Computed NMR Shielding Effects over Fused Aromatic / Antiaromatic Hydrocarbons

Abstract
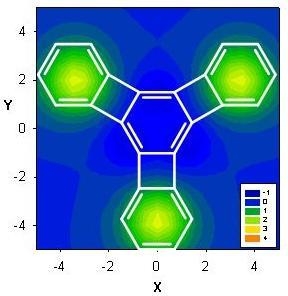
:
1. Introduction
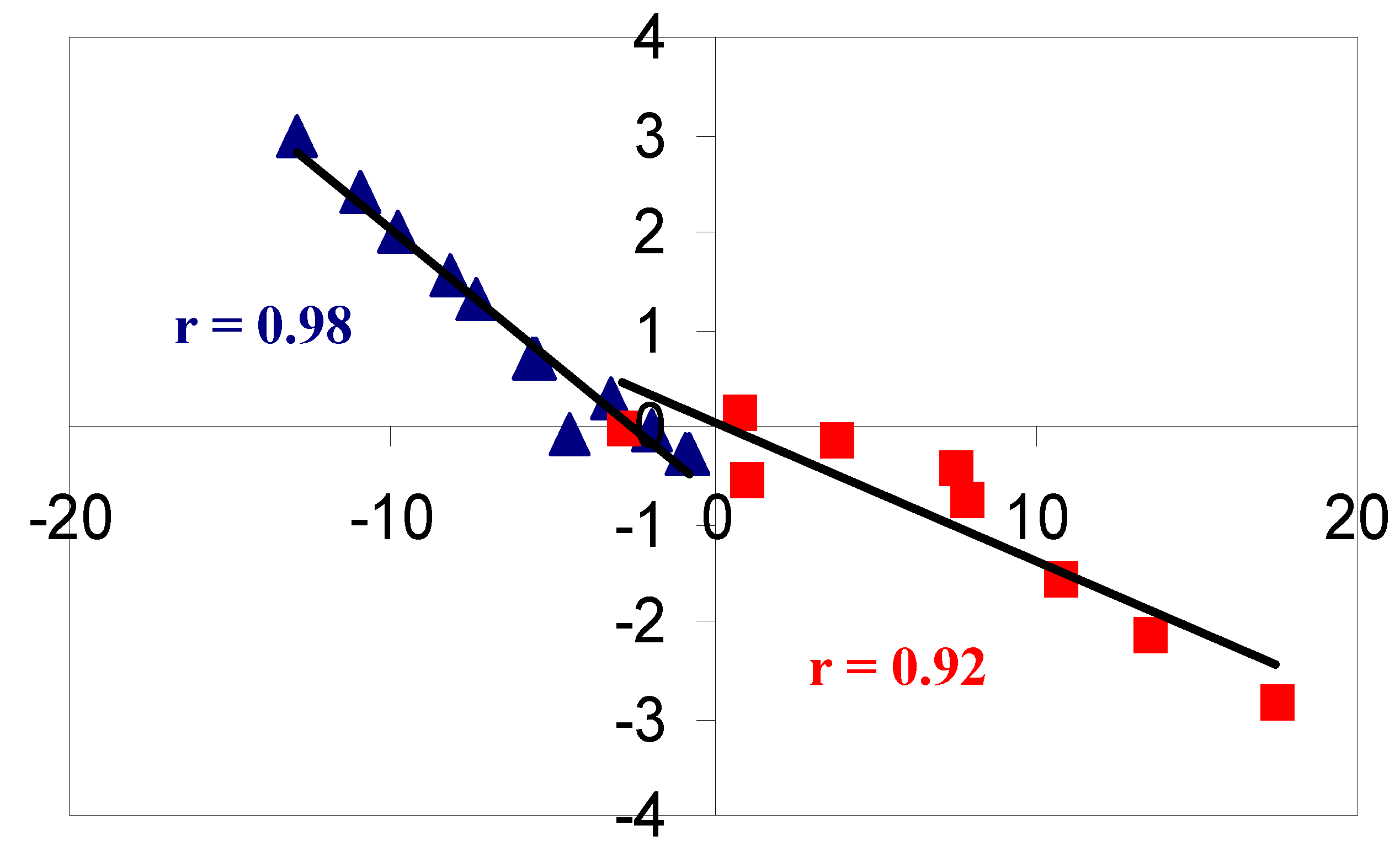
2. Results and Discussion
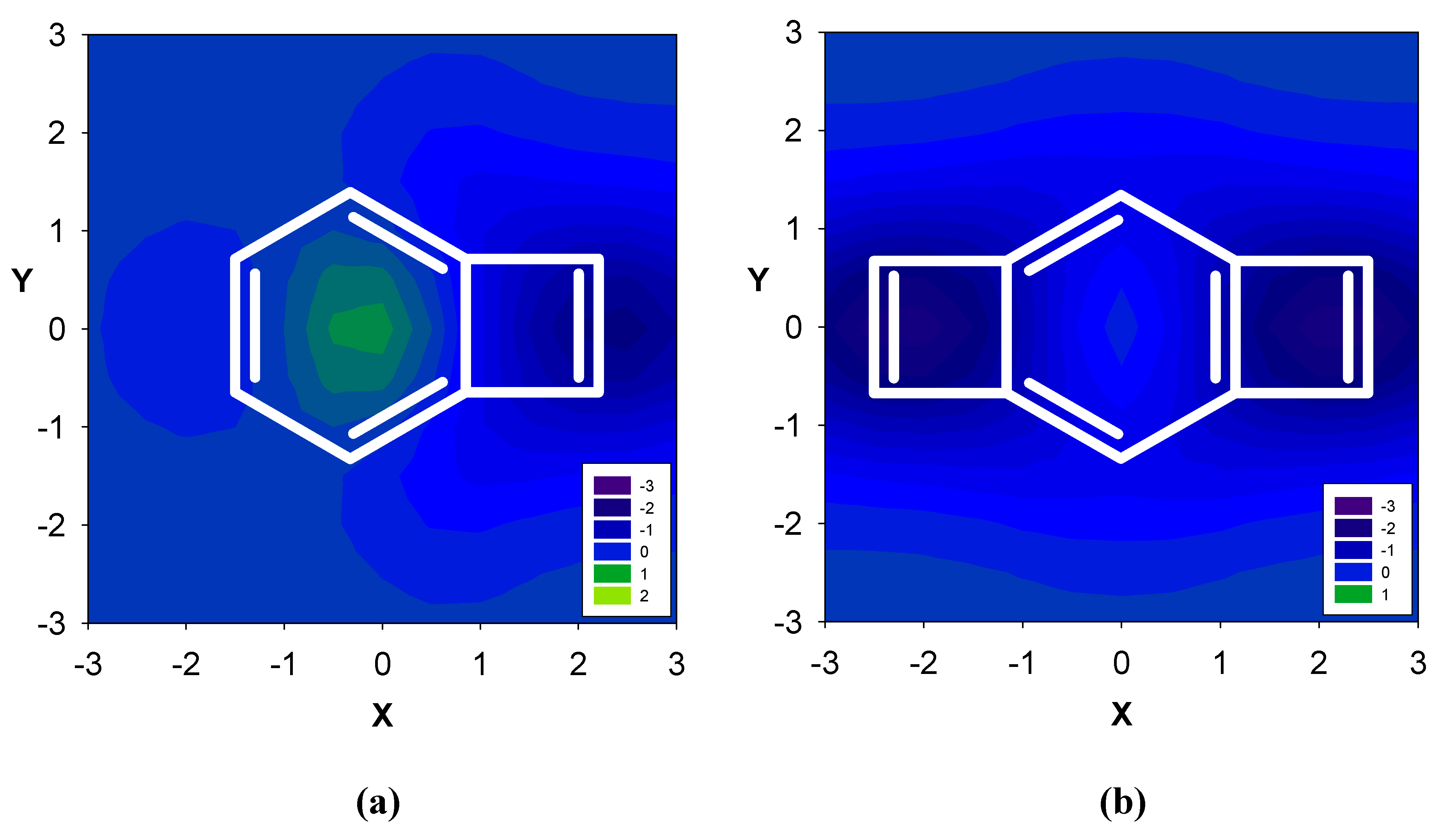
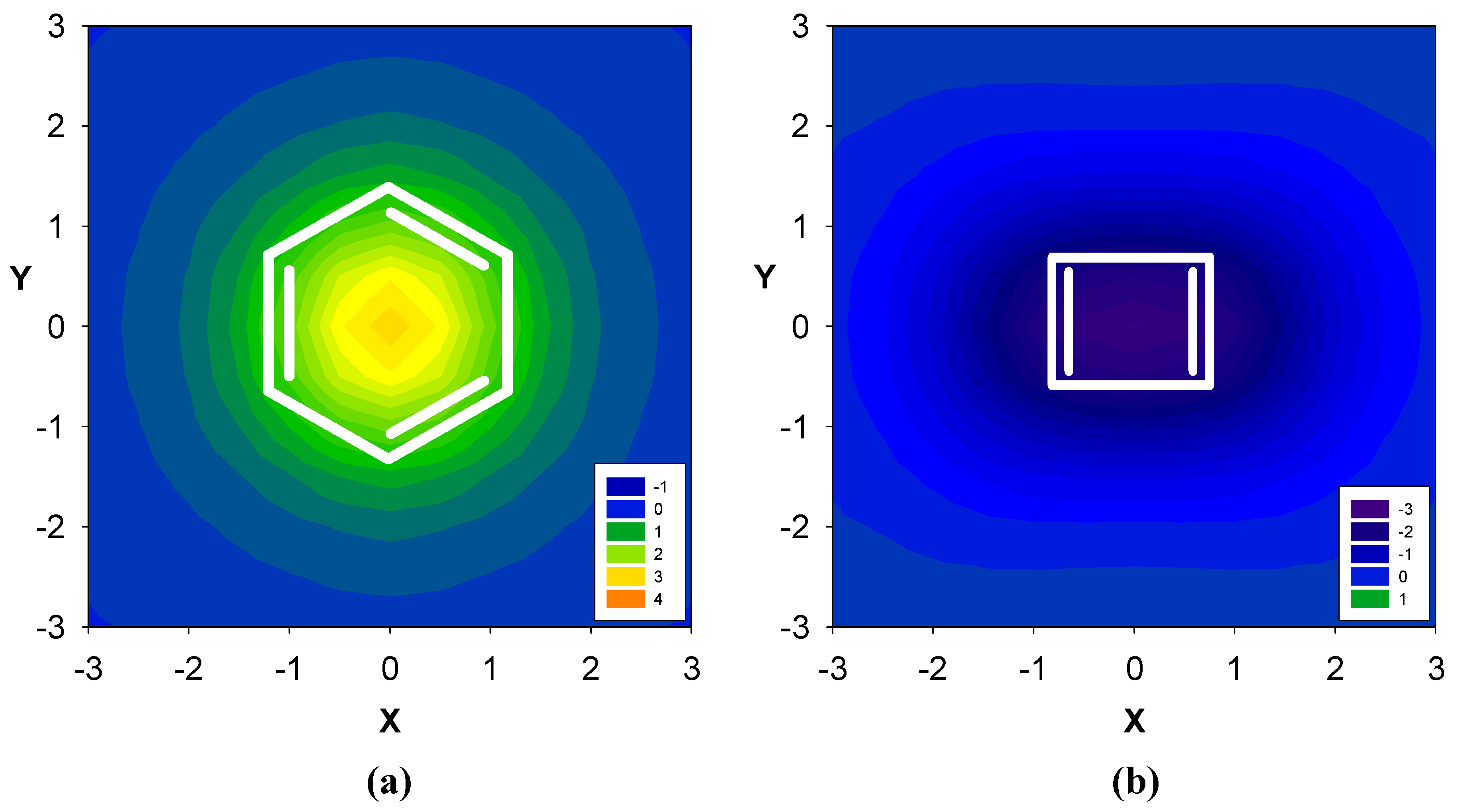
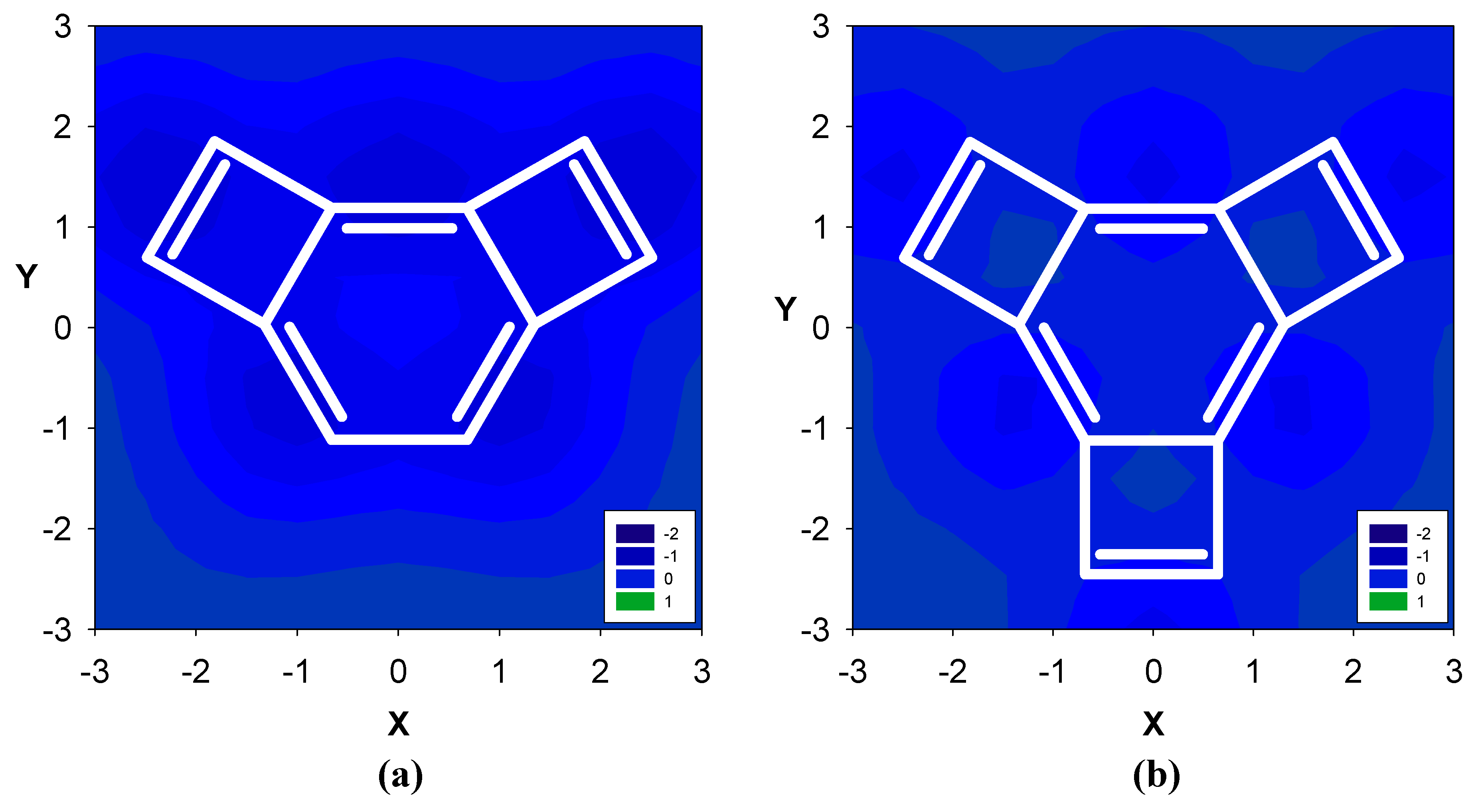
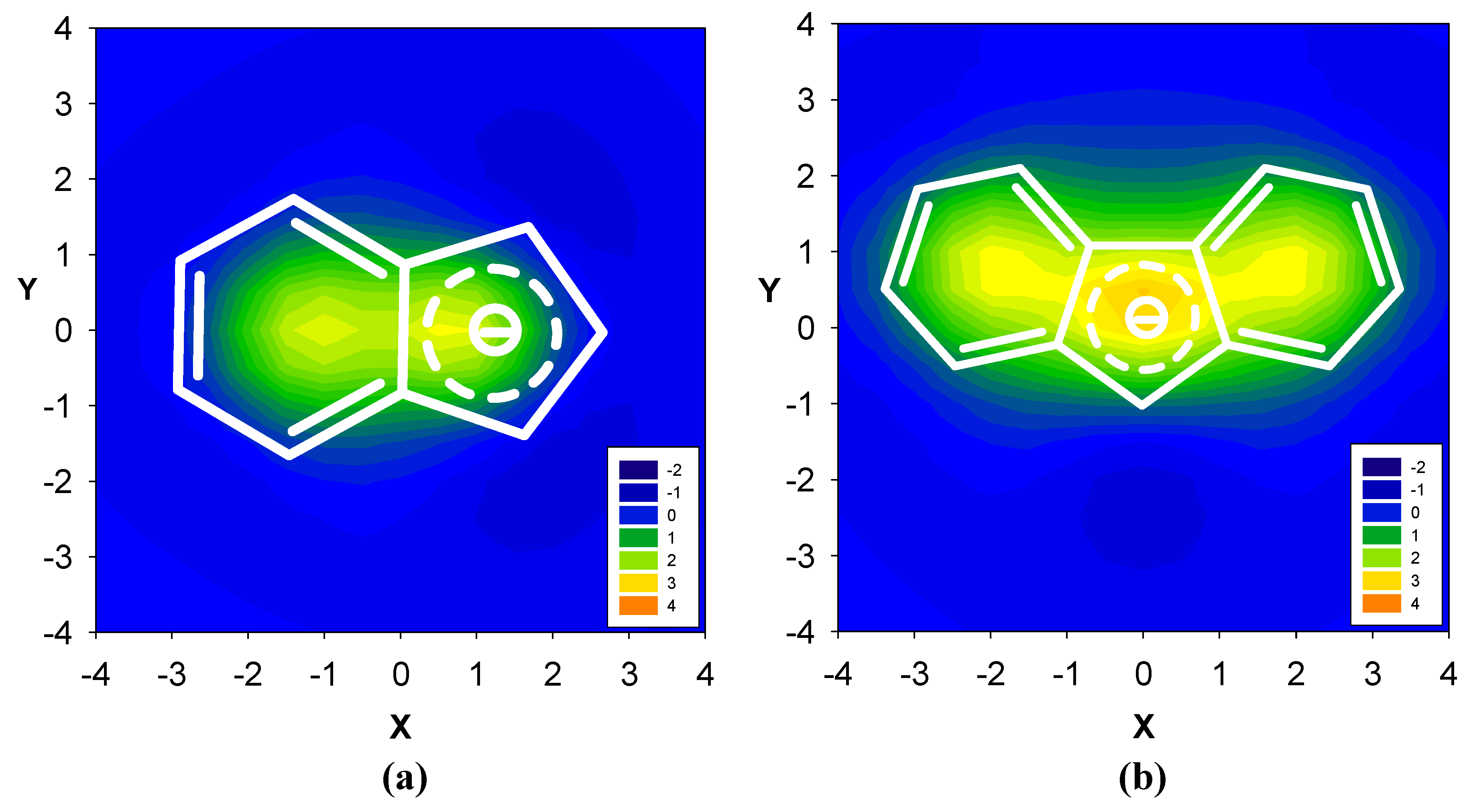
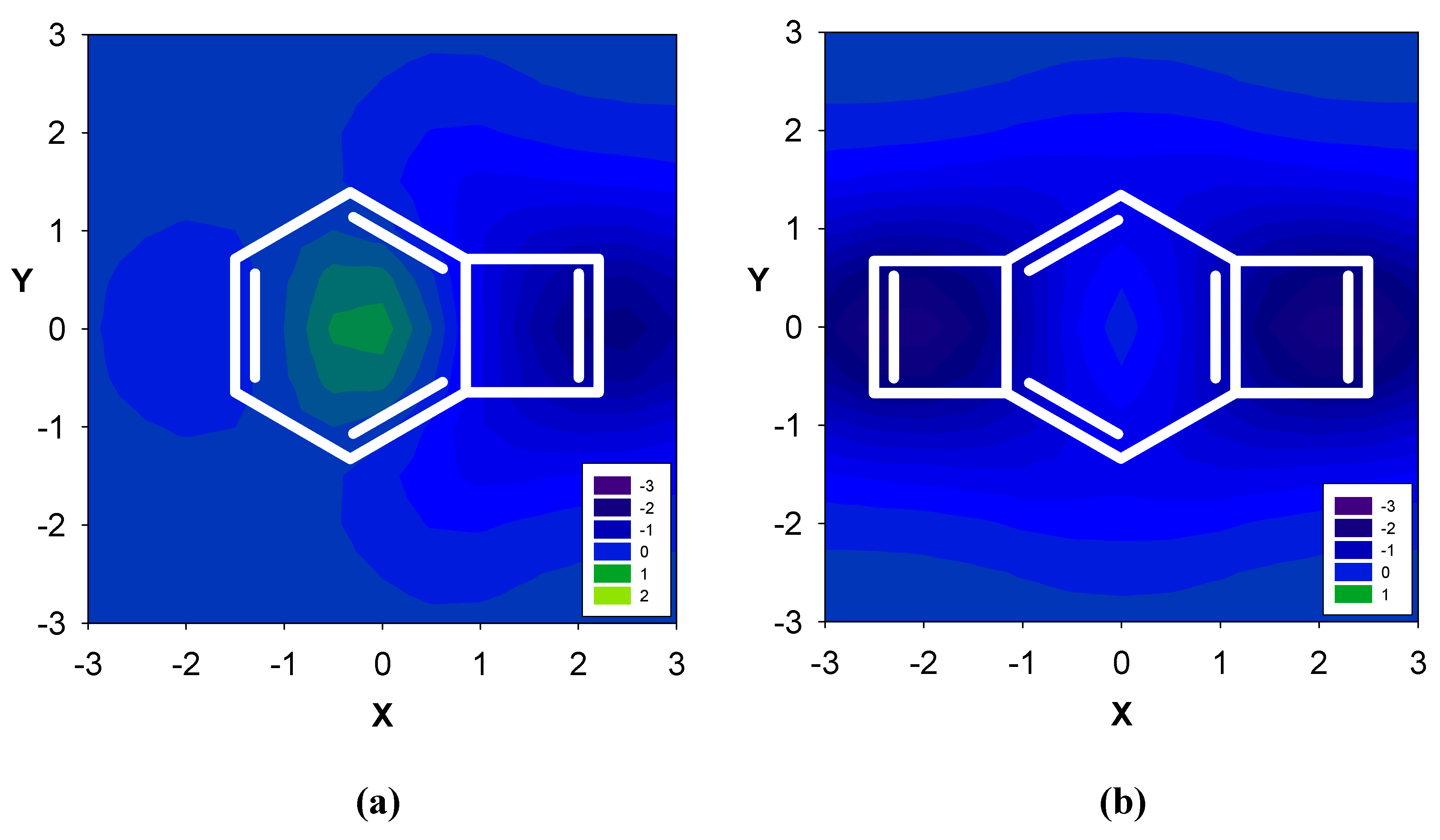
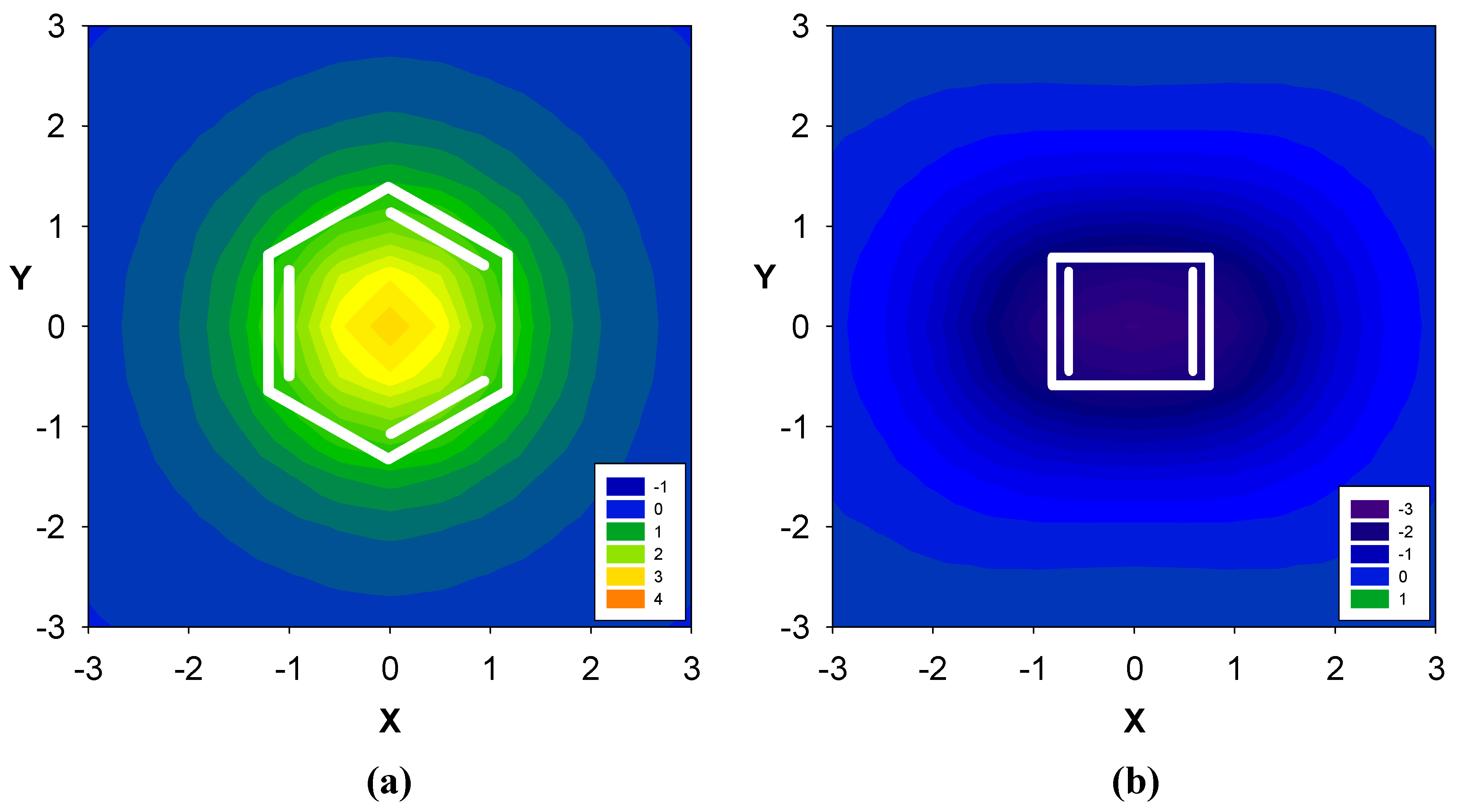
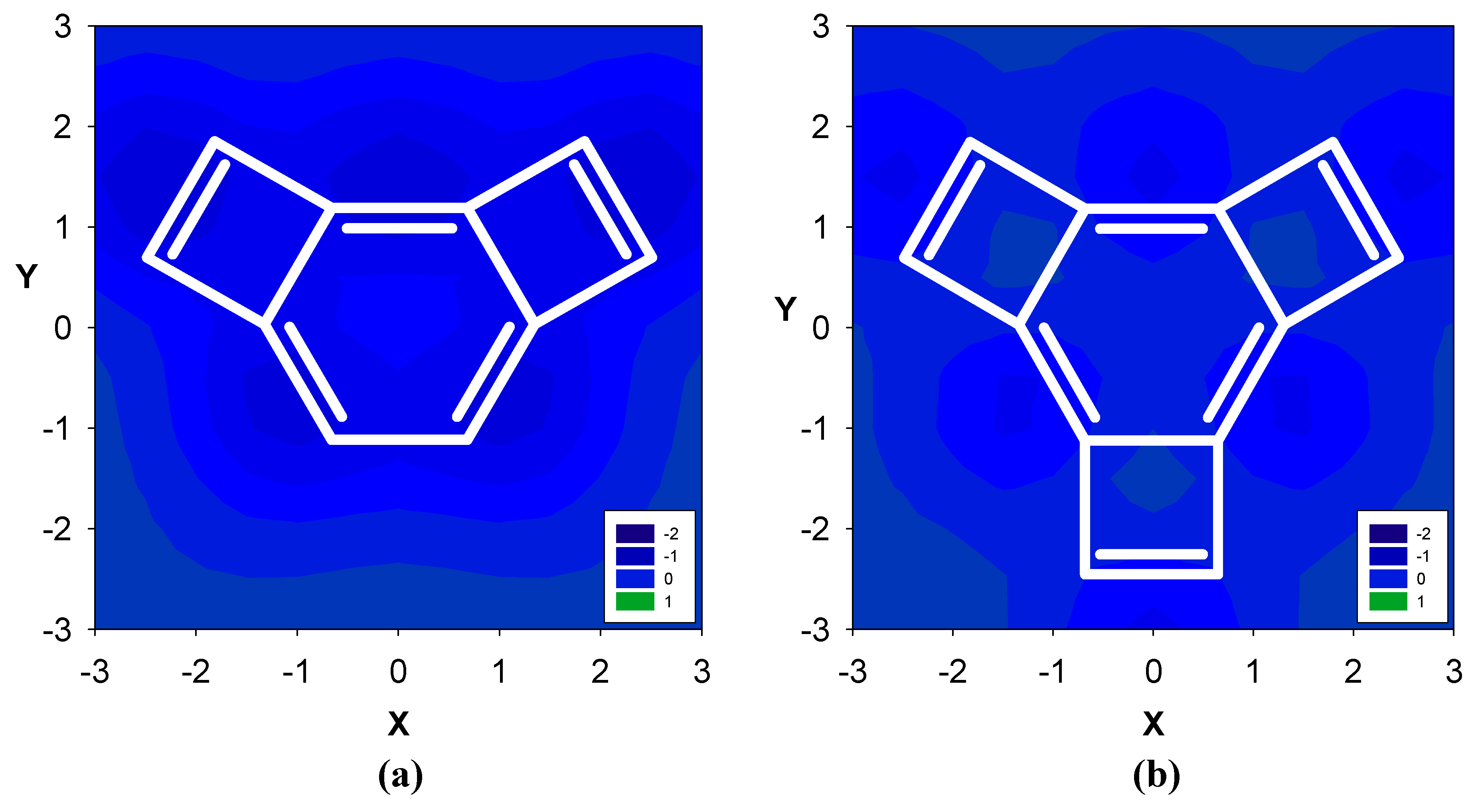
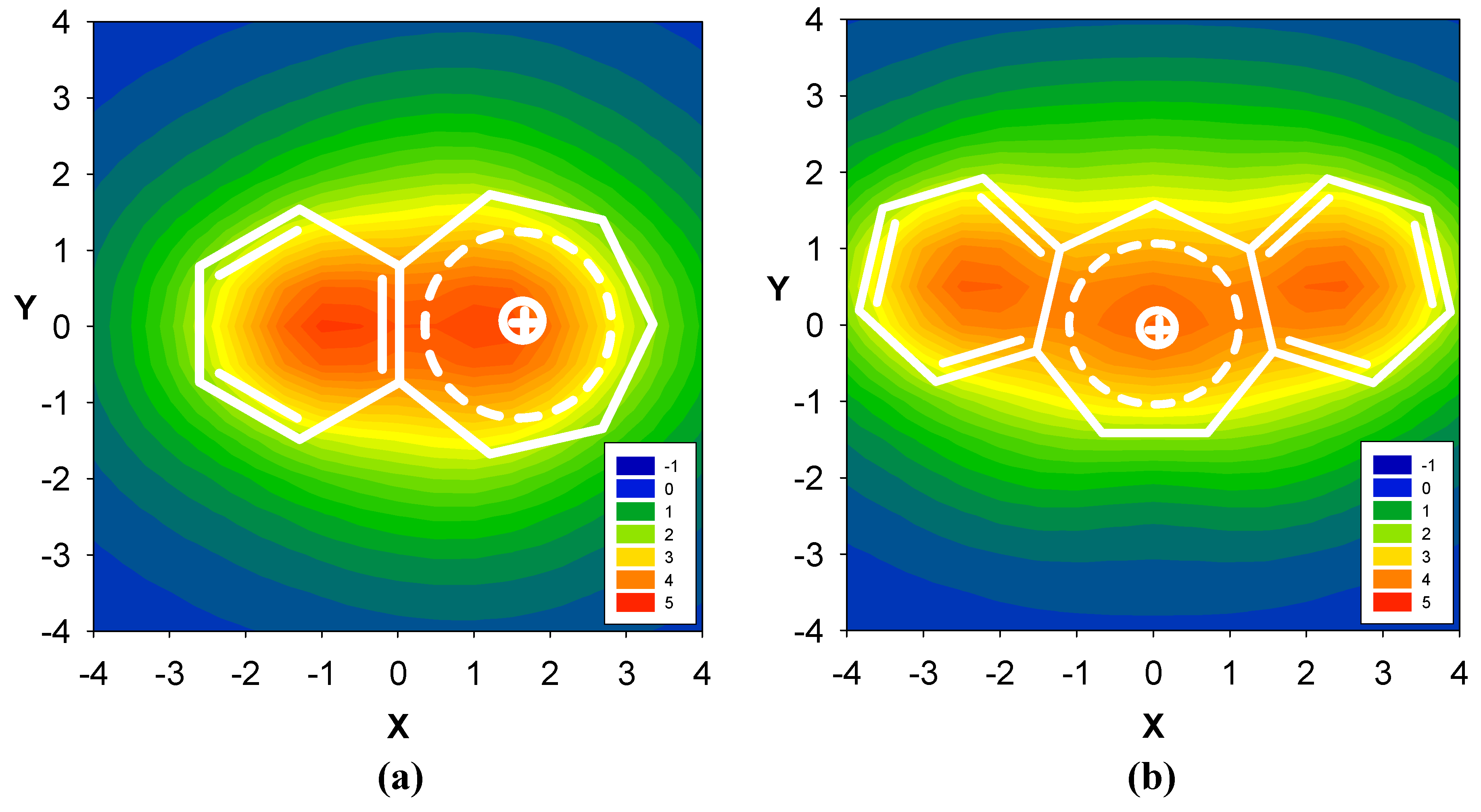
2.1. Benzene fused to cyclobutadiene
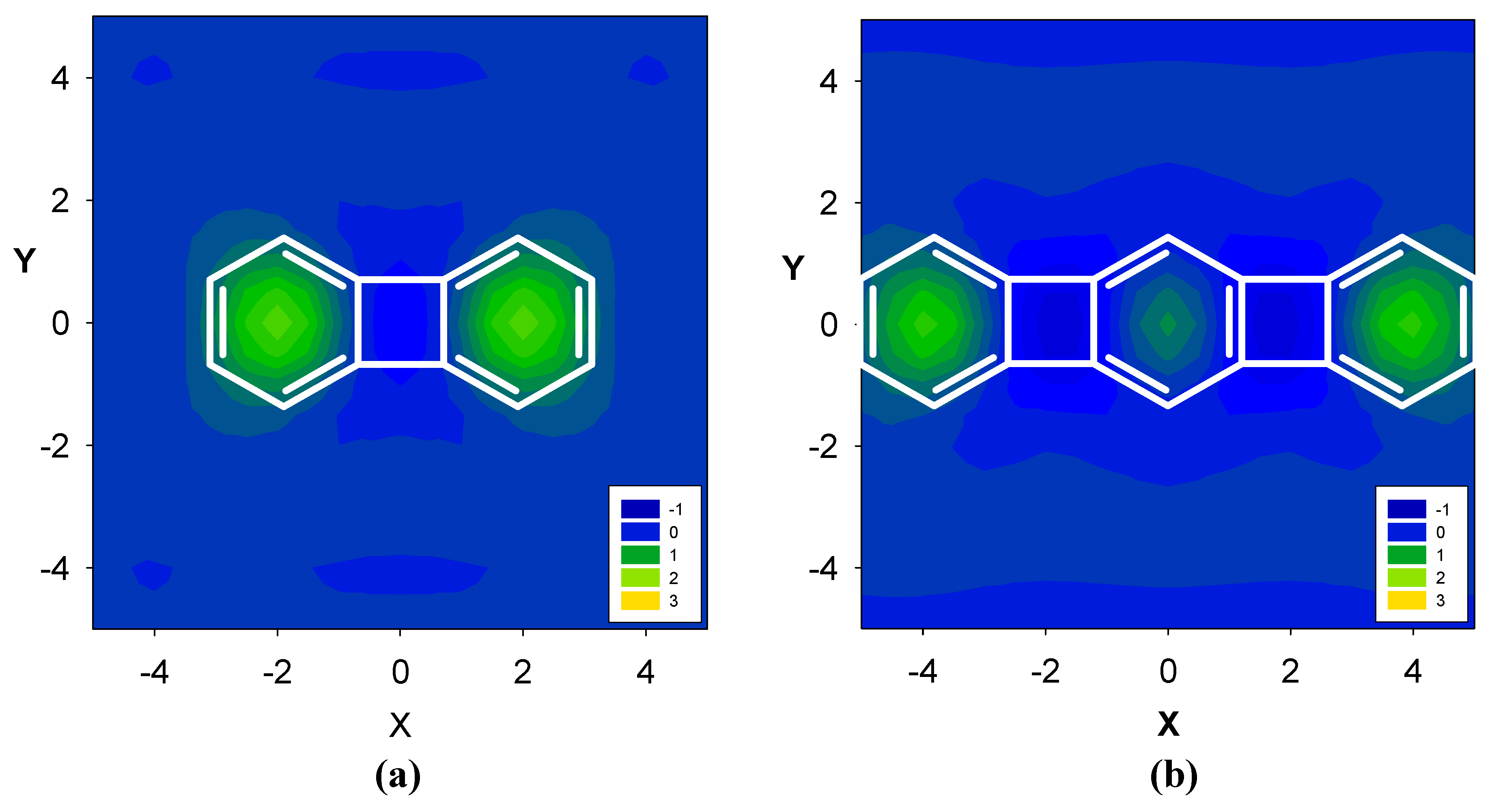
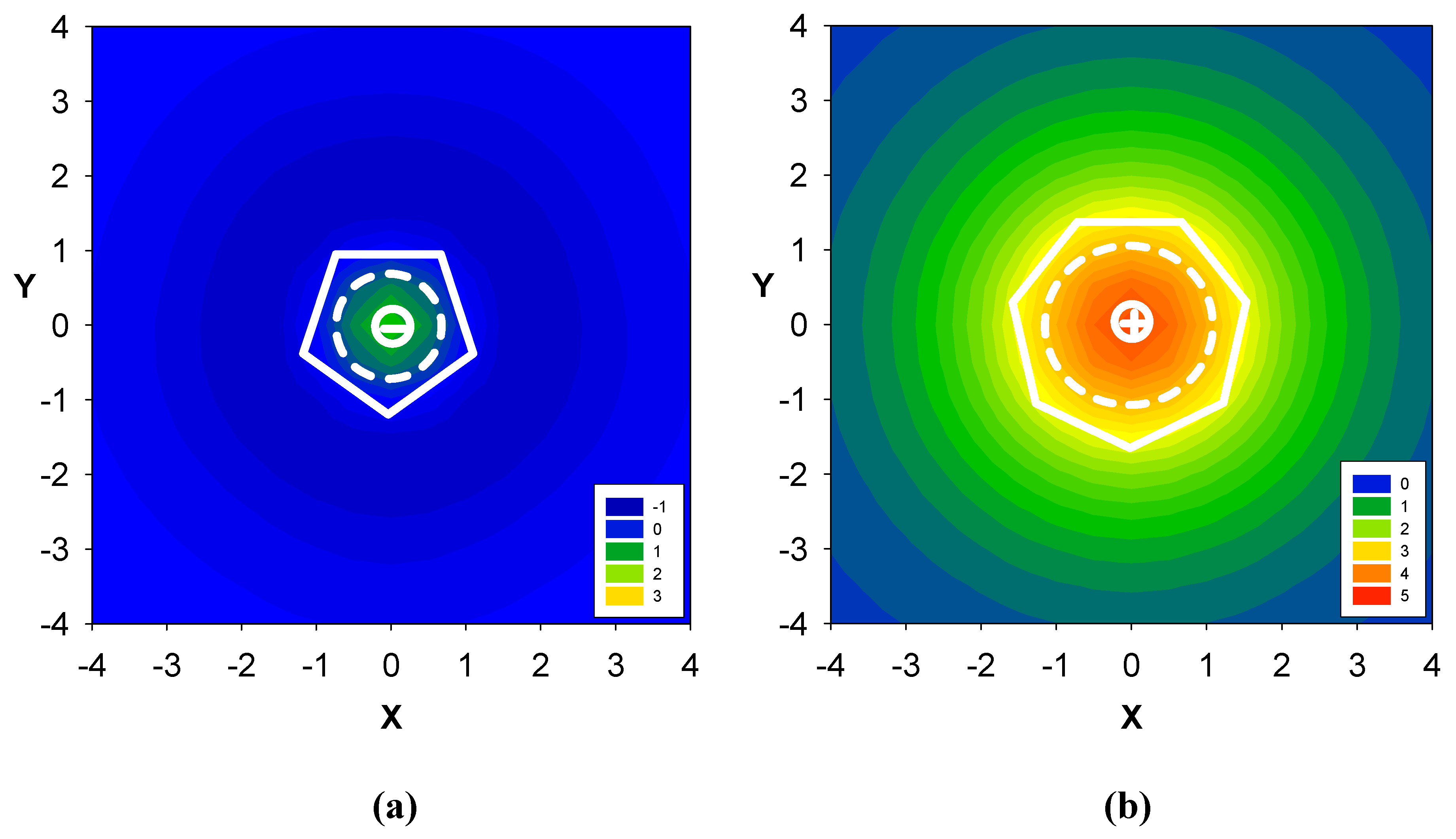
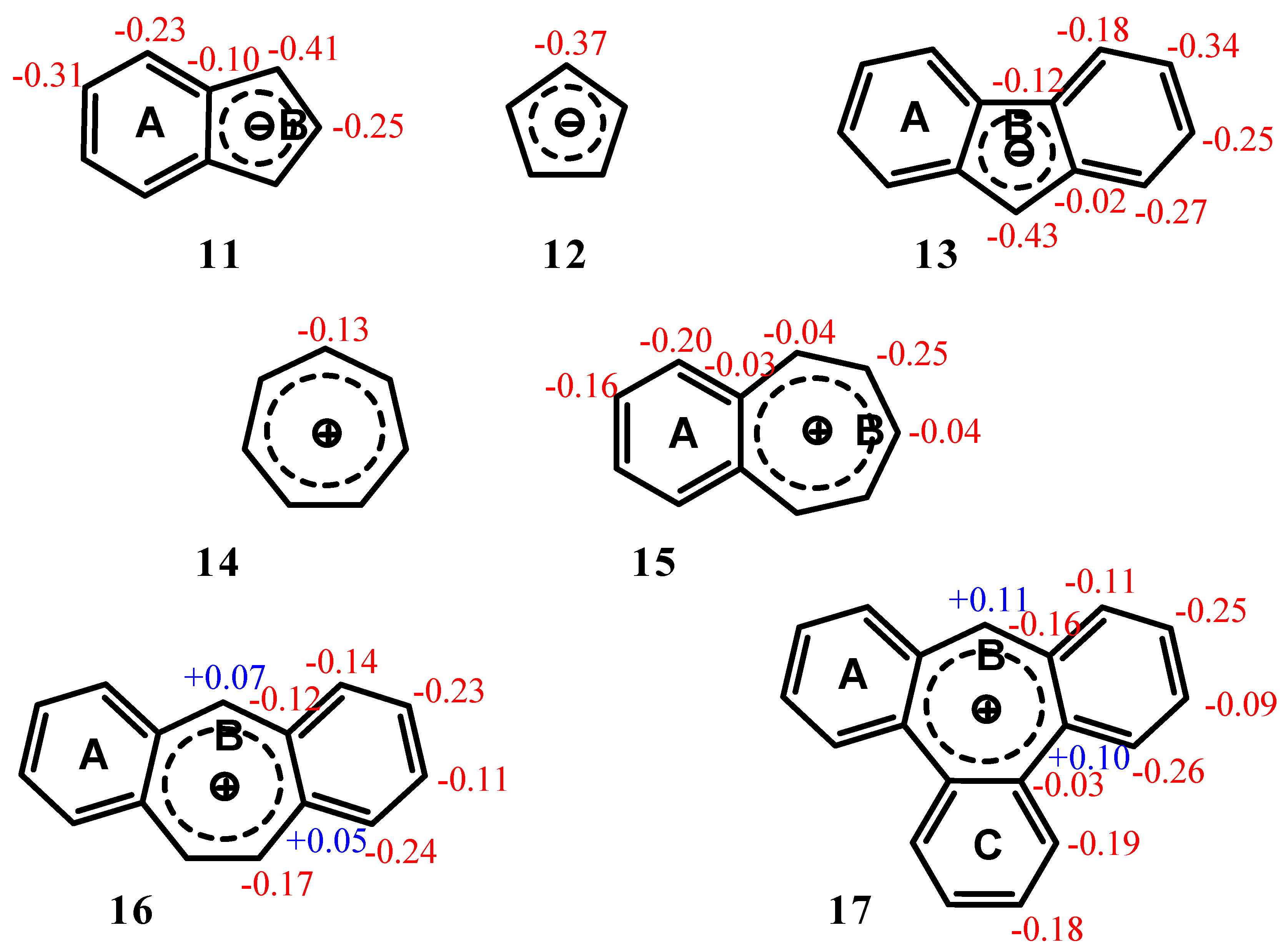
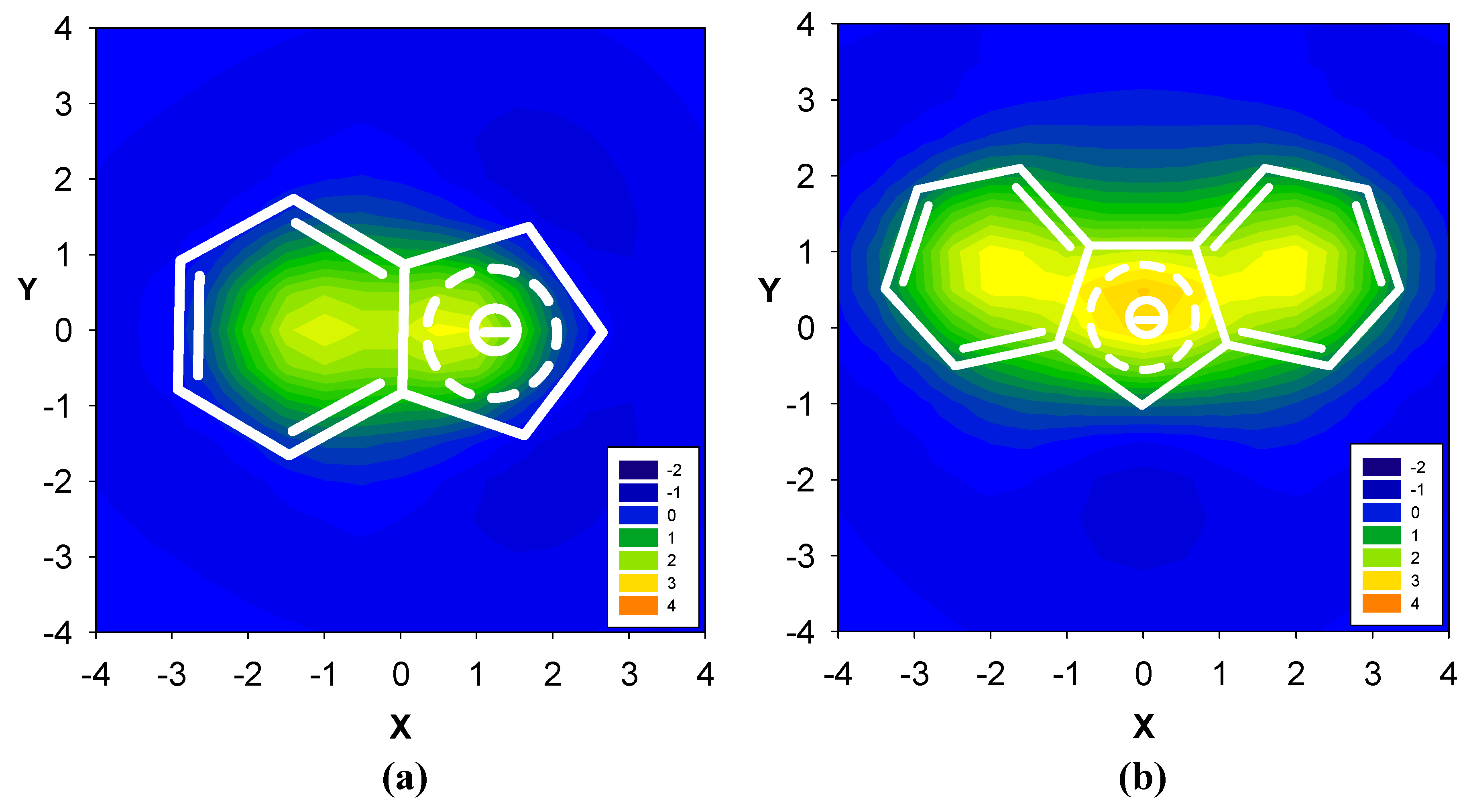
2.2. Benzene fused to cyclopentadienyl anion
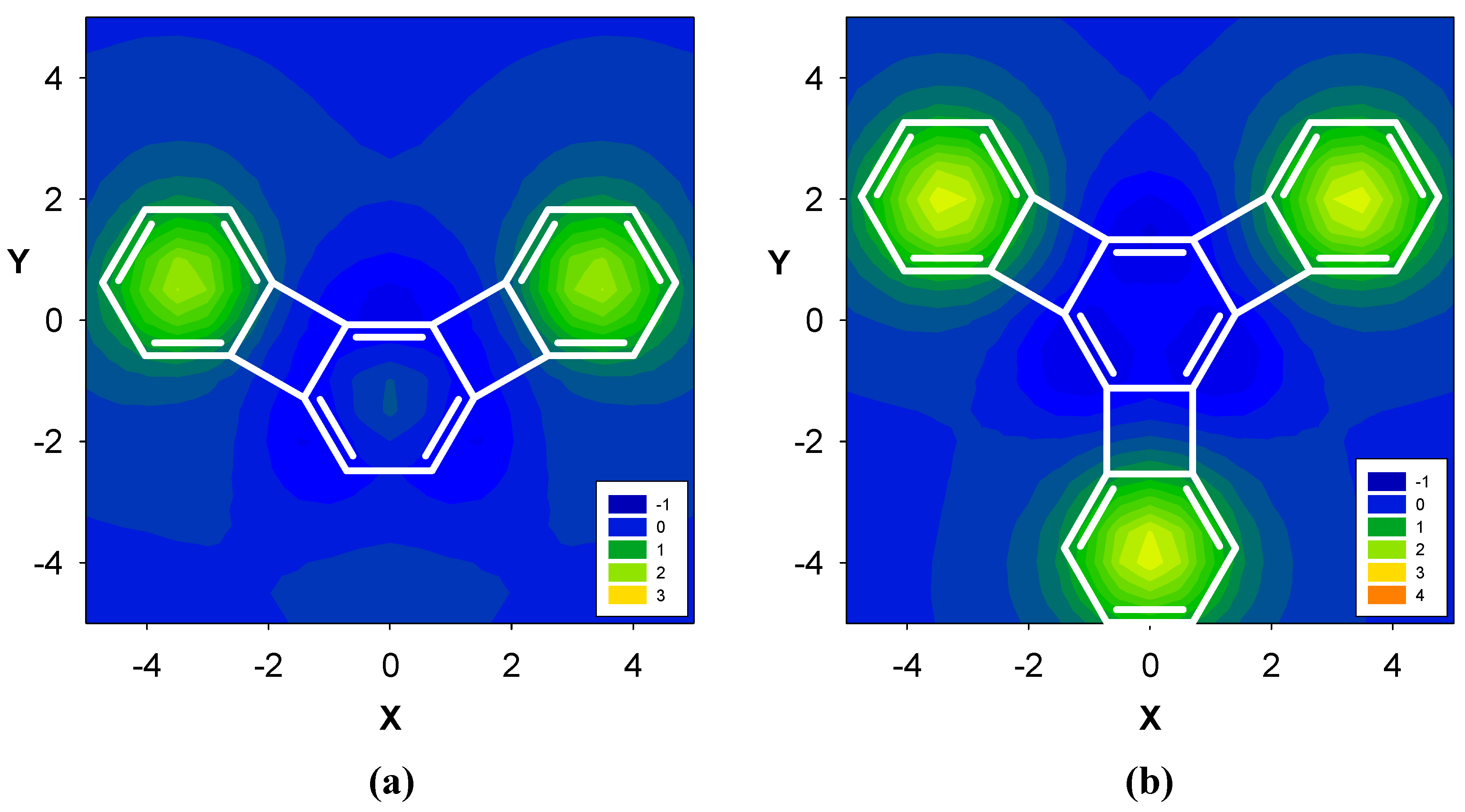
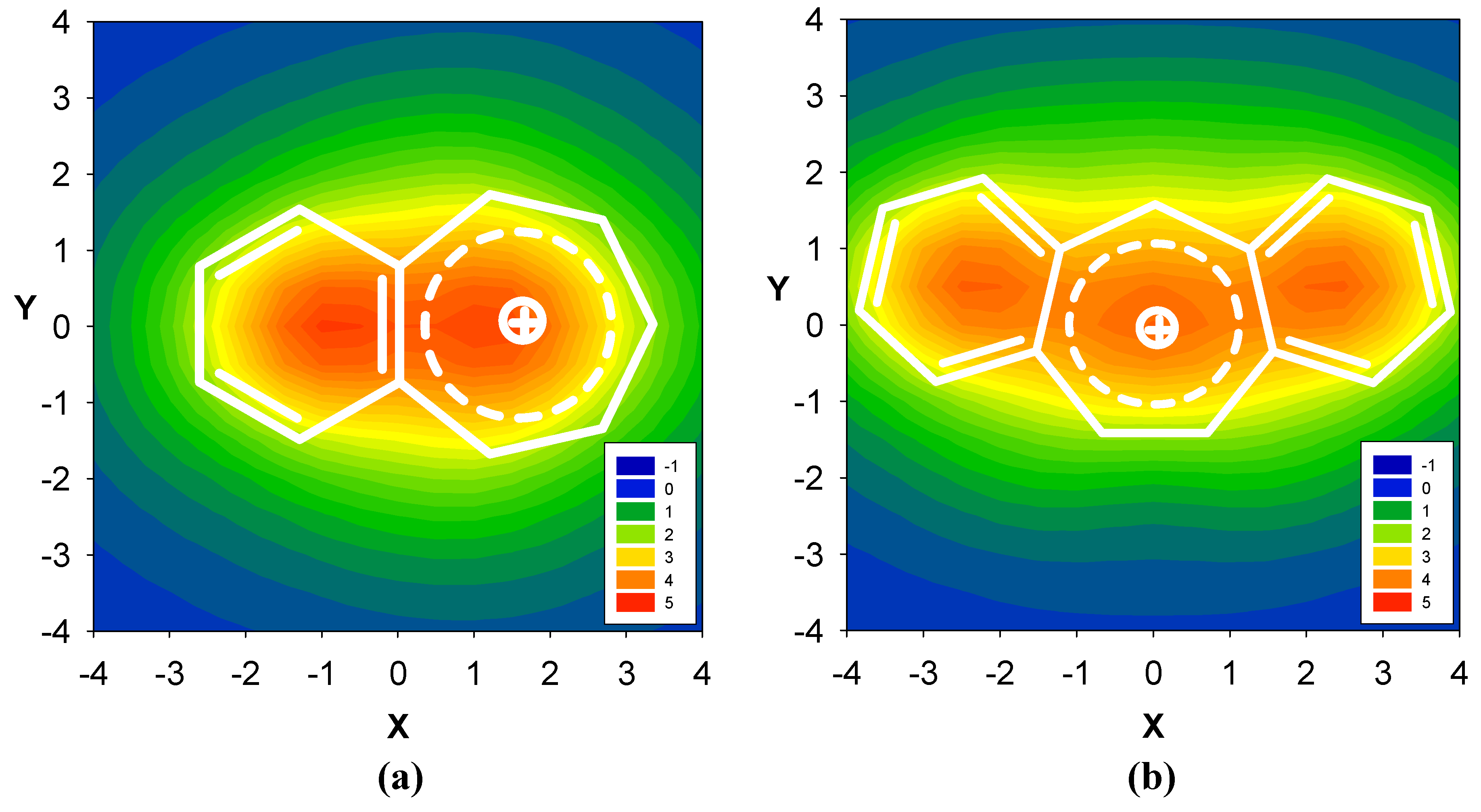
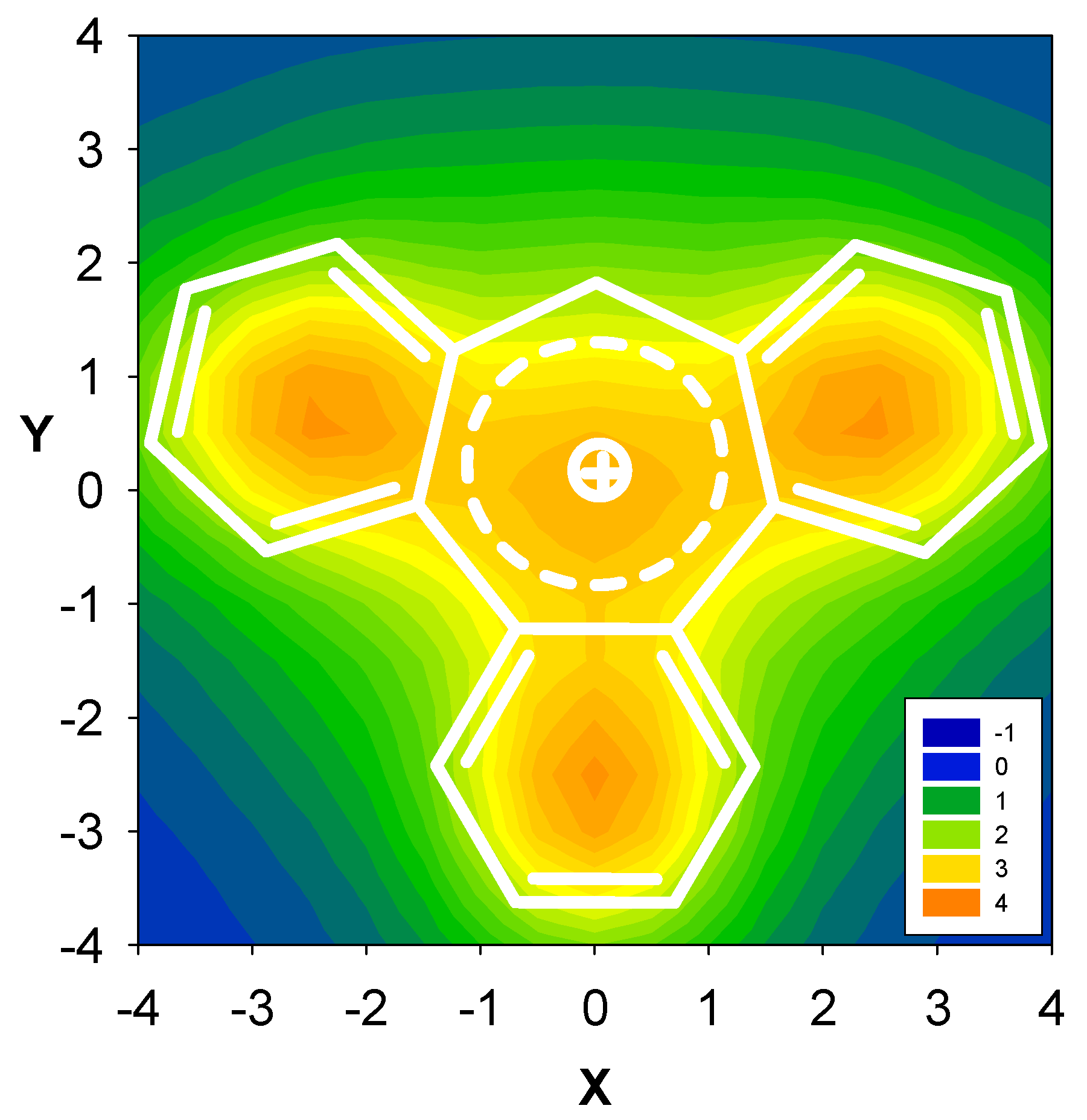
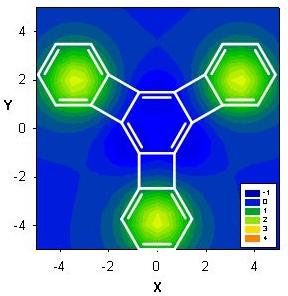
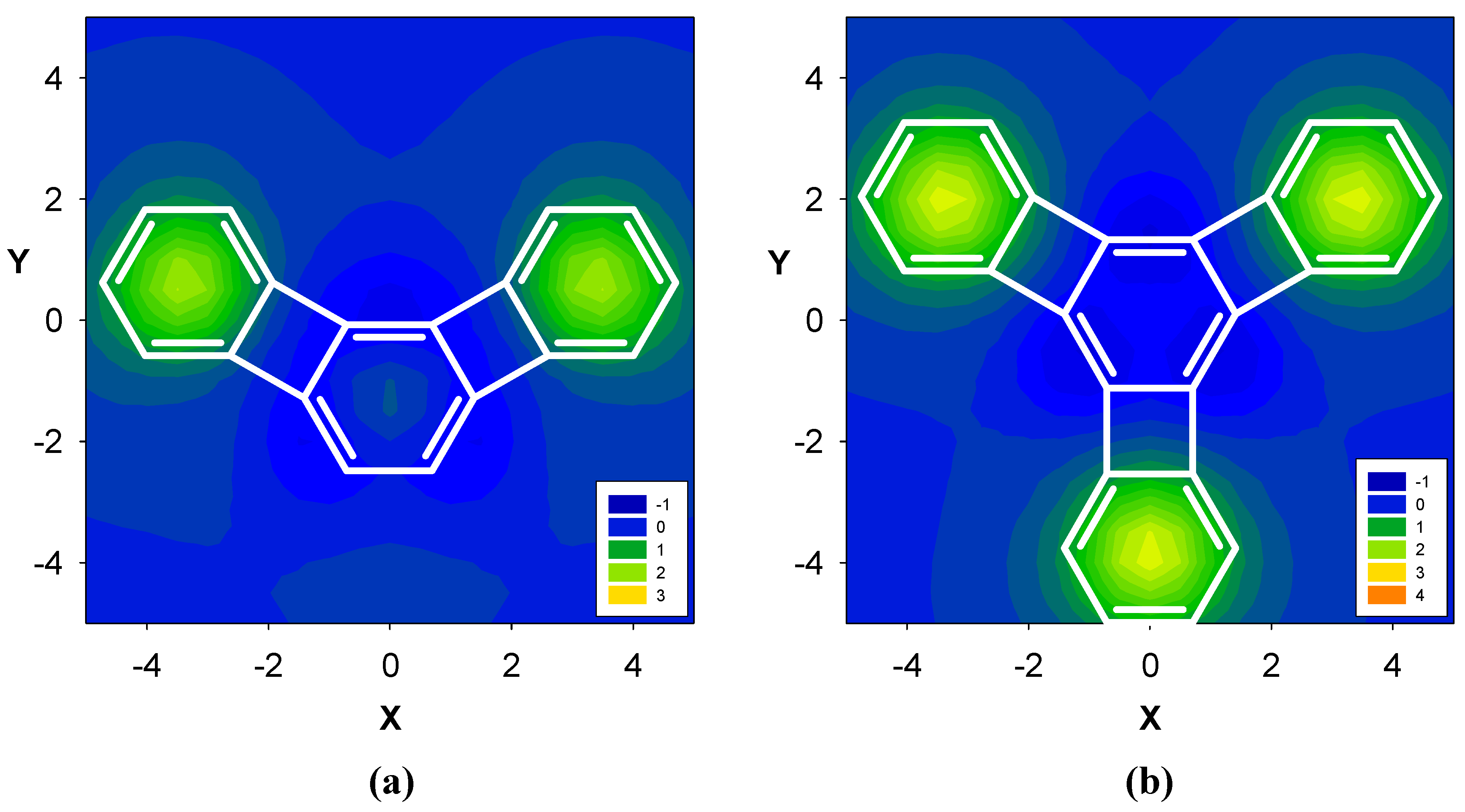
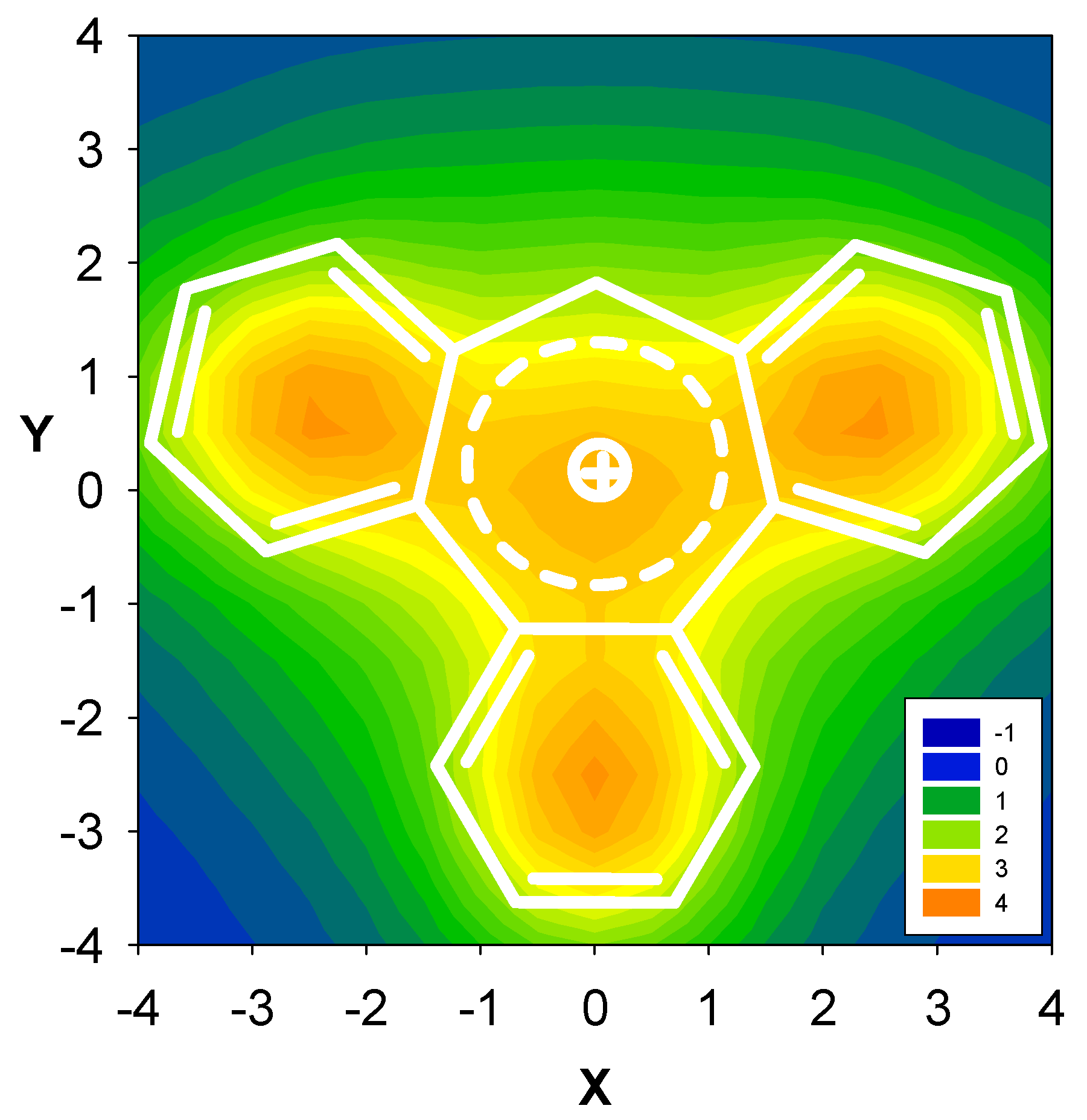
2.3. Benzene fused to tropylium ion
3. Computational Section
