
On the Charge Density Refinement of Odd-Order Multipoles Invariant under Crystal Point Group Symmetry



{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Structure Factor
- The Isotropy group of is the subgroup of G containing the elements which map x onto itself:
- The Orbit of under G, is the subset of X consisting of all elements with g running through G:
2.2. The Multipolar Structure Factor
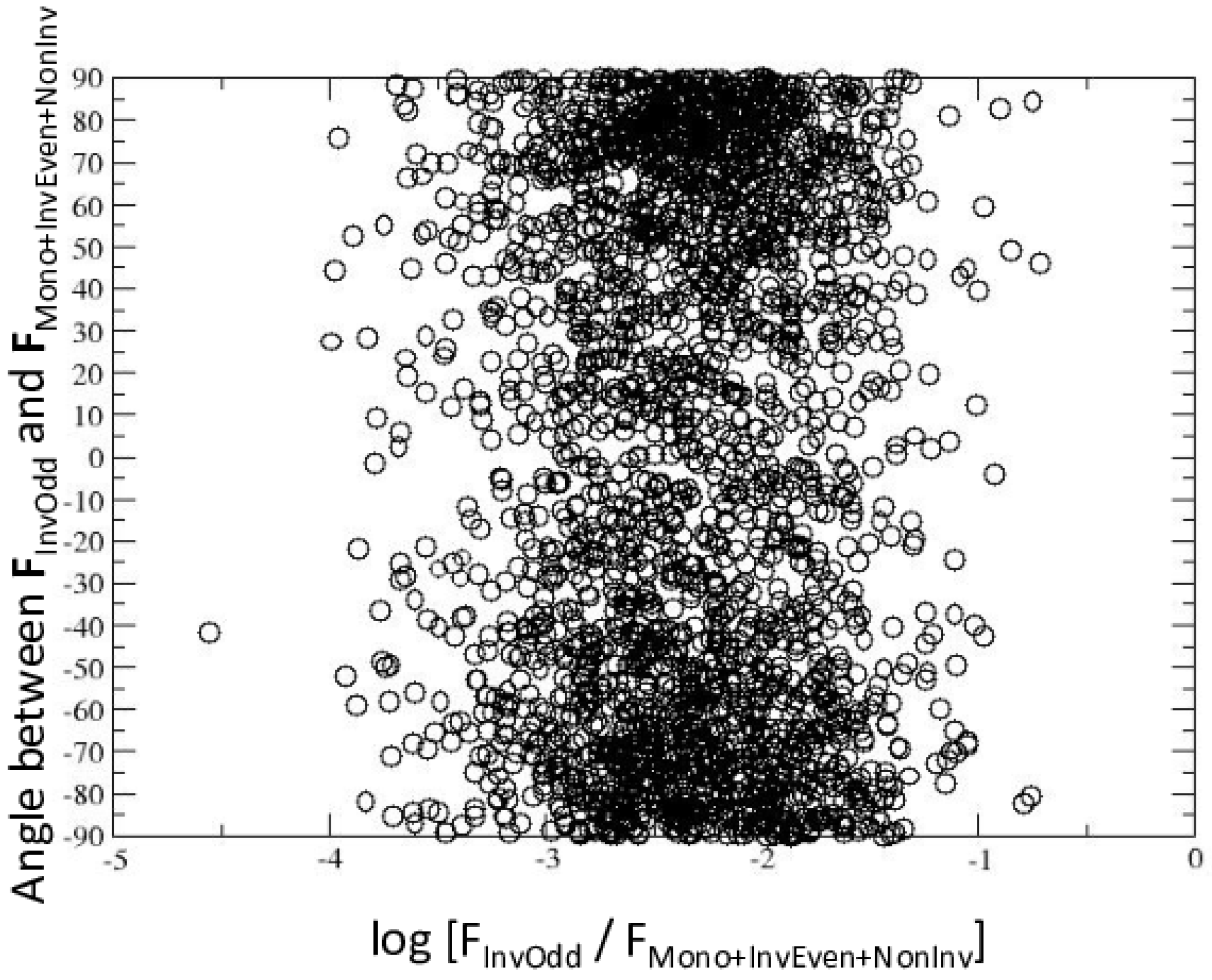
2.3. Multipoles Invariant under Point-Group Symmetry
3. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schmidt, A.; Jelsch, C.; Ostergaard, P.; Rypniewski, W.; Lamzin, V.S. Trypsin revisited: Crystallography at (sub)atomic resolution and quantum chemistry revealing details of catalysis. J. Biol. Chem. 2003, 278, 43357–43362. [Google Scholar] [CrossRef] [PubMed]
- Guillot, B.; Jelsch, C.; Podjarny, A.; Lecomte, C. Charge-density analysis of a protein structure at subatomic resolution: The human aldose reductase case. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 567–588. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Bendeif, E.E.; Guillot, B.; Podjarny, A.; Lecomte, C.; Jelsch, C. Charge density and electrostatic interactions of fidarestat, an inhibitor of human aldose reductase. J. Am. Chem. Soc. 2009, 131, 10929–10941. [Google Scholar] [CrossRef] [PubMed]
- Zarychta, B.; Lyubimov, A.; Ahmed, M.; Munshi, P.; Guillot, B.; Vrielink, A.; Jelsch, C. Cholesterol oxidase: Ultrahigh-resolution crystal structure and multipolar atom model-based analysis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Takeda, K.; Miki, K. Charge-density analysis of an iron-sulfur protein at an ultra-high resolution of 0.48 Å. Nature 2016, 534, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.F. Electron population parameters with rigid pseudoatoms. Acta Crystallogr. 1976, A32, 565–574. [Google Scholar] [CrossRef]
- Hansen, N.K.; Coppens, P. Testing aspherical atoms sefinements on small–molecule data sets. Acta Crystallogr. 1978, A34, 909–921. [Google Scholar] [CrossRef]
- Yeates, T.O.; Kent, S.B.H. Racemic protein crystallography. Annu. Rev. Biophys. 2012, 41, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, M.; Craven, B.M.; Stewart, R. Hexamethylenetetramine at 298 K: New refinements. Acta Crystallogr. 1993, A49, 685–692. [Google Scholar] [CrossRef]
- Takata, M.; Kubota, Y.; Sakata, M. The electron density distribution in be metal obtained from synchrotron–radiation powder data by the maximum–entropy method. Z. Naturforsch. A Phys. Sci. 1993, 48, 75–80. [Google Scholar] [CrossRef]
- El Haouzi, A.; Hansen, N.; Le Hénaff, C.; Protas, J. The phase problem in the analysis of X–ray diffraction data in terms of electron–density distributions. Acta Crystallogr. 1996, A52, 291–301. [Google Scholar] [CrossRef]
- Van Beek, C.; Overeem, J.; Ruble, J.; Craven, B. Electrostatic properties of ammonium fluoride and deuterated ice–Ih. Can. J. Chem. 1996, 74, 943–950. [Google Scholar] [CrossRef]
- Bricogne, G. Fourier Transforms in crystallography: Theory, algorithms, and applications. In International Tables for Crystallography; Reciprocal Space; Shmueli, U., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1993; Volume B, pp. 23–106. [Google Scholar]
- Stewart, R.F.; Spackman, M. VALRAY Users Manual, 1st ed.; Carnegie–Mellon University: Pittsburgh, PA, USA, 1983. [Google Scholar]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in protein and small-molecule charge-density refinement methods using MoPro. J. Appl. Crystallogr. 2005, 38, 38–54. [Google Scholar] [CrossRef]
