Facile and Convenient One-Pot Process for the Synthesis of Spirooxindole Derivatives in High Optical Purity Using (−)-(S)-Brevicolline as an Organocatalyst



Abstract
:
1. Introduction
2. Results and Discussion
3. Experimental
3.1. General Data
3.2. General Procedure for Preparing of Compounds (5)
4. Conclusions
Acknowledgements
References and Notes
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Baden-Württemberg, Germany, 2005; p. 440. [Google Scholar]
- Dondoni, A.; Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef] [PubMed]
- Carlos, F.B., III. Organocatalysis Lost: Modern Chemistry, Ancient Chemistry, and an Unseen Biosynthetic Apparatus. Angew. Chem. Int. Ed. 2007, 47, 42–47. [Google Scholar]
- Dalko, P.I.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric Aminocatalysis-Gold Rush in Organic Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Asymmetric Construction of Stereogenic Carbon Centers Featuring a Trifluoromethyl Group from Prochiral Trifluoromethylated Substrates. Chem. Rev. 2011, 111, 455–529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W. Novel Syntheses of Bridge-Containing Organic Compounds. Chem. Rev. 2010, 110, 1706–1745. [Google Scholar] [CrossRef] [PubMed]
- Kizirian, J.-C. Chiral Tertiary Diamines in Asymmetric Synthesis. Chem. Rev. 2008, 108, 140–205. [Google Scholar] [CrossRef] [PubMed]
- France, S.; Guerin, D.J.; Miller, S.J.; Lectka, T. Nucleophilic Chiral Amines as Catalysts in Asymmetric Synthesis. Chem. Rev. 2003, 103, 2985–3012. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Marcelli, T.; van Maarseveen, J.H.; Hiemstra, H. Cupreines and Cupreidines: An Emerging Class of Bifunctional Cinchona Organocatalysts. Angew. Chem. Int. Ed. 2006, 45, 7496–7504. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-B.; Wu, Z.-J.; Pei, Q.-L.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Highly enantioselective construction of spiro [4H-pyran-3,3′-oxindoles] through a domino Knoevenagel/Michael/Cyclization sequence catalyzed by cupreine. Org. Lett. 2010, 12, 3132–3135. [Google Scholar] [CrossRef] [PubMed]
- Farina, V.; Reeves, J.T.; Senanayake, C.H.; Song, J.J. Asymmetric Synthesis of Active Pharmaceutical Ingredients. Chem. Rev. 2006, 106, 2734–2793. [Google Scholar] [CrossRef]
- Vember, P.A.; Terenteva, I.V.; Lazurevskij, G. The structure of brevikolline. Khim. Prir. Soedin. 1967, 3, 249. [Google Scholar]
- Terenteva, I.V.; Lazurevskij, G.V.; Shirshova, T.I. About the structure of brevikarine. Khim. Prir. Soedin. 1969, 5, 397. [Google Scholar]
- Vember, P.A.; Terenteva, I.V.; Uljanova, A.V. About the structure of brevikolline. Khim. Prir. Soedin. 1968, 4, 98. [Google Scholar]
- The ee measurement was confirmed by HPLC using Chiralpak IA [n-hexane/i-PrOH = 80/20, 1.0 mL/min, t = 5.536 min].
