Chiral Symmetry Breaking Phenomenon Caused by a Phase Transition

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
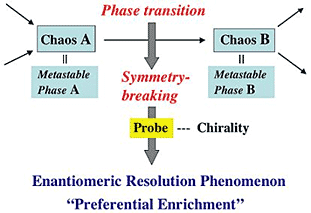
1. Introduction
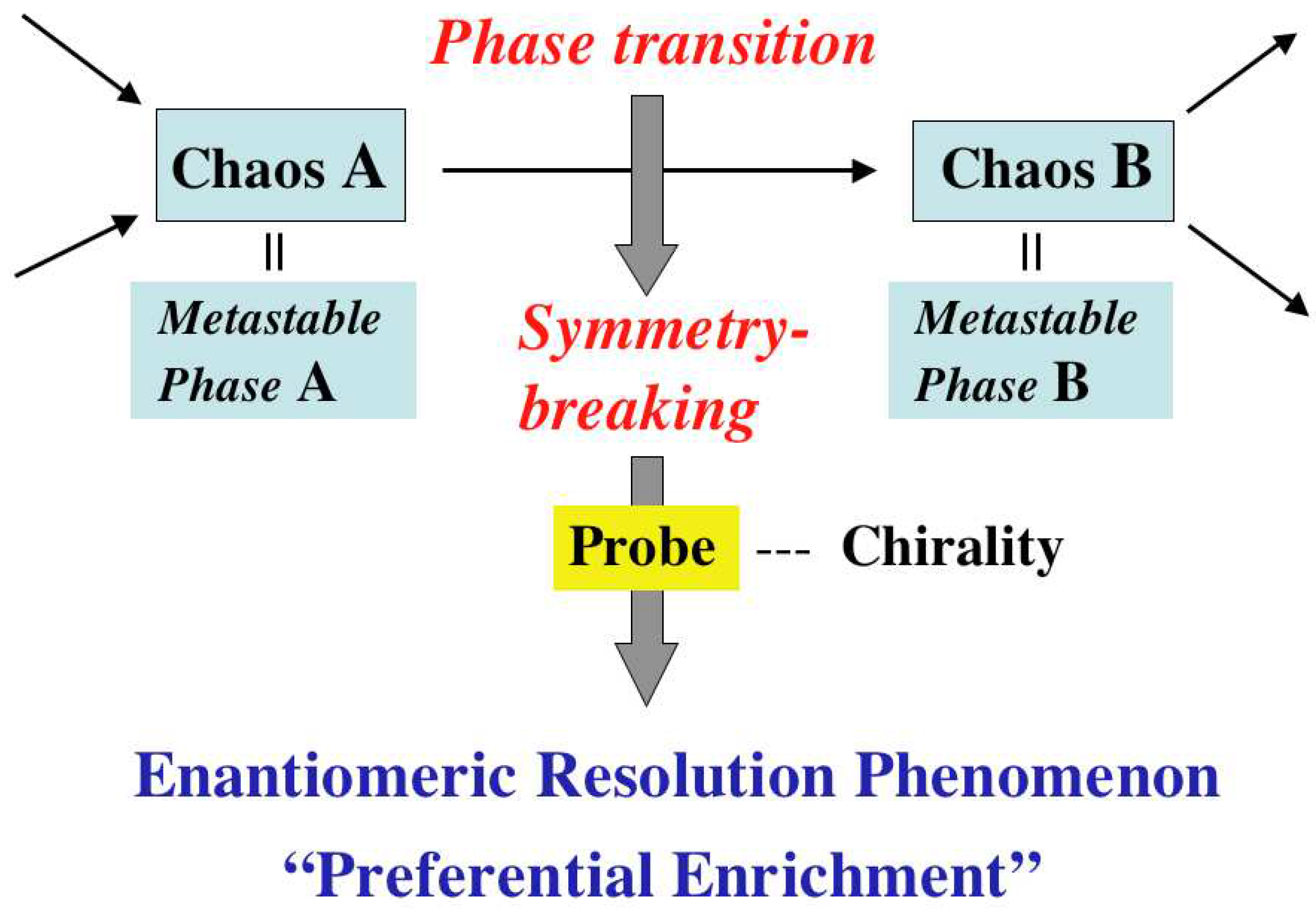
2. Features of Preferential Enrichment
- 1)
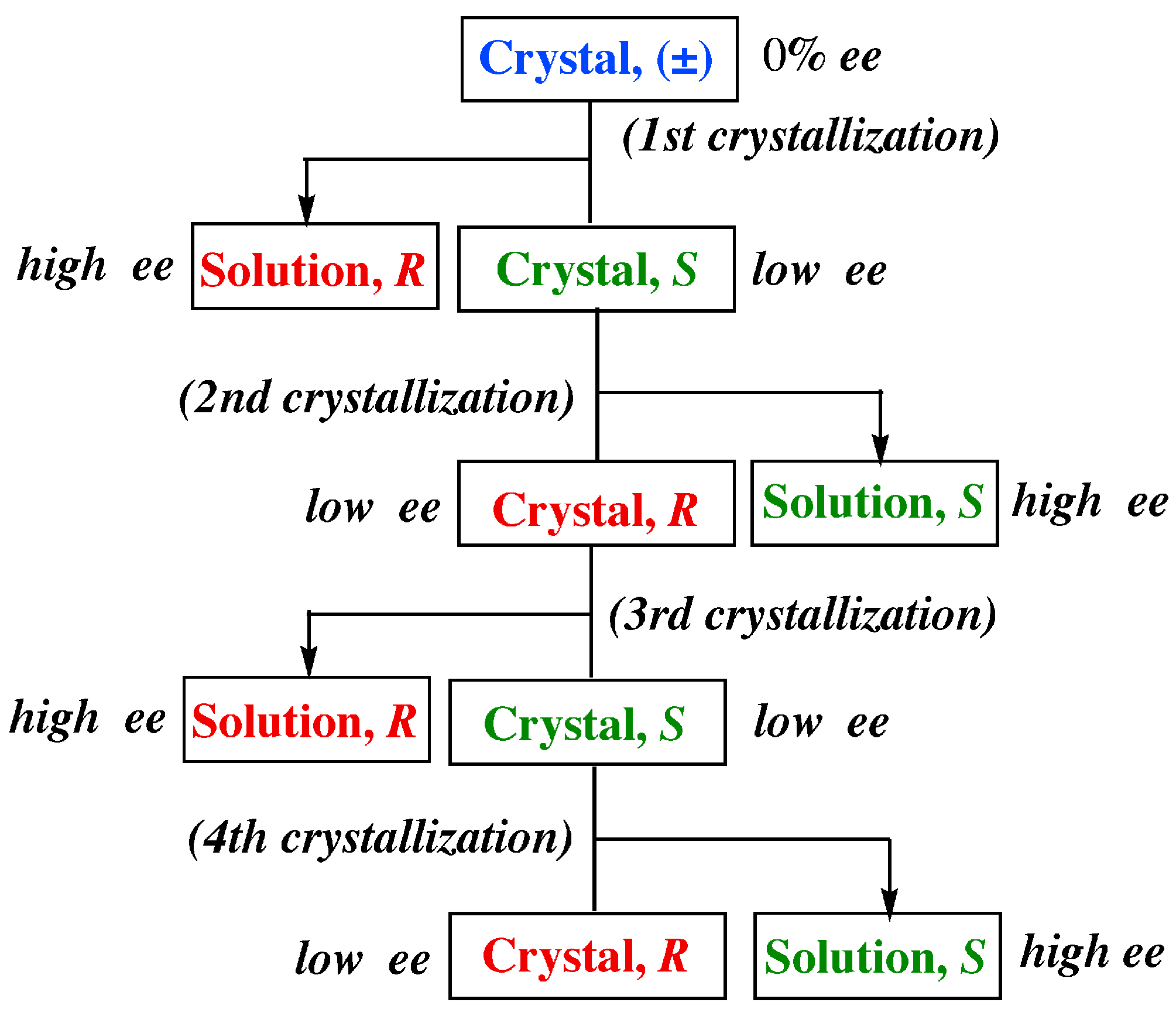
- Usual standard recrystallization conditions with neither vigorous stirring nor abrasive grinding are applied to the preferential enrichment experiment, except that approximately 4- to 25-fold supersaturated solutions are employed because the supersolubility (a solubility obtained by dissolving the sample in a solvent on heating followed by being cooled) of the racemates showing preferential enrichment is considerably higher than that of the solubility at 25 °C. The attainment of such a high supersolubility is closely associated with the preferential formation of homochiral 1D R and S chains even in the racemic solution. At the lower supersaturated concentrations, preferential enrichment does not efficiently occur.
- 2)
- Racemic or nonracemic samples of less than 10% ee are more suitable for the preferential enrichment experiment than those of higher ee values to achieve a very efficient resolution.
