preparation.
- Xiao, Y.K.; Yinming, Z.; Qingzhong, W.; Haizhen, W.; Weiguo, L.; Eastoe, C.J. A secondary isotopic reference material of chlorine from selected seawater. Chem. Geol. 2002, 182, 655–661. [Google Scholar] [CrossRef]
- Wei, H.-Z.; Jiang, S.-Y.; Xiao, Y.-K.; Wang, J.; Lu, H.; Wu, B.; Wu, H.-P.; Li, Q.; Luo, C.-G. Precise determination of the absolute isotopic abundance ratio and the atomic weight of chlorine in three international reference materials by the positive thermal ionization mass spectrometer-Cs2Cl+-graphite method. Anal. Chem. 2012, 84, 10350–10358. [Google Scholar] [CrossRef] [PubMed]
- Kempe, U.; Götze, J. Cathodoluminescence (CL) behaviour and crystal chemistry of apatite from rare-metal deposits. Mineral. Mag. 2002, 66, 151–172. [Google Scholar] [CrossRef]
- Gaft, M.; Reisfeld, R.; Panczer, G.; Blank, P.; Boulon, G. Laser-induced time-resolved luminescence of minerals. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1998, 54, 2163–2175. [Google Scholar] [CrossRef]
- Blanc, P.; Baumer, A.; Cesbron, F.; Ohnenstetter, D.; Panczer, G.; Rémond, G. Systematic cathodoluminescence spectral analysis of synthetic doped minerals: Anhydrite, apatite, calcite, fluorite, scheelite and zircon. In Cathodoluminescence in Geosciences; Springer: Berlin/Heidelberg, Germany, 2000; pp. 127–160. [Google Scholar]
- Putnis, A. Mineral replacement reactions: From macroscopic observations to microscopic mechanisms. Mineral. Mag. 2002, 66, 689–708. [Google Scholar] [CrossRef]
- Putnis, A. Mineral replacement reactions. Rev. Mineral. Geochem. 2009, 70, 87–124. [Google Scholar] [CrossRef]
- Kusebauch, C.; John, T.; Whitehouse, M.J.; Klemme, S.; Putnis, A. Distribution of halogens between fluid and apatite during fluid-mediated replacement processes. Geochim. Cosmochim. Acta 2015, 170, 225–246. [Google Scholar] [CrossRef]
- Brès, E.F.; Hutchison, J.L.; Voegel, J.-C.; Frank, R.M. Observation of a low angle grain boundary in tooth enamel crystals using HREM. Le J. Phys. Colloq. 1990, 51, C1-97. [Google Scholar] [CrossRef]
- Cherniak, D.J. Uranium and manganese diffusion in apatite. Chem. Geol. 2005, 219, 297–308. [Google Scholar] [CrossRef]
- Stormer, J.C.; Pierson, M.L.; Tacker, R.C. Variation of F and Cl X-ray intensity due to anisotropic diffusion in apatite during electron microprobe analysis. Am. Mineral. 1993, 78, 641–648. [Google Scholar]
- Gulbrandsen, R.A. Physical and chemical factors in the formation of marine apatite. Econ. Geol. 1969, 64, 365–382. [Google Scholar] [CrossRef]
- Krajewski, K.P.; van Cappellen, P.; Trichet, J.; Kuhn, O.; Lucas, J.; Martin-Algarra, A.; Prevot, L.; Tewari, V.C.; Gaspar, L.; Knight, R.I.; et al. Biological processes and apatite formation in sedimentary environments. Eclogae Geol. Helv. 1994, 87, 701–746. [Google Scholar]
- Ayers, J.C.; Watson, E.B. Apatite/fluid partitioning of rare-earth elements and strontium: Experimental results at 1.0 GPa and 1000 °C and application to models of fluid-rock interaction. Chem. Geol. 1993, 110, 299–314. [Google Scholar] [CrossRef]
- Chu, M.-F.; Wang, K.-L.; Griffin, W.L.; Chung, S.-L.; O’Reilly, S.Y.; Pearson, N.J.; Iizuka, Y. Apatite composition: Tracing petrogenetic processes in Transhimalayan granitoids. J. Petrol. 2009, 50, 1829–1855. [Google Scholar] [CrossRef]