- Gaunt, M.J.; Johansson, C.C. Recent Developments in the Use of Catalytic Asymmetric Ammonium Enolates in Chemical Synthesis. Chem. Rev. 2007, 107, 5596–5605. [Google Scholar] [CrossRef] [PubMed]
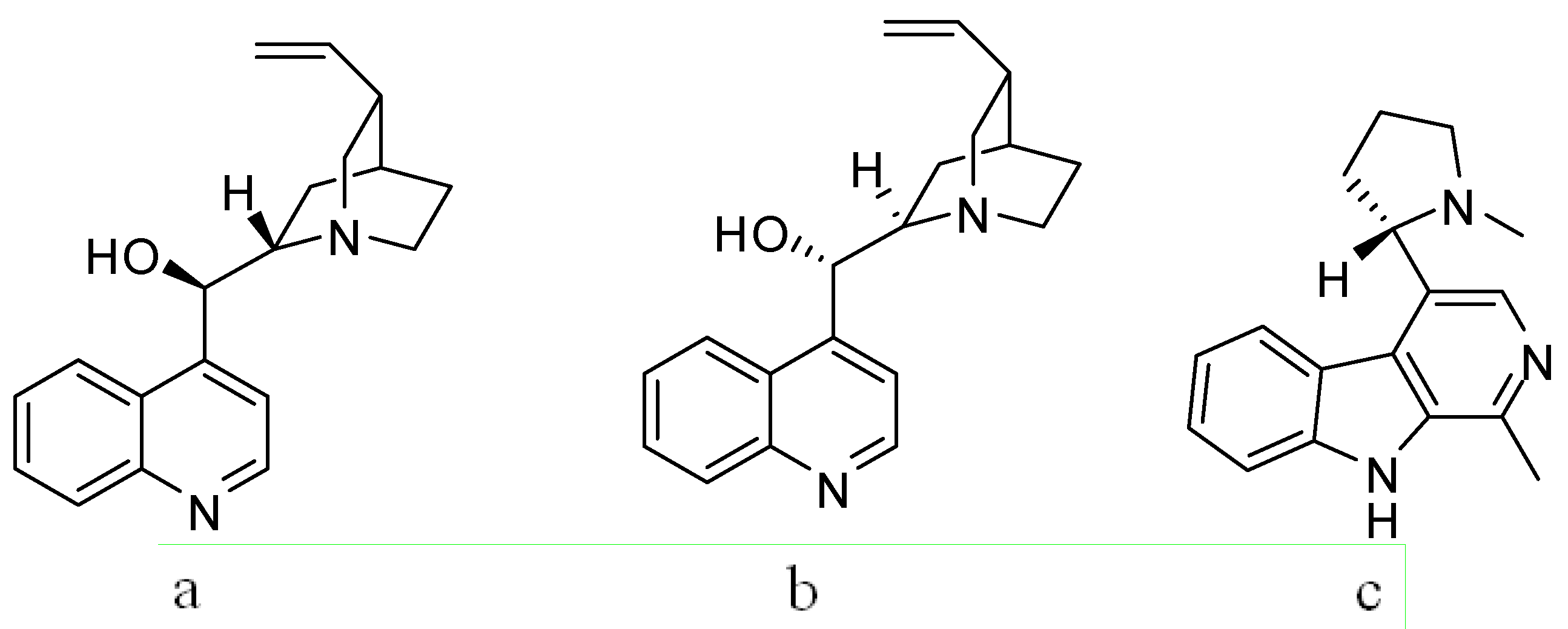
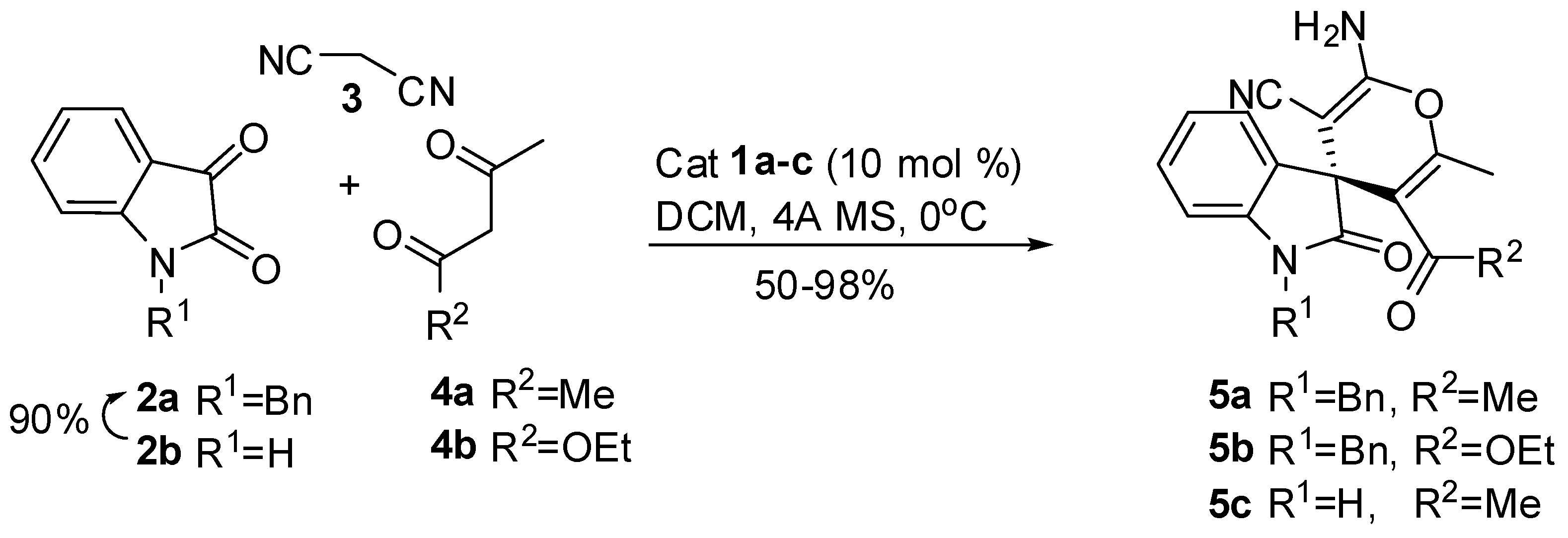

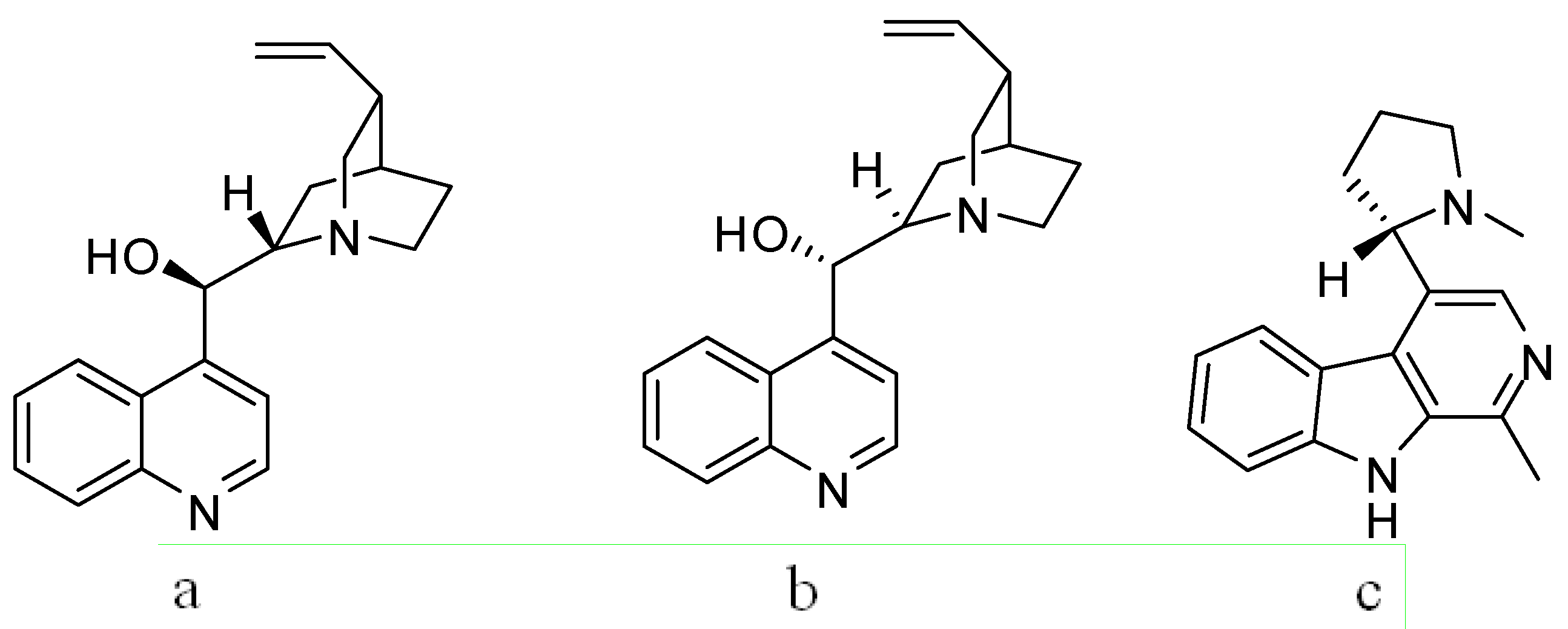
{kind=link}
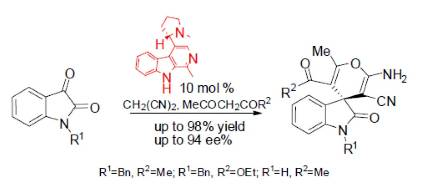
{kind=link}
{kind=link}
| Entry | Catalyst | Isatine | Dicarbonyl compound | Product | aYield (%) | [α]D | b/cee (%) |
|---|---|---|---|---|---|---|---|
| 1 | 1a | 2a | 4a | 5a | 80 | +2 (c 0.35) | b23 |
| 2 | 1b | 2a | 4a | 5a | 77 | +1.8 (c 0.84) | b21/ |
| e15 | |||||||
| 3 | 1c | 2a | 4a | 5a | 50 | +3.7 (c 0.6) | b43 |
| 4 | 1a | 2a | 4b | 5b | 87 | +2.27 (c 0.9) | c8 |