4. Conclusions
Acknowledgements
References and Notes
- Cyrañsky, M.K.; Krygowski, T.M.; Katritsky, A.R.; Schleyer, P.v.R. To what extent can aromaticity be defined uniquely? J. Org. Chem. 2002, 67, 1333–1338. [Google Scholar] [CrossRef]
- Hückel, E. Quantentheoretische Beiträge zum Benzolproblem I. Die Elektronenkonfiguration des Benzols und verwandter Verbindungen. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.J.; Hommes, N.J.R.v.E. Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Manoharan, M.; Wang, Z.-X.; Kiran, B.; Jiao, H.; Puchta, R.; Hommes, N.J.R.v.E. Dissected Nucleus-Independent Chemical Shift Analysis of π-Aromaticity and Antiaromaticity. Org. Lett. 2001, 3, 2465–2468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Which NICS Aromaticity Index for Planar π Rings Is Best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Corminboeuf, C.; Heine, T.; Seifert, G.; Schleyer, P.v.R.; Weber, J. Induced magnetic fields in aromatic [n]-annulenes−interpretation of NICS tensor components. Phys. Chem. Chem. Phys. 2004, 6, 273–276. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Klod, S. Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes-application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. 2001, 2, 1893–1898. [Google Scholar]
- Martin, N.H.; Caldwell, B.C.; Carlson, K.P.; Teague, M.R. Ab initio calculation of through-space magnetic shielding of linear polycyclic aromatic hydrocarbons (acenes): Extent of aromaticity. J. Mol. Graph. Modell. 2009, 27, 689–692. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. An ab initio study of the NMR properties (absolute shieldings and NICS) of a series of significant aromatic and antiaromatic compounds. Tetrahedron 2001, 57, 6043–6049. [Google Scholar] [CrossRef]
- Schulman, J.M.; Disch, R.L.; Jiao, H.; Schleyer, P.v.R. Chemical Shifts of the [N]Phenylenes and Related Compounds. J. Chem. Phys. A 1998, 102, 8051–8055. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Klod, S.; Koch, A. Visualization of through space NMR shieldings of aromatic and anti-aromatic molecules and a simple means to compare and estimate aromaticity. J. Mol. Struct. (THEOCHEM) 2007, 811, 45–60. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.H.; Loveless, D.M.; Main, K.L.; Wade, D.C. Computation of through-space NMR shielding effects by small-ring aromatic and antiaromatic hydrocarbons. J. Mol. Graph. Modell. 2006, 25, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.H.; Rowe, J.E.; Pittman, E.L. Computed NMR shielding increments over unsaturated five-membered ring heterocyclic compounds as a measure of aromaticity. J. Mol. Graph. Modell. 2009, 28, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.H.; Rowe, J.E.; Pittman, E.L. Computed NMR shielding increments over benzo-analogs of unsaturated five-membered ring heterocyclic compounds as a measure of aromaticity. J. Mol. Graph. Modell. 2010, 28, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.S. Mills-Nixon Effect: Wherefore Art Thou? Angew. Chem. Int. Ed. Engl. 1994, 33, 1721–1723, and references cited therein. [Google Scholar] [CrossRef]
- Neidlein, R.; Christen, D.; Poignée, V.; Boese, R.; Bläser, D.; Gieren, A.; Ruiz-Perez, C.; Hübner, T. The Structures of 1H-Cyclopropabenzene and Its 1,1-Bis(triisopropylsilyl) Derivative. Angew. Chem. Int. Ed. Engl. 1988, 27, 294–295. [Google Scholar] [CrossRef]