- Coppens, P. The structure factor. In International Tables for Crystallography; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; Volume B, pp. 10–22. [Google Scholar]
- Altmann, S.L.; Bradley, C.J. On the symmetries of spherical harmonics. Philos. Trans. R. Soc. Lond. 1963, 255, 199–215. [Google Scholar] [CrossRef]
- Janssen, T. Representations of crystallographic groups. In International Tables for Crystallography; Physical properties of crystals; Authier, A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2006; Volume D, pp. 34–71. [Google Scholar]
- Coppens, P. The structure factor. In International Tables for Crystallography; Reciprocal Space; Shmueli, U., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1993; Volume B, pp. 10–23. [Google Scholar]
- Becka, L.; Cruickshank, D. The crystal structure of hexamethylenetetramine. II. The lattice vibrations of a simple molecular crystal. Proc. R. Soc. Lond. 1963, A273, 455–465. [Google Scholar] [CrossRef]
- Stevens, E.; Hope, H. Accurate positional and thermal parameters of hexamethylenetetramine from K–shell X–ray diffraction data. Acta Crystallogr. 1975, A31, 494–498. [Google Scholar] [CrossRef]
- Kampermann, S.; Craven, B.M.; Ruble, J.R. The charge density distribution in hexamethylenetetramine at 120 K. Acta Crystallogr. 1994, B50, 737–741. [Google Scholar] [CrossRef]
- Levy, H.A.; Corey, R.B. The crystal structure of dl-alanine. J. Am. Chem. Soc. 1941, 63, 2095–2108. [Google Scholar] [CrossRef]
- Donohue, J. The crystal structure of dl-alanine. II. Revision of parameters by three-dimensional Fourier analysis. J. Am. Chem. Soc. 1950, 72, 949–953. [Google Scholar] [CrossRef]
- Simpson, H.J.; Marsh, R.E. The crystal structure of l-alanine. Acta Crystallogr. 1966, 20, 550–555. [Google Scholar] [CrossRef]
- Destro, R.; Soave, R.; Barzaghi, M. Physicochemical properties of zwitterionic l- and dl-alanine crystals from their experimental and theoretical charge densities. J. Phys. Chem. B 2008, 112, 5163–5174. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Guillot, B.; Ruiz-Lopez, M.F.; Genoni, A. Libraries of extremely localized molecular orbitals. 1. Model molecules approximation and molecular orbitals transferability. J. Chem. Theory Comput. 2016, 12, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Guillot, B.; Ruiz-Lopez, M.F.; Jelsch, C.; Genoni, A. Libraries of extremely localized molecular orbitals. 2. Comparison with the pseudoatoms transferability. J. Chem. Theory Comput. 2016, 12, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roversi, P.; Destro, R. On the Charge Density Refinement of Odd-Order Multipoles Invariant under Crystal Point Group Symmetry. Symmetry 2017, 9, 63. https://doi.org/10.3390/sym9050063
Roversi P, Destro R. On the Charge Density Refinement of Odd-Order Multipoles Invariant under Crystal Point Group Symmetry. Symmetry. 2017; 9(5):63. https://doi.org/10.3390/sym9050063
Chicago/Turabian StyleRoversi, Pietro, and Riccardo Destro. 2017. "On the Charge Density Refinement of Odd-Order Multipoles Invariant under Crystal Point Group Symmetry" Symmetry 9, no. 5: 63. https://doi.org/10.3390/sym9050063
APA StyleRoversi, P., & Destro, R. (2017). On the Charge Density Refinement of Odd-Order Multipoles Invariant under Crystal Point Group Symmetry. Symmetry, 9(5), 63. https://doi.org/10.3390/sym9050063