| 5 | 1b | 2a | 4b | 5b | 98 | +0.98 (c 1.16) | c3 |
| 6 | 1c | 2a | 4b | 5b | 68 | +3.5 (c 0.33) | c12 |
| 7 | 1a | 2b | 4a | 5c | 60 | +6.32 (c 0.17) | d22 |
| 8 | 1b | 2b | 4a | 5c | 67 | +2.16 (c 0.35) | e8 |
| 9 | 1c | 2b | 4a | 5c | 62 | +46.8 (c 0.12) | e94 |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macaev, F.; Sucman, N.; Shepeli, F.; Zveaghintseva, M.; Pogrebnoi, V. Facile and Convenient One-Pot Process for the Synthesis of Spirooxindole Derivatives in High Optical Purity Using (−)-(S)-Brevicolline as an Organocatalyst. Symmetry 2011, 3, 165-170. https://doi.org/10.3390/sym3020165
Macaev F, Sucman N, Shepeli F, Zveaghintseva M, Pogrebnoi V. Facile and Convenient One-Pot Process for the Synthesis of Spirooxindole Derivatives in High Optical Purity Using (−)-(S)-Brevicolline as an Organocatalyst. Symmetry. 2011; 3(2):165-170. https://doi.org/10.3390/sym3020165
Chicago/Turabian StyleMacaev, Fliur, Natalia Sucman, Felix Shepeli, Marina Zveaghintseva, and Vsevolod Pogrebnoi. 2011. "Facile and Convenient One-Pot Process for the Synthesis of Spirooxindole Derivatives in High Optical Purity Using (−)-(S)-Brevicolline as an Organocatalyst" Symmetry 3, no. 2: 165-170. https://doi.org/10.3390/sym3020165
APA StyleMacaev, F., Sucman, N., Shepeli, F., Zveaghintseva, M., & Pogrebnoi, V. (2011). Facile and Convenient One-Pot Process for the Synthesis of Spirooxindole Derivatives in High Optical Purity Using (−)-(S)-Brevicolline as an Organocatalyst. Symmetry, 3(2), 165-170. https://doi.org/10.3390/sym3020165