- Boese, R.; Bläser, D. Structures and Deformation Electron Densities of 1,2-Dihydrocyclobuta-benzene and 1,2,4,5-Tetrahydrodicyclobuta[a,d]benzene. Angew. Chem. Int. Ed. Engl. 1988, 27, 304–305. [Google Scholar] [CrossRef]
- Stanger, A. Is the Mills-Nixon Effect Real? J. Am. Chem. Soc. 1991, 113, 8277–8280. [Google Scholar] [CrossRef]
- Soncini, A.; Havenith, R.W.A.; Fowler, P.W.; Jenneskens, L.W.; Steiner, E. Control of the Diatropic π Ring Current in Strained Benzenes: Effects of Annelation with Cyclopropa, Cyclobuta, and Cyclobutadieno Clamping Groups. J. Org. Chem. 2002, 67, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.N.; Jana, D.F.; Wu, J.I.-C.; Schleyer, P.v.R.; Mo, Y.; Corminboeuf, C. Direct Assessment of Electron Delocalization Using NMR Chemical Shifts. Angew. Chem. Int. Ed. 2009, 48, 9828–9833. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J. Ab initio hybrid DFT-GIAO calculations of the shielding produced by carbon-carbon bonds and aromatic rings in 1H-NMR spectroscopy. New J. Chem. 1998, 381–385. [Google Scholar] [CrossRef]