- Rendon-Angeles, J.C.; Yanagisawa, K.; Ishizawa, N.; Oishi, S. Conversion of calcium fluorapatite into calcium hydroxyapatite under alkaline hydrothermal conditions. J. Solid State Chem. 2000, 151, 65–72. [Google Scholar] [CrossRef]
- Rendon-Angeles, J.C.; Yanagisawa, K.; Ishizawa, N.; Oishi, S. Effect of metal ions of chlorapatites on the topotaxial replacement by hydroxyapatite under hydrothermal conditions. J. Solid State Chem. 2000, 154, 569–578. [Google Scholar] [CrossRef]
- Harlov, D.E.; Forster, H.-J.; Nijland, T.G. Fluid-induced nucleation of (Y+REE)-phosphate minerals within apatite: Nature and experiment. Part I. Chlorapatite. Am. Mineral. 2002, 87, 245–261. [Google Scholar] [CrossRef]
- Harlov, D.E.; Forster, H.-J. Fluid-induced nucleation of (Y+REE)-phosphate minerals within apatite: Nature and experiment. Part II. Fluorapatite. Am. Mineral. 2003, 88, 1209–1229. [Google Scholar] [CrossRef]
- Elliott, J.C. Structure and Chemistry of the Apatites and other Calcium Orthophosphates; Elsevier: Amsterdam, The Netherland, 1994; pp. 1–389. [Google Scholar]
- Hughes, J.M.; Harlov, D.; Kelly, S.R.; Rakovan, J.; Wilke, M. Solid solution in the apatite OH-Cl binary system: Compositional dependence of solid-solution mechanisms in calcium phosphate apatites along the Cl-OH binary. Am. Mineral. 2016, 101, 1783–1791. [Google Scholar] [CrossRef]
- Hughes, J.M.; Harlov, D.; Rakovan, J.F. Structural variations along the apatite F-OH join. Am. Mineral. 2018, 103, 1981–1987. [Google Scholar] [CrossRef]
- Marks, M.A.W.; Wenzel, T.; Whitehouse, M.J.; Loose, M.; Zack, T.; Barth, M.; Worgard, L.; Krasz, V.; Eby, G.N.; Stosnach, H.; et al. The volatile inventory (F, Cl, Br, S, C) of magmatic apatite: An integrated analytical approach. Chem. Geol. 2012, 291, 241–255. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Host Rock 1,2 | Mineral Composition of Host Rock 1,2 | REE Profile of Apatite and Corresponding Apatite Origin 1,2 | Apatite Type 1 | Volatile Concentrations in Apatite [wt %] 1 | δDVSMOW [‰] in Apatite 1 | |||
|---|---|---|---|---|---|---|---|---|---|
| Major Phases | Minor Phases | [Cl] | [F] | [H2O] 3 | |||||
| AL4-1 | banded iron formation | quartz, magnetite, cummingtonite | stilpnomelane, calcite, apatite, pyrite | flat (sedimentary) or LREE-depleted (mafic) | --- | nd 4 | nd | nd | nd |
| AL10-1 | banded iron formation | quartz, magnetite, cummingtonite | pyrite, Fe-rich dolomite, apatite, monazite | MREE-enriched (metasomatic) | hydroxylapatite | 1.38 ± 0.06 | 1.20 ± 0.14 | 0.83 ± 0.07 | −57 |
| AL10-2 | banded iron formation | quartz, magnetite, actinolite | pyrite, Fe-rich dolomite, apatite | flat (sedimentary) | hydroxylapatite | 1.76 ± 0.06 | 1.06 ± 0.14 | 0.81 ± 0.06 | −56 |
| AL13 | banded iron formation | quartz, magnetite, cummingtonite | Fe-rich dolomite, apatite | MREE-enriched (metasomatic) | hydroxylapatite | 1.17 ± 0.25 | 1.51 ± 0.14 | 0.74 ± 0.06 | −71 |
| AL15B | banded iron formation | grunerite, magnetite, quartz | actinolite, chlorite, apatite | flat (sedimentary) | fluorapatite | 0.16 ± 0.02 | 2.82 ± 0.16 | 0.41 ± 0.07 | −62 |