- 3)
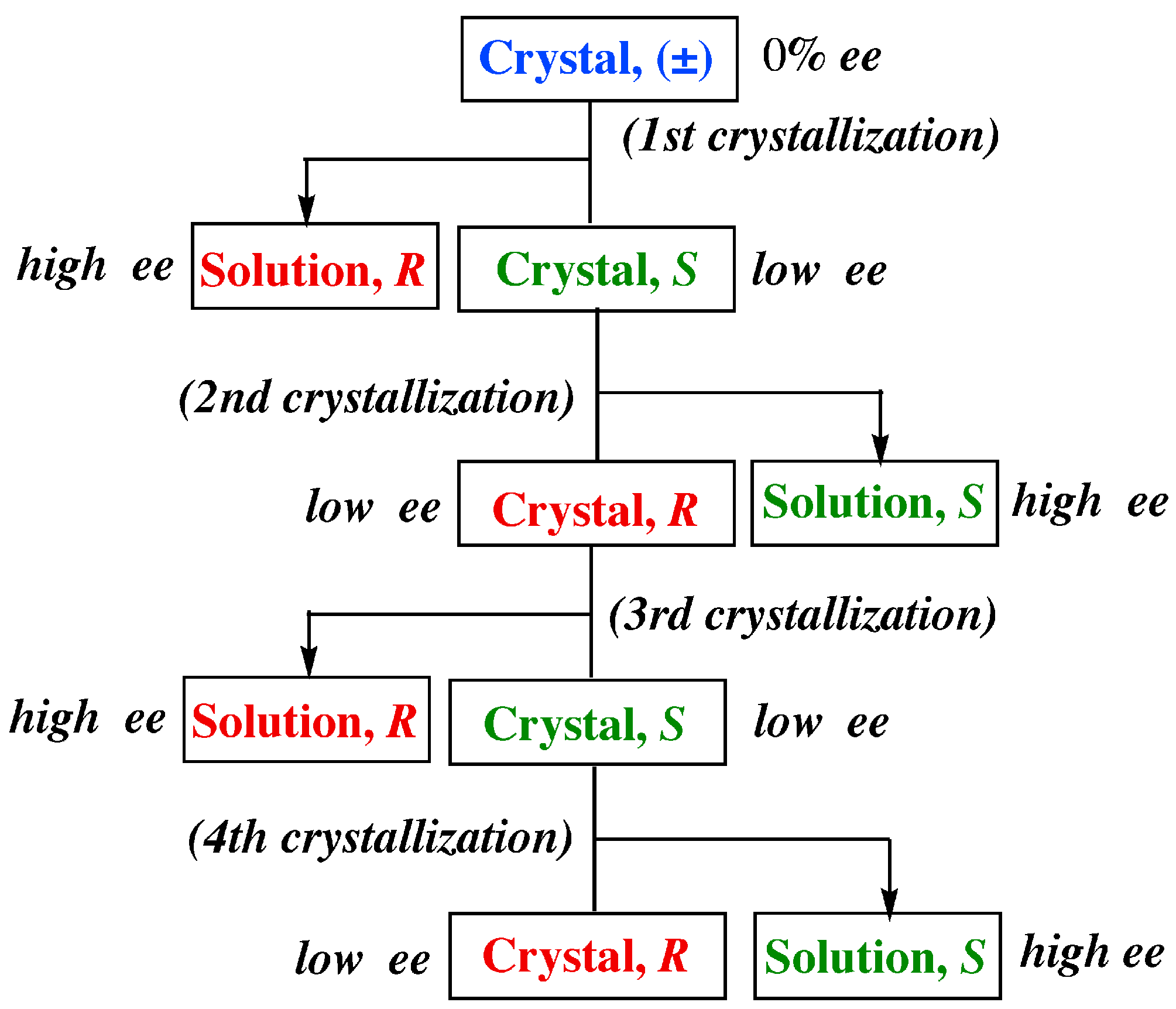
- Recrystallization of the nonracemic sample of less than 10% ee from the supersaturated solution leads to a remarkable enrichment of the excess enantiomer up to 100% ee in the mother liquors (a considerable enrichment of the excess enantiomer in the mother liquor). At the same time, the resulting deposited crystals always display the opposite chirality at around 5% ee (a slight enrichment of the opposite enantiomer in the deposited crystals). These phenomena are fully reproducible.