- Fawcett, J. K.; Trotter, J. A refinement of the structure of biphenylene. Acta Crystallogr. 1966, 20, 87–93. [Google Scholar] [CrossRef]
- Toda, F.; Garratt, P. Four-Membered Ring Compounds Containing Bis(methy1ene)cyclobutene or Tetrakis(methylene)cyclobutane Moieties. Benzocyclobutadiene, Benzodicyclobutadiene, Biphenylene, and Related Compounds. Chem. Rev. 1992, 92, 1685–1707. [Google Scholar] [CrossRef]
- Berris, B.C.; Hovakeemian, G.H.; Lai, Y.-H.; Mestdagh, H.; Vollhardt, K.P.C. A New Approach to the Construction of Biphenylenes by the Cobalt-Catalyzed Cocyclization of o-Diethynyl-benzenes with Alkynes. Application to an Iterative Approach to [3]Phenylene, the First Member of a Novel Class of Benzocyclobutadienoid Hydrocarbons. J. Am. Chem. Soc. 1985, 107, 5670–5687. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Koch, A. Trisannelated benzenes – Aromatic molecules or 1,3,5-cyclohexatriene derivatives subjected to magnetic properties. J. Mol. Struct. (THEOCHEM) 2008, 857, 89–94. [Google Scholar] [CrossRef]