| AL35-6 | banded iron formation | quartz, magnetite | Fe-rich dolomite, apatite | flat (sedimentary) | hydroxylapatite | 0.06 ± 0.02 | 1.70 ± 0.18 | 0.95 ± 0.09 | −87 |
| AL26 | metachert | quartz | magnetite, apatite, cummingtonite, pyrite | MREE-enriched (metasomatic), flat (sedimentary) or LREE-depleted (mafic) | --- | nd | nd | nd | nd |
| AL8-1 | metacarbonate (carbonate-rich layer in mafic rock) | chlorite, magnetite, Mg–Mn-rich siderite, Fe-rich dolomite, cummingtonite | quartz, pyrite, graphite, apatite | MREE-enriched (metasomatic) | fluorapatite | 0.73 ± 0.04 | 2.19 ± 0.19 | 0.55 ± 0.08 | −93 |
| AL17 | metacarbonate (carbonate-rich layer in metachert) | quartz, Fe-rich dolomite, calcite, cummingtonite | graphite, chlorite, stilpnomelane, apatite | MREE-enriched (metasomatic) | fluorapatite | 0.64 ± 0.16 | 2.07 ± 0.28 | 0.62 ± 0.10 | −60 |
| AL1-2 | mafic dyke | chlorite, quartz, stilpnomelane | calcite, titanite, allanite, monazite, apatite, zircon | LREE-depleted (magmatic) | --- | nd | nd | nd | nd |
| AL21-2 | mafic dyke | chlorite, actinolite-hornblende series, quartz | pyrite, apatite, titanite, zircon | LREE-depleted (magmatic) | hydroxylapatite | 1.49 ± 0.13 | 1.13 ± 0.12 | 0.85 ± 0.07 | −69 |
| Reference Material | Cl Content (wt.%) | Recommended δ37ClSMOC (‰) 3 |
|---|---|---|
| Eagle County (Colorado, USA) 1 | 0.95 ± 0.03 (1 s) | 0.22 |
| TUBAF#37 (Bamble, Norway) 2 | 0.26 ± 0.04 (1 s) | 0.20 ± 0.13 (1 s) |
| TUBAF#50 (Spain) 2 | 0.56 ± 0.07 (1 s) | 0.32 ± 0.25 (1 s) |
| TUBAF#40 (Kragerø, Norway) 2 | 1.37 ± 0.11 (1 s) | 0.20 ± 0.24 (1 s) |
| Sample ID | Rock Type | Mean δ37ClSMOC [‰] | δ37ClSMOC Value Range [‰] | Number of Crystals | Number of Measurements |
|---|---|---|---|---|---|
| AL10-2 | BIF | 1.40 | 1.18–1.60 | 18 | 20 |
| AL13 | BIF | 0.48 | 0.00–0.69 | 13 | 27 |
| AL15B | BIF | −0.34 | −0.82–0.16 | 15 | 15 |
| AL8-1 | metacarbonate | 0.32 | 0.16–0.48 | 14 | 15 |
| AL17 | metacarbonate | 0.29 | −0.08–0.68 | 27 | 37 |
| AL21-2 | mafic dyke | 0.17 | −0.10–0.38 | 12 | 25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wudarska, A.; Słaby, E.; Wiedenbeck, M.; Birski, Ł.; Wirth, R.; Götze, J.; Lepland, A.; Kusebauch, C.; Kocjan, I. Chlorine Isotope Composition of Apatite from the >3.7 Ga Isua Supracrustal Belt, SW Greenland. Minerals 2020, 10, 27. https://doi.org/10.3390/min10010027
Wudarska A, Słaby E, Wiedenbeck M, Birski Ł, Wirth R, Götze J, Lepland A, Kusebauch C, Kocjan I. Chlorine Isotope Composition of Apatite from the >3.7 Ga Isua Supracrustal Belt, SW Greenland. Minerals. 2020; 10(1):27. https://doi.org/10.3390/min10010027
Chicago/Turabian StyleWudarska, Alicja, Ewa Słaby, Michael Wiedenbeck, Łukasz Birski, Richard Wirth, Jens Götze, Aivo Lepland, Christof Kusebauch, and Izabela Kocjan. 2020. "Chlorine Isotope Composition of Apatite from the >3.7 Ga Isua Supracrustal Belt, SW Greenland" Minerals 10, no. 1: 27. https://doi.org/10.3390/min10010027
APA StyleWudarska, A., Słaby, E., Wiedenbeck, M., Birski, Ł., Wirth, R., Götze, J., Lepland, A., Kusebauch, C., & Kocjan, I. (2020). Chlorine Isotope Composition of Apatite from the >3.7 Ga Isua Supracrustal Belt, SW Greenland. Minerals, 10(1), 27. https://doi.org/10.3390/min10010027