- 4)
- The solubility of the sample of a high ee value is much higher than that of a low ee one. This property allows dissolution of the excess enantiomer from the just-made crystals into solution until a slight enrichment of the opposite enantiomer in the deposited crystals occurs together with a considerable enrichment of the excess enantiomer in the mother liquor.
- 5)
- When the original supersaturated solution is strictly racemic, the probability for either the R or S enantiomer to be enriched in the mother liquor after crystallization was 50%. In the resulting deposited crystals, the opposite enantiomer is enriched up to around 5% ee.
- 6)
- Only racemic or nonracemic samples have to be crystalline to implement the preferential enrichment experiment efficiently. It does not matter whether the enantiomerically enriched samples with high ee values exist as solids or oils, in sharp contrast to preferential crystallization of a racemic conglomerate.
- 7)
- Seed crystals are not necessary at all. Addition of seed crystals may accelerate or inhibit the occurrence of preferential enrichment, depending on the kind of added seed crystals.
- 8)
- The features of preferential enrichment are completely opposed to those of preferential crystallization of a racemic conglomerate, where a substantial enantiomeric enrichment does not occur in the mother liquor but in the deposited crystals.
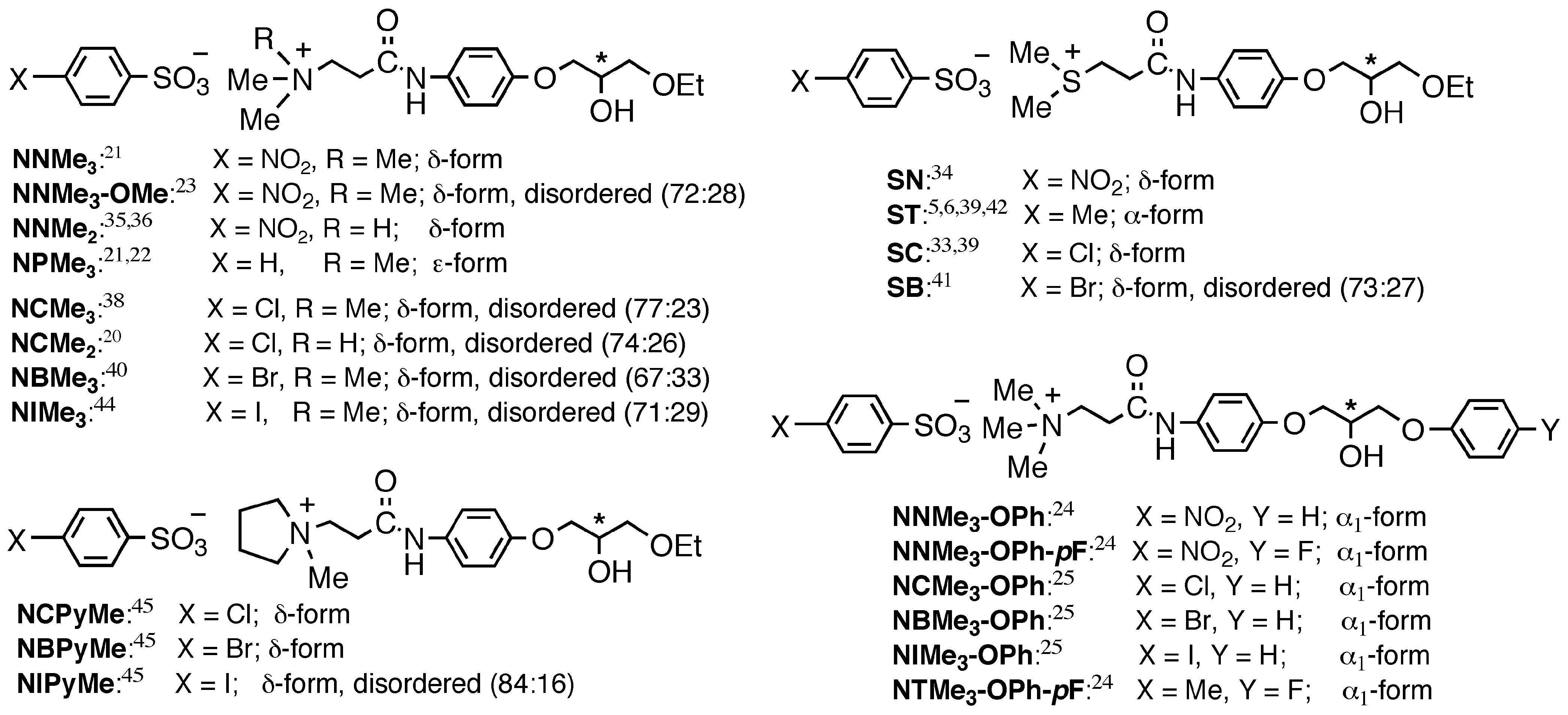
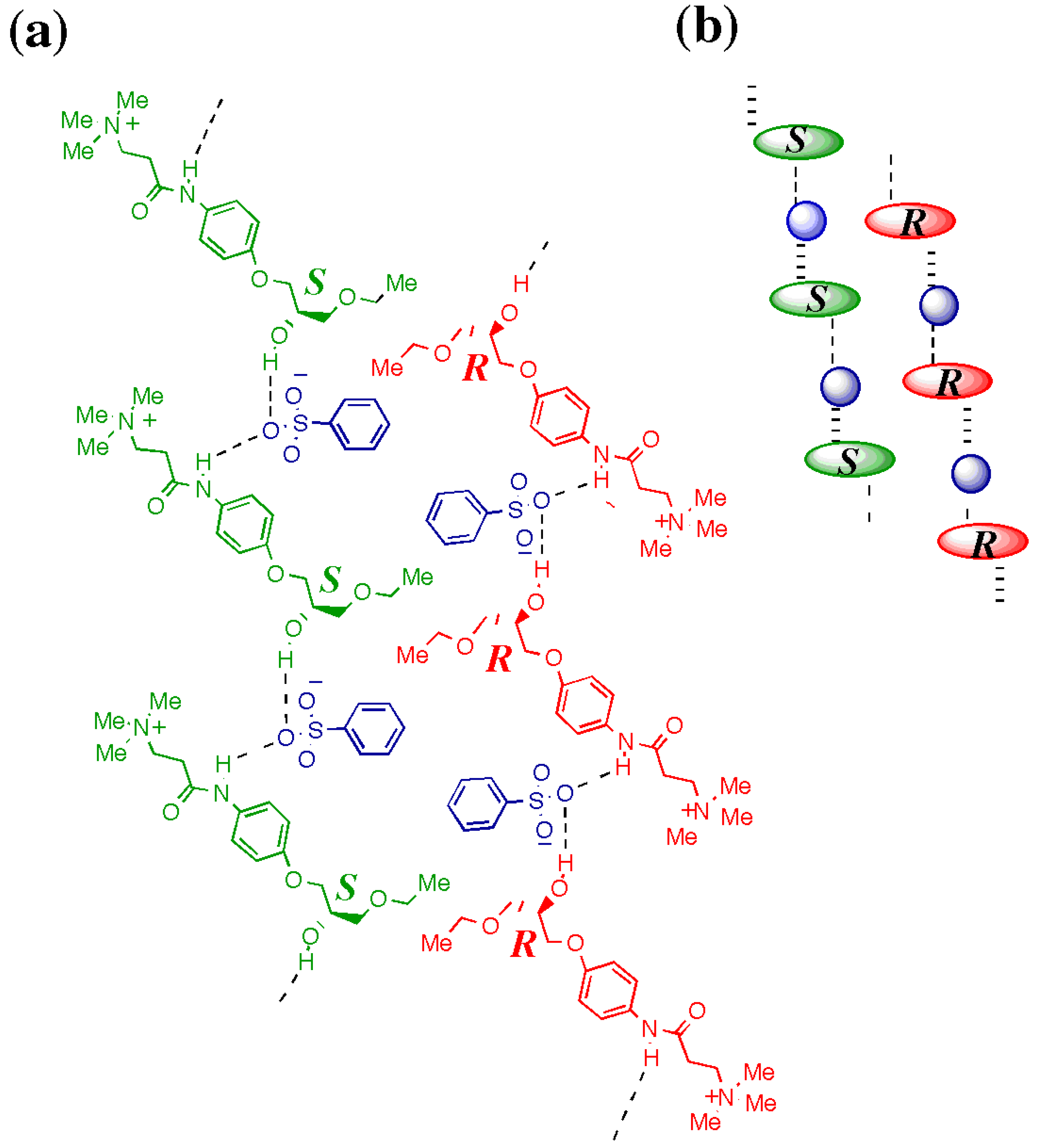
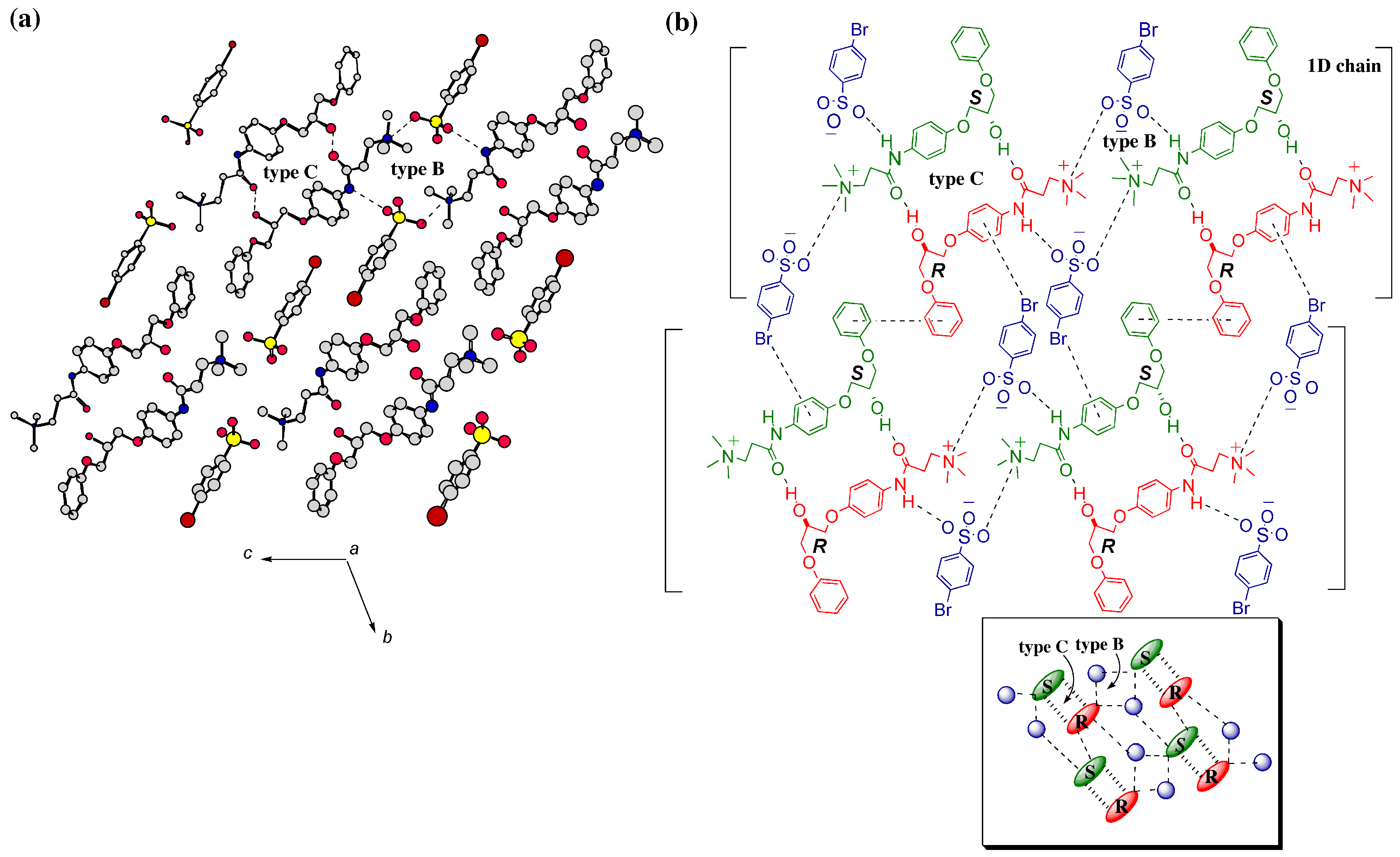
3. Mechanism of Preferential Enrichment
3.1. Association Mode of Enantiomers in Solution
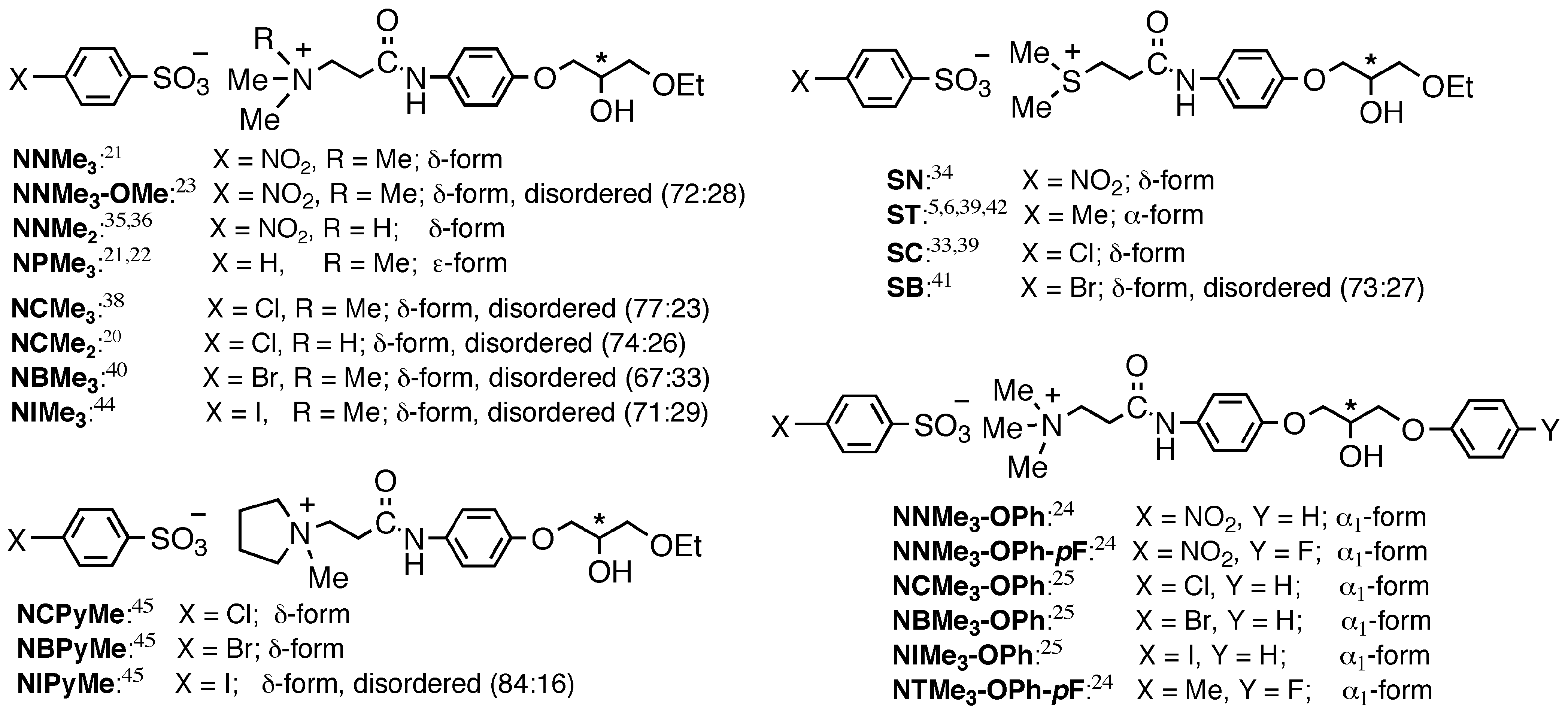
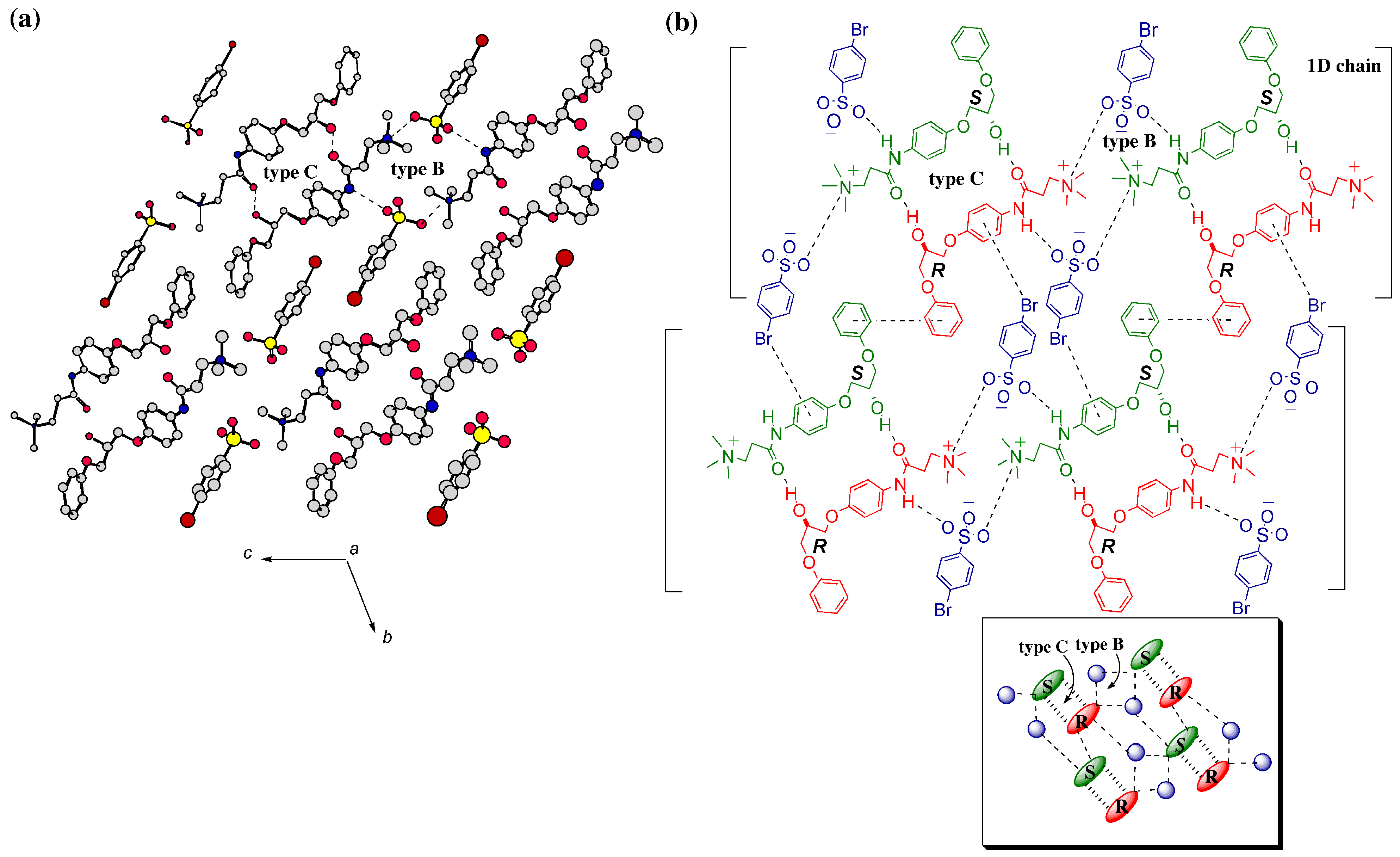
3.2. Crystal Structures
3.2.1. Metastable γ-Form
3.2.2. Stable Forms
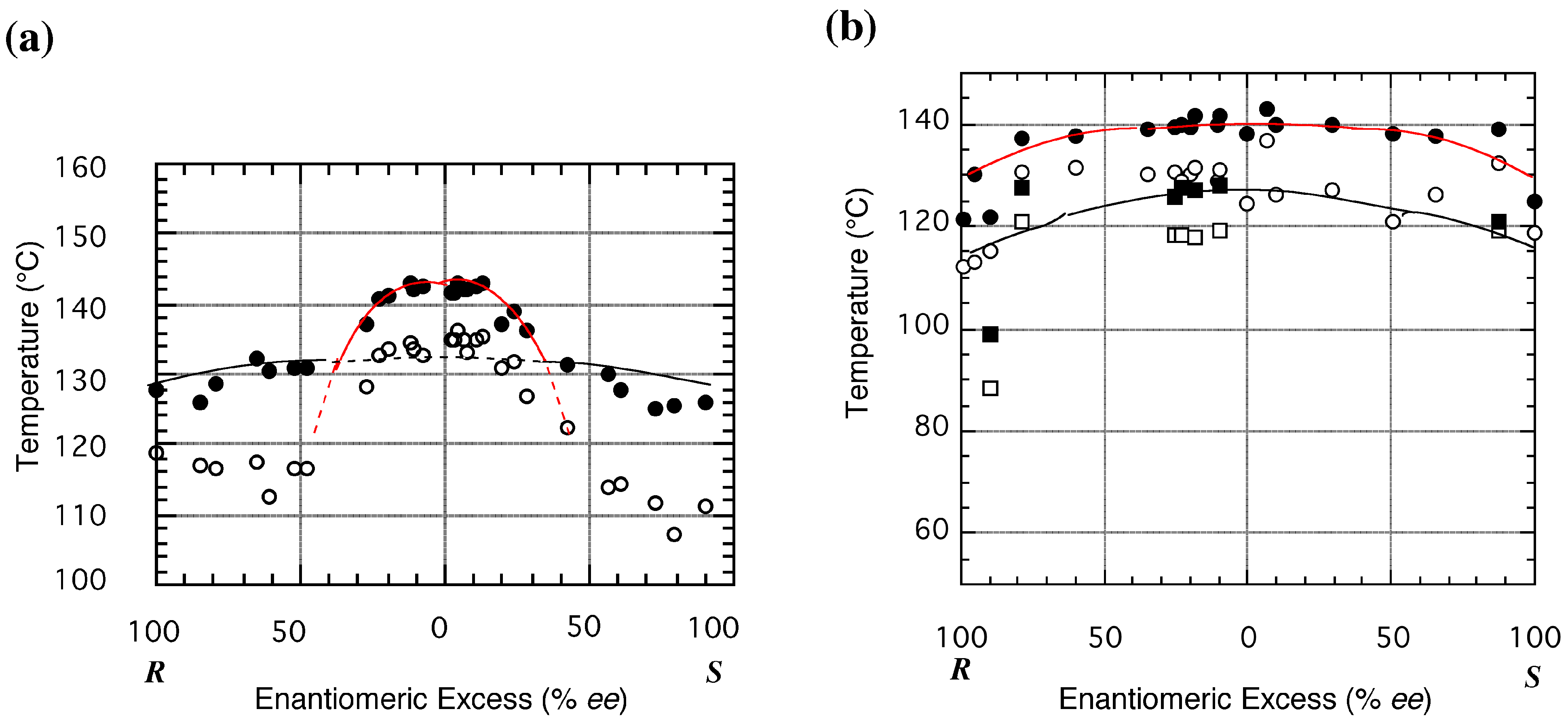
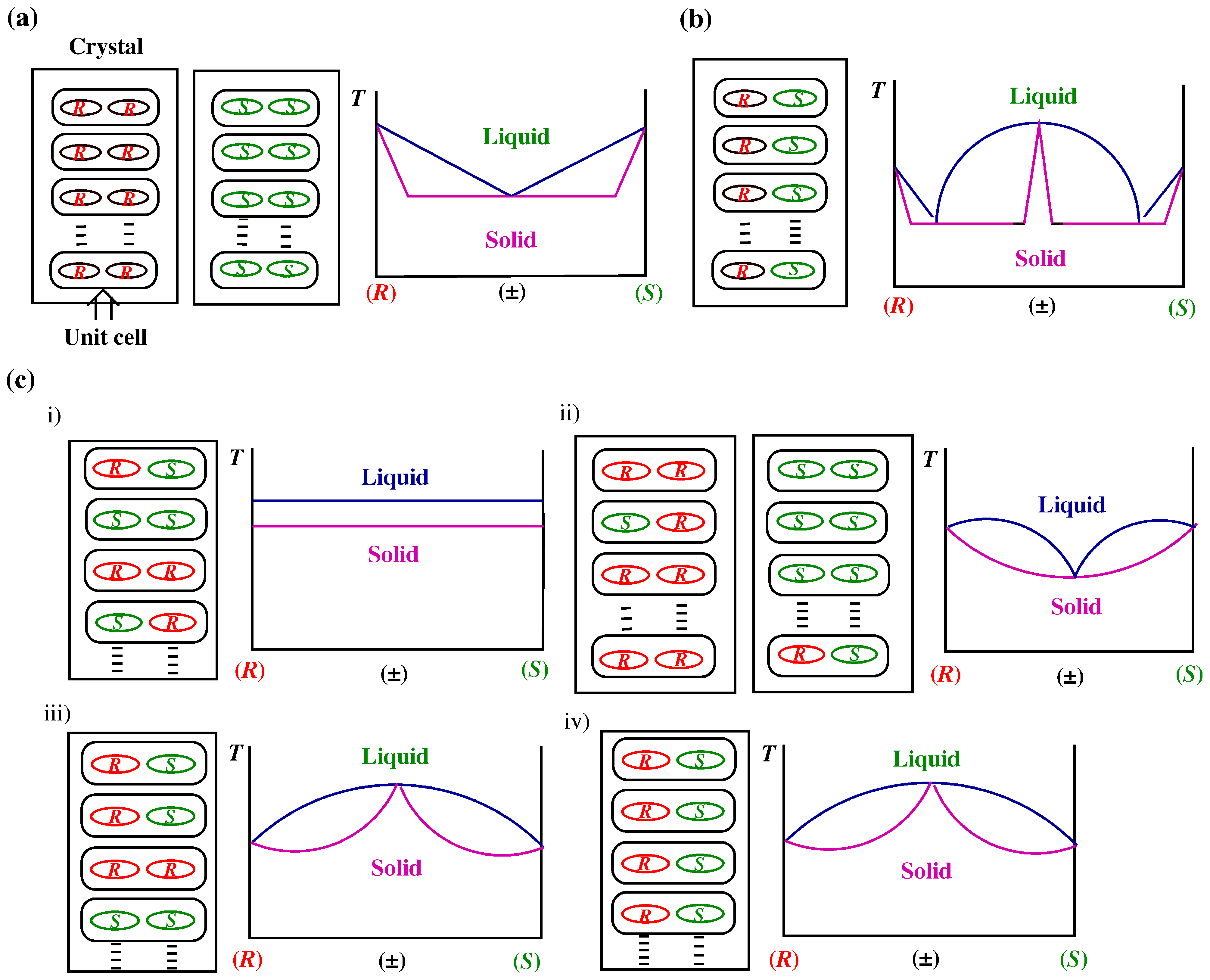