- Diercks, R.; Vollhardt, K.P.C. Novel Synthesis of the Angular [3]Phenylene (Terphenylene) by Cobalt-Catalyzed Cyclization of Bis(2-ethynylphenyl)ethyne: a Molecule With an Internal Cyclohexatriene Ring. Angew. Chem., Int. Ed. Engl. 1986, 25, 266–268. [Google Scholar] [CrossRef]
- Fowler, P.W.; Havenith, R.W.A.; Jenneskens, L.W.; Soncini, A.; Steiner, E. Survival and extinction of delocalised ring currents in clamped benzenes. Chem. Commun. 2001, 22, 2386–2387. [Google Scholar] [CrossRef]
- Diercks, R.; Vollhardt, K.P.C. Tris(benzocyclobutadieno)benzene, the Triangular [4]Phenylene with a Completely Bond-Fixed Cyclohexatriene Ring: Cobalt-Catalyzed Synthesis From Hexaethynylbenzene and Thermal Ring Opening to 1,2:5,6:9,10-Tribenzo-3,4,7,8,11,12-hexadehydro[12]annulene. J. Am. Chem. Soc. 1986, 108, 3150–3152. [Google Scholar] [CrossRef]
- Martin, N.H.; Loveless, D.M.; Wade, D.C. A comparison of calculated NMR shielding probes. J. Mol. Graph. Modell. 2004, 23, 285–290, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Titan; Version 1.0.1; Wavefunction Inc., Schrödinger, Inc.: Portland, OR, 1999.
- Hehre, W.J.; Radom, L.; Schleyer, P.v.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, USA, 1986. [Google Scholar]
- Martin, N.H.; Allen, N.W., III; Minga, E.K.; Ingrassia, S.T. An Empirical Proton NMR Shielding Equation for Alkenes based on Ab Initio Calculations. Struct. Chem. 1998, 9, 403–410. [Google Scholar] [CrossRef]
- Martin, N.H.; Brown, J.D.; Nance, K.H.; Schaefer, H.F., Jr.; Schleyer, P.v.R.; Wang, Z.-X.; Woodcock, H.L. Analysis of the Origin of Through-Space Proton NMR Deshielding by Selected Organic Functional Groups. Org. Lett. 2001, 3, 3823–3826. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03; Revision B.01; Gaussian Inc.: Pittsburgh, PA, 2003. [Google Scholar]
- Martin, N.H.; Floyd, R.M.; Woodcock, H.L.; Huffman, S.; Lee, C.-K. Computation of through-space NMR shielding effects in aromatic ring pi-stacked complexes. J. Mol. Graph. Modell. 2008, 26, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculations of small molecular interaction by the difference of separate total energies. Some procedures with reduced error. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- SigmaPlot 2004 for Windows; Version 9.01; Systat Software, Inc.: San Jose, CA, 2004.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
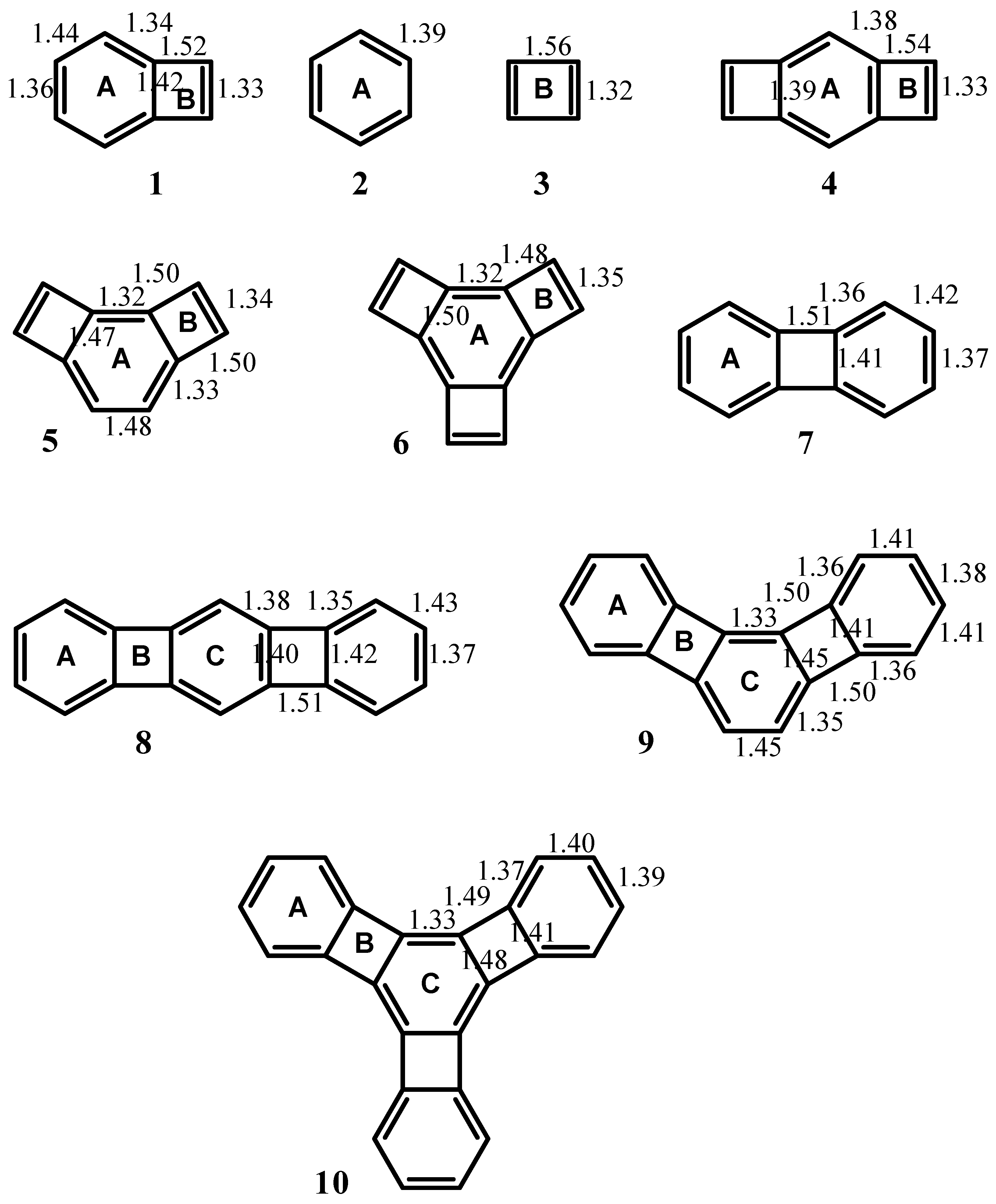
| Δσ and (NICS(1)) in ppm above center of ring: | |||
|---|---|---|---|
| Structure | A | B | C |
| 1 | 0.71 (-5.52) | -1.56 (10.70) | -- |
| 2 | 2.96 (-12.90) | -- | -- |
| 3 | -- | -2.83 (17.42) | -- |
| 4 | -0.08 (-4.43) | -2.11 (13.46) | -- |
| 5 | -0.29 (-0.91) | -0.54 (1.01) | -- |
| 6 | -0.05 (-1.92) | -0.01 (-2.85) | -- |
| 7 | 1.55 (-8.13) | -0.41 (7.50) | -- |
| 8 | 1.30 (-7.33) | -0.73 (7.86) | 0.69 (-5.63) |
| 9 | 2.01 (-9.82) | -0.13 (3.74) | 0.28 (-3.17) |
| 10 | 2.39 (-10.99) | 0.18 (0.72) | -0.27 (-0.79) |
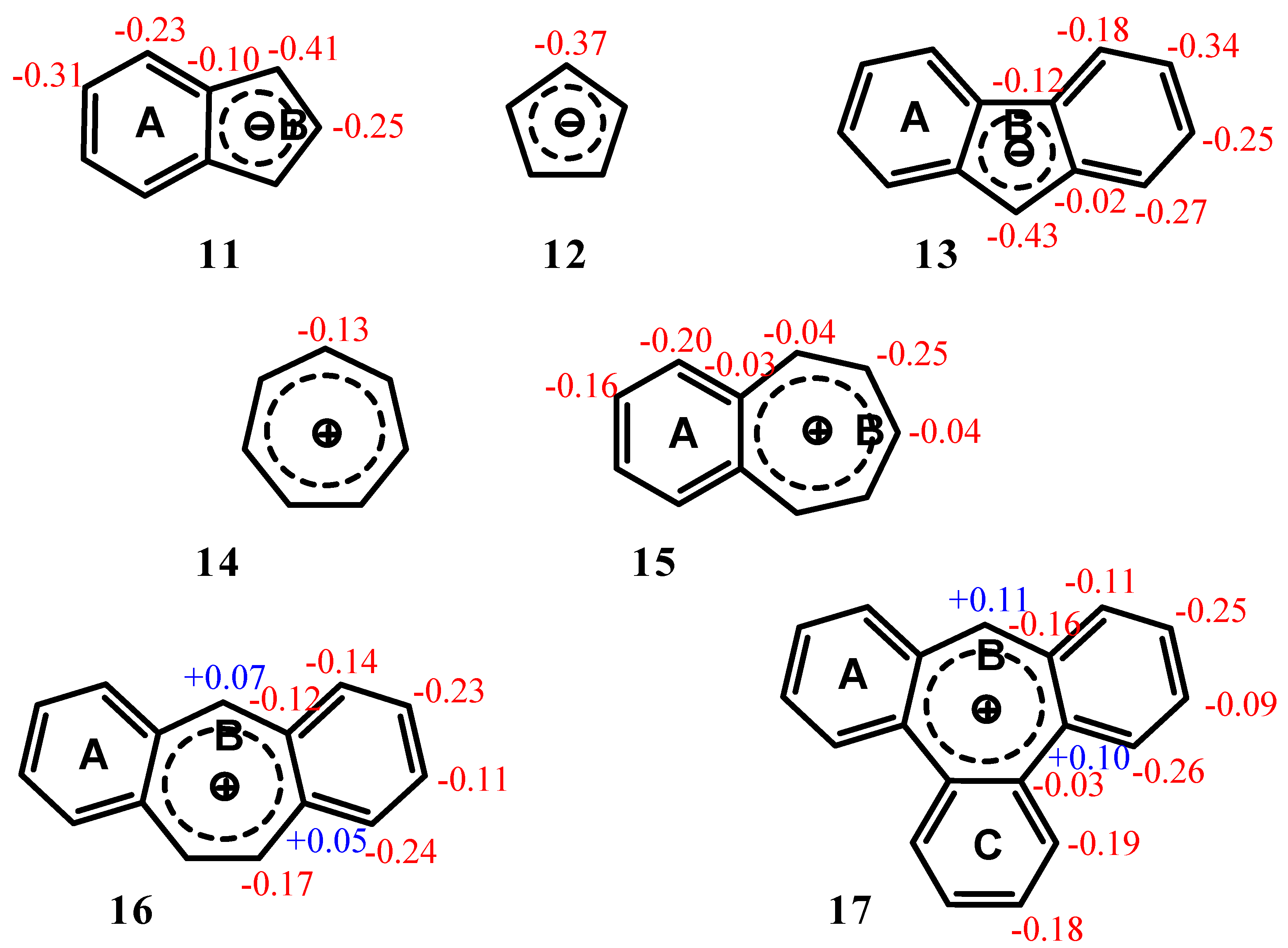
| Δσ and (NICS(1)) in ppm above center of ring: | |||
|---|---|---|---|
| Structure | A | B | C |
| 11 | 2.36 (-12.35) | 2.32 (-16.97) | -- |
| 12 | -- | 1.17 (-14.03) | |
| 13 | 2.93 (-12.56) | 2.61 (-15.76) | -- |
| 14 | -- | 4.52 (-11.22) | |
| 15 | 4.51 (-14.35) | 4.52 (-10.56) | -- |
| 16 | 4.18 (-13.41) | 4.22 (-8.46) | -- |
| 17 | 3.64 (-11.91) | 3.40 (-4.72) | 3.66 (-12.13) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martin, N.H.; Teague, M.R.; Mills, K.H. Computed NMR Shielding Effects over Fused Aromatic / Antiaromatic Hydrocarbons. Symmetry 2010, 2, 418-436. https://doi.org/10.3390/sym2010418
Martin NH, Teague MR, Mills KH. Computed NMR Shielding Effects over Fused Aromatic / Antiaromatic Hydrocarbons. Symmetry. 2010; 2(1):418-436. https://doi.org/10.3390/sym2010418
Chicago/Turabian StyleMartin, Ned H., Mathew R. Teague, and Katherine H. Mills. 2010. "Computed NMR Shielding Effects over Fused Aromatic / Antiaromatic Hydrocarbons" Symmetry 2, no. 1: 418-436. https://doi.org/10.3390/sym2010418
APA StyleMartin, N. H., Teague, M. R., & Mills, K. H. (2010). Computed NMR Shielding Effects over Fused Aromatic / Antiaromatic Hydrocarbons. Symmetry, 2(1), 418-436. https://doi.org/10.3390/sym2010418