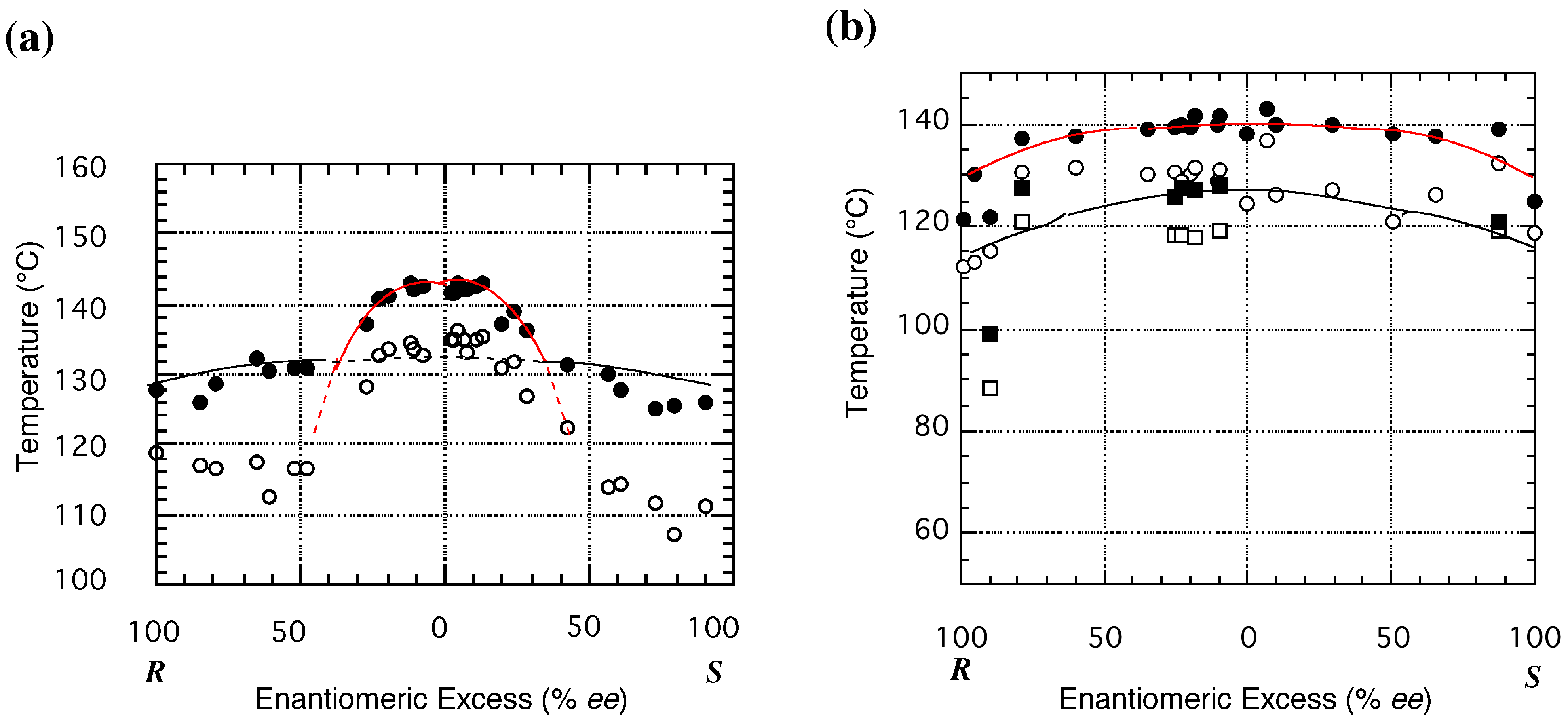
3.3. Melting Point Diagram
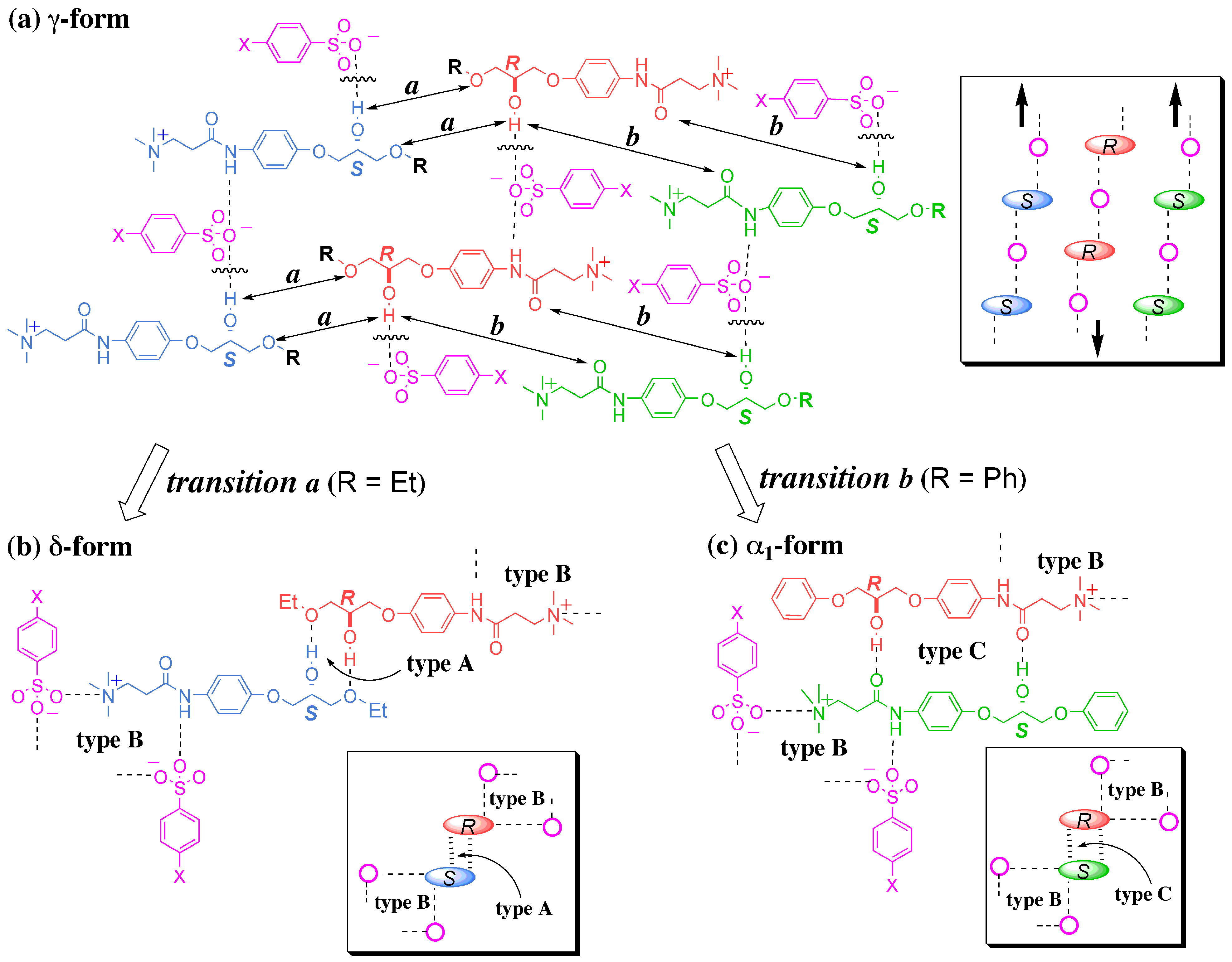
3.4. Flexible Modes of Polymorphic Transition
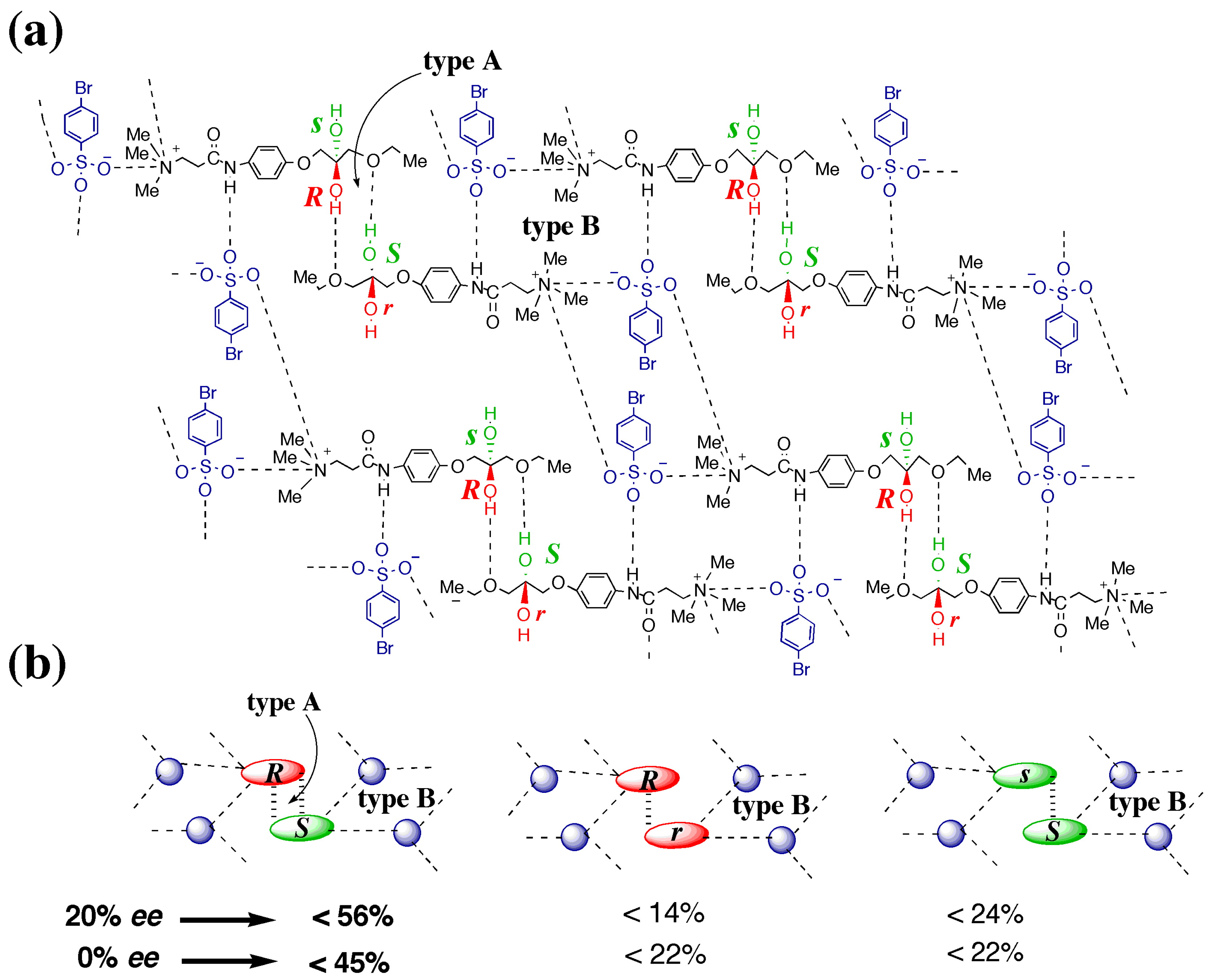
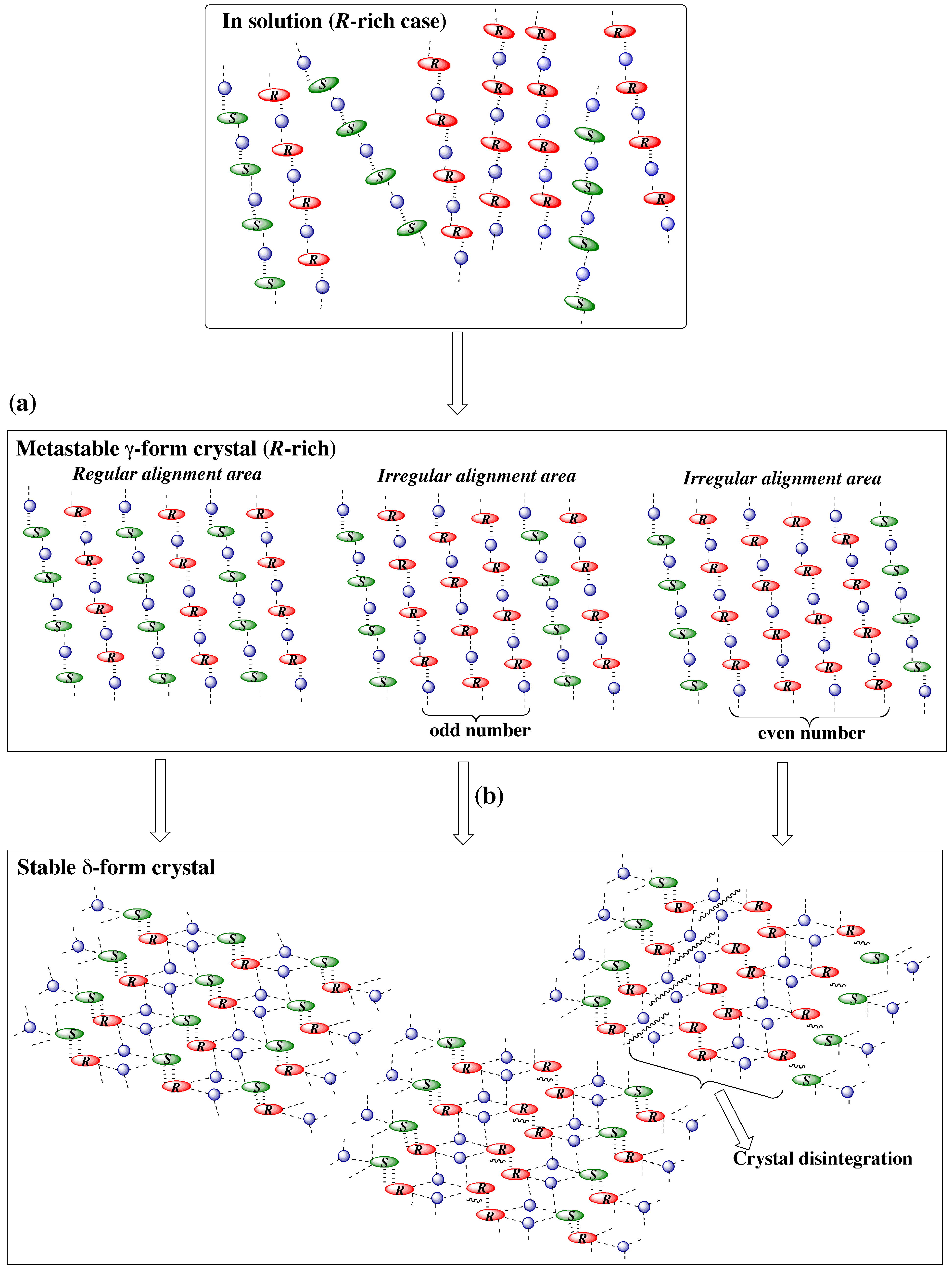
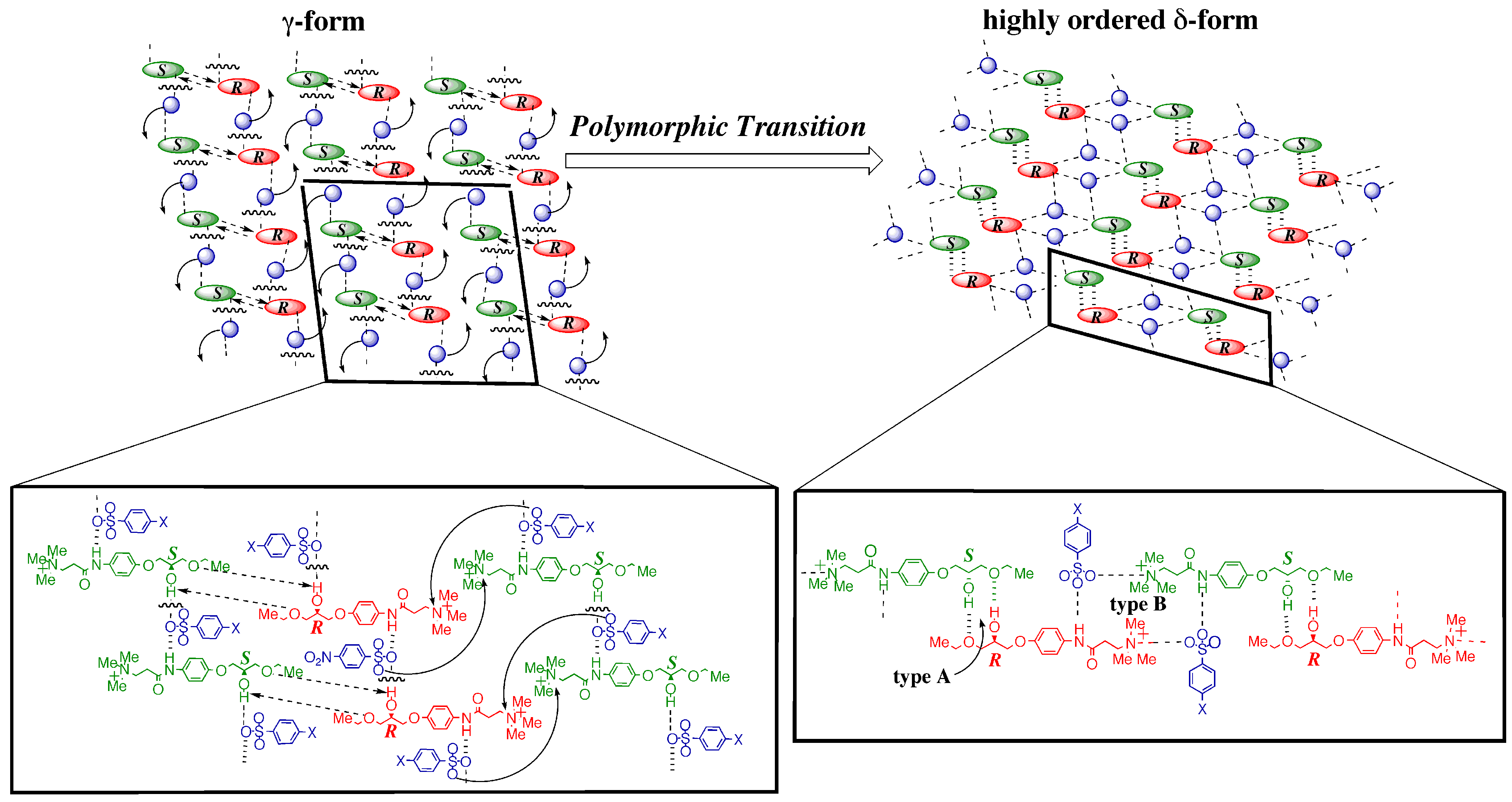
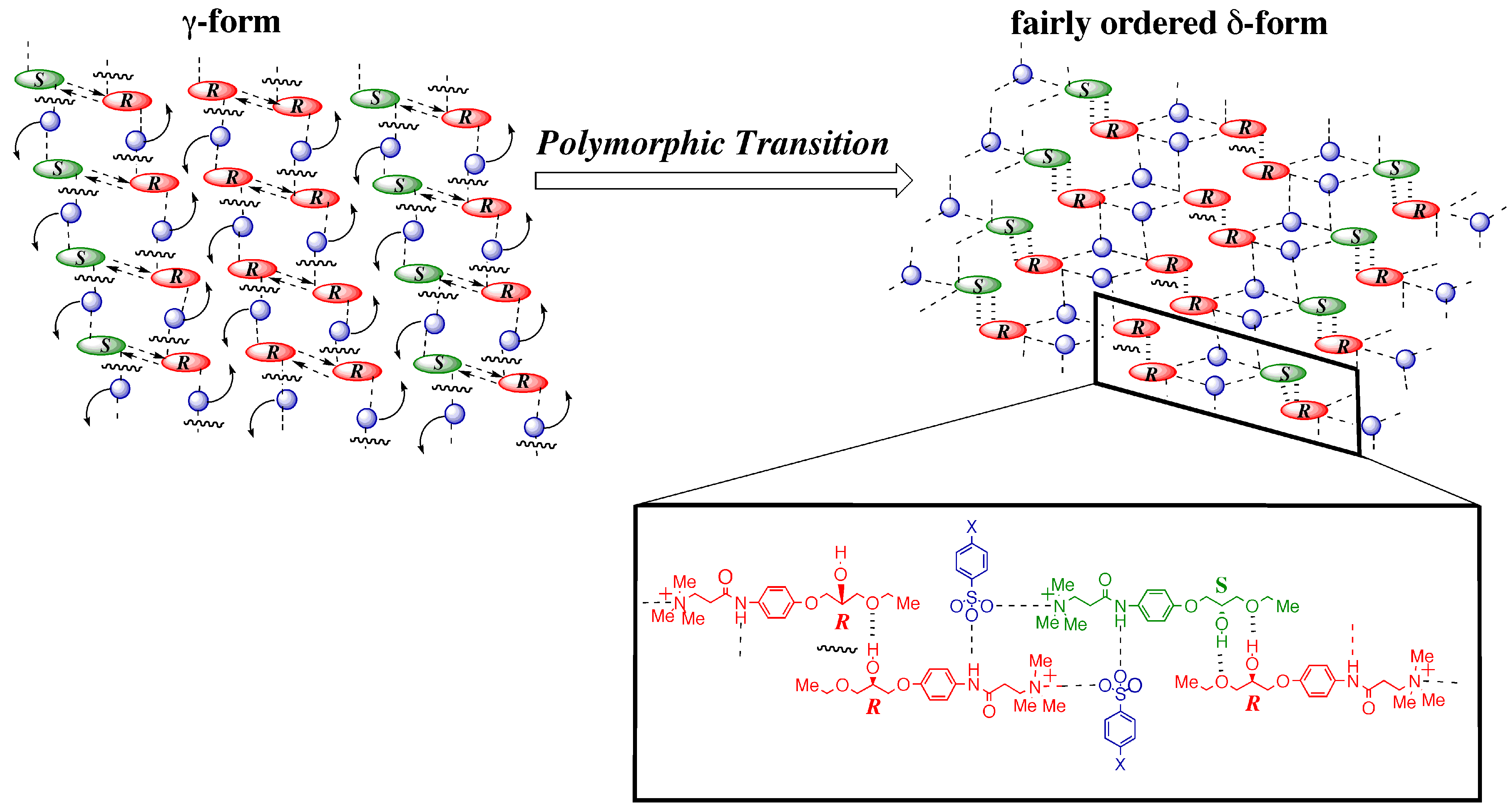
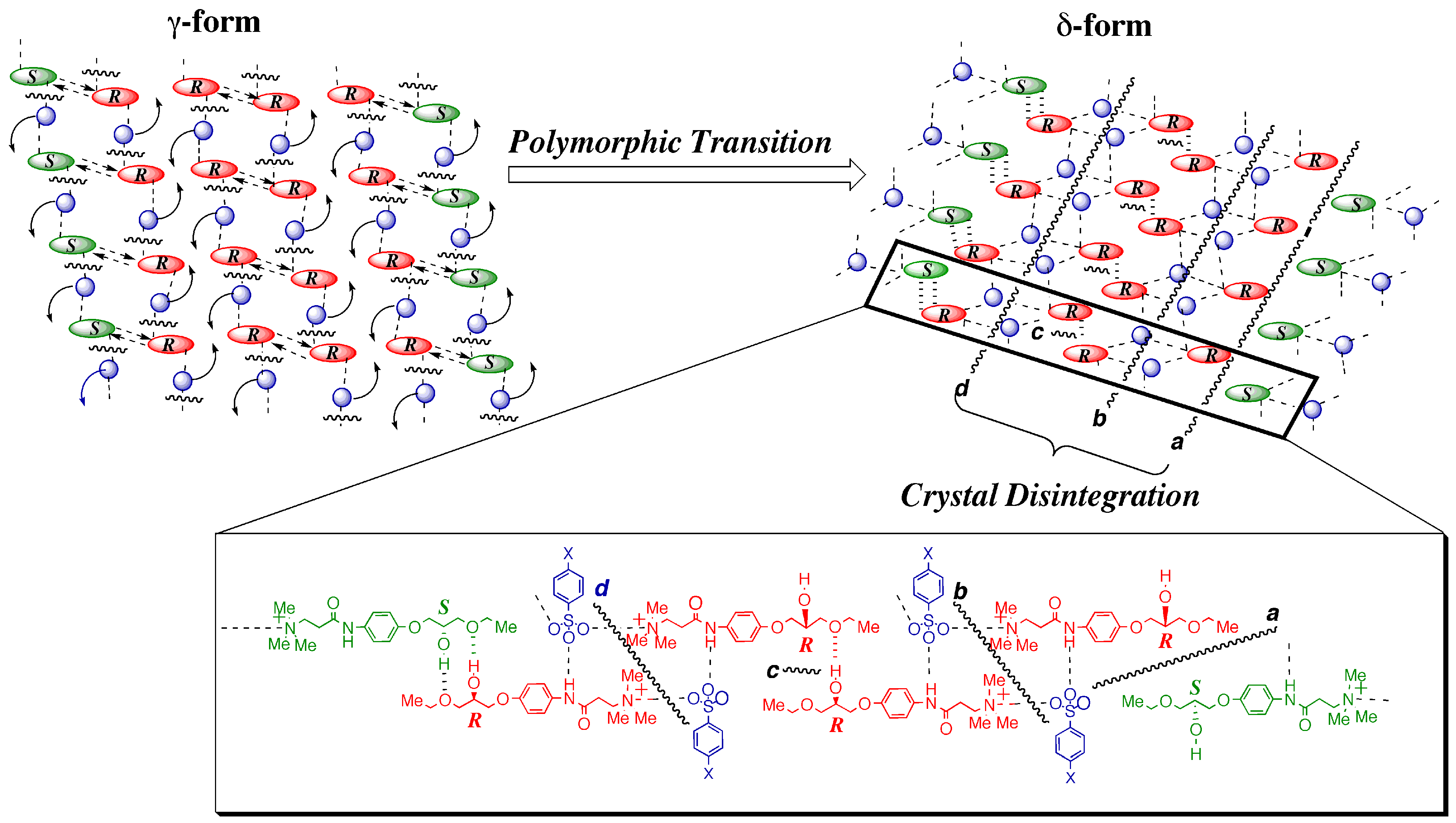
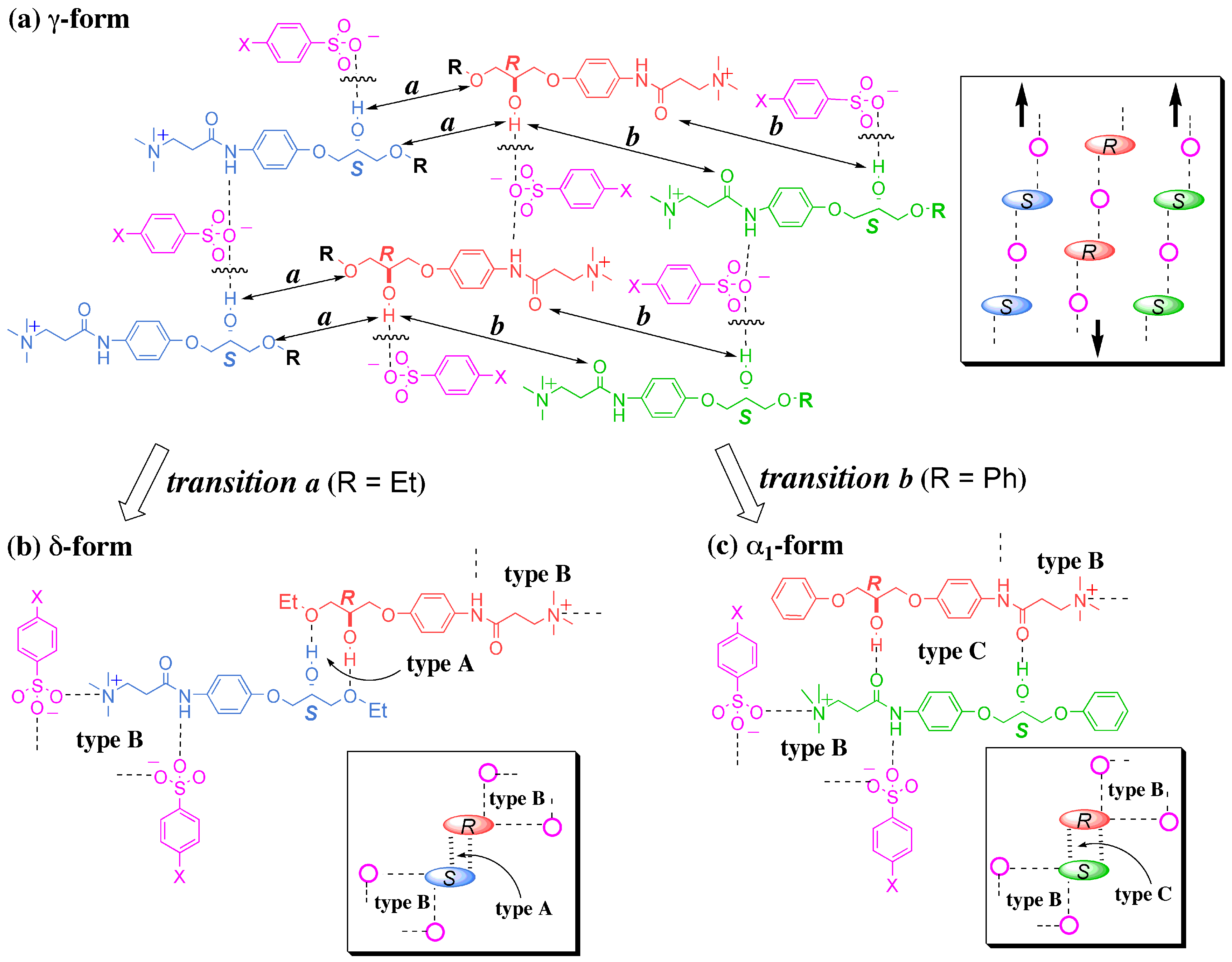
3.4.1. γ to δ transition
- 1)
- 2)
- 3)
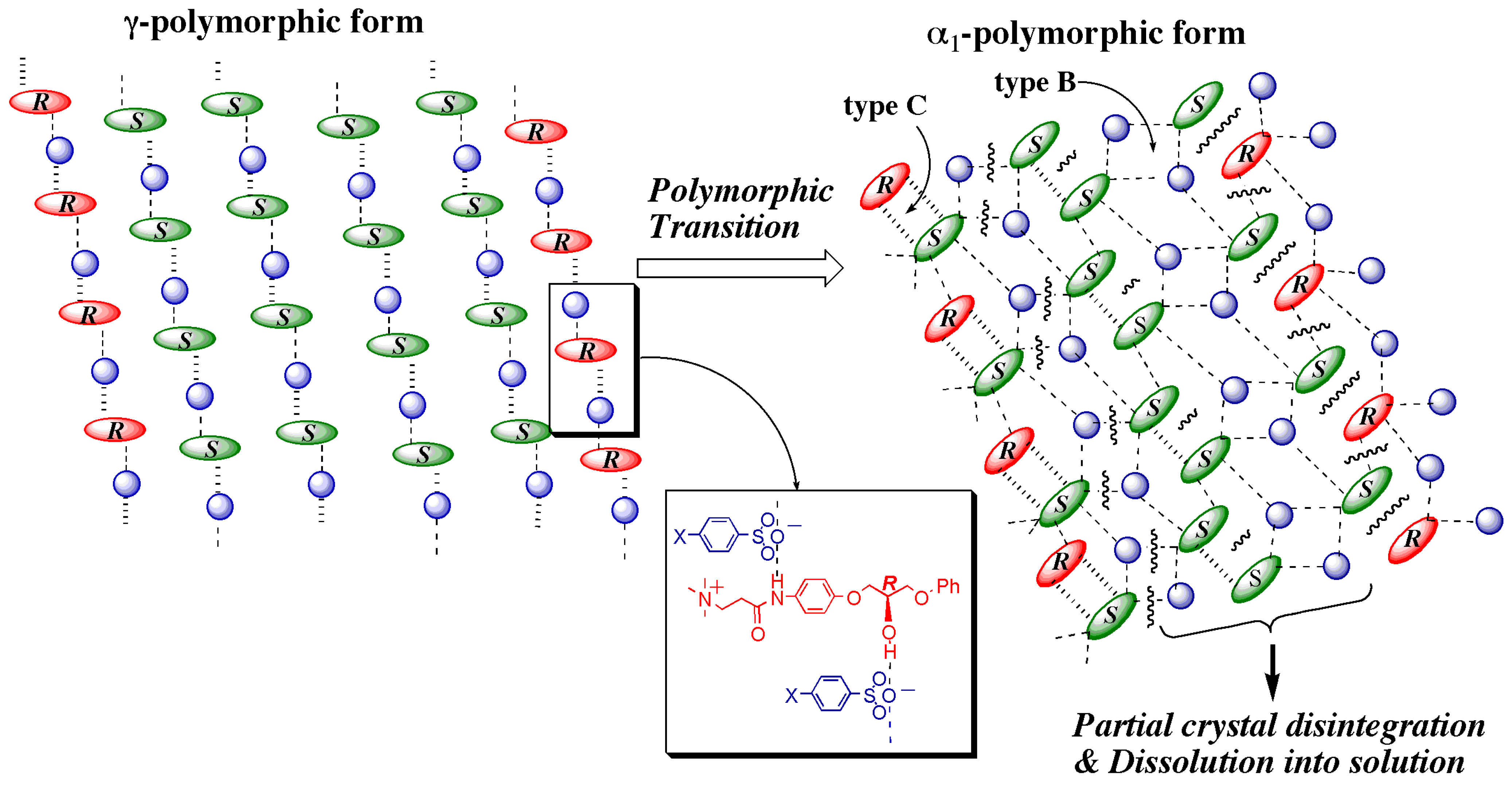
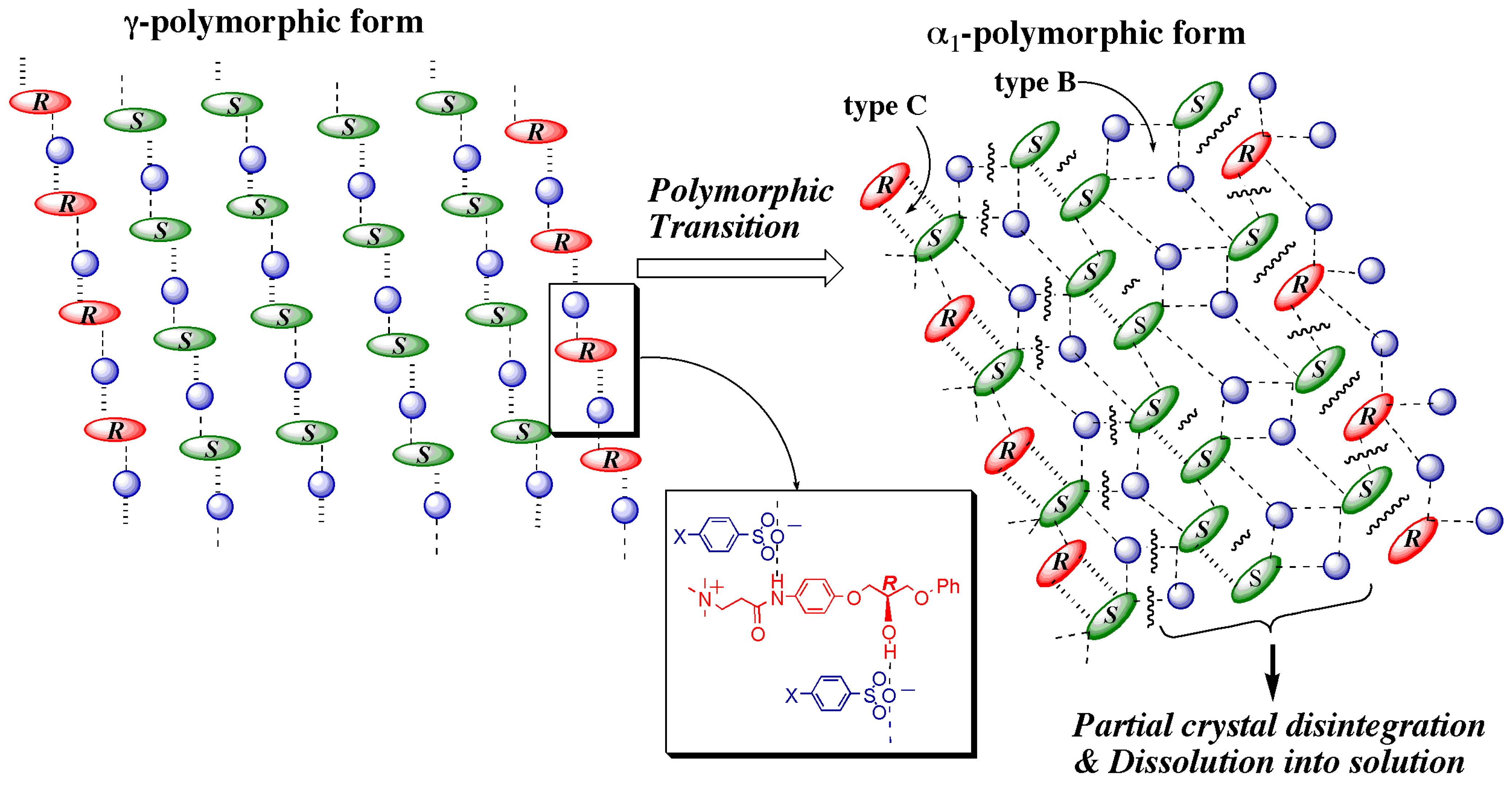
- At the sites where an even number of homochiral R chains are aligned between two S chains, after the polymorphic transition, partial crystal disintegration occurs in the resulting δ-form crystal to release the R-rich area into solution, because the R-rich area was surrounded by the sites where the OH⋅⋅⋅OEt interaction cannot be formed at all as well as the sites with weak electrostatic interactions (Figure 10b and Figure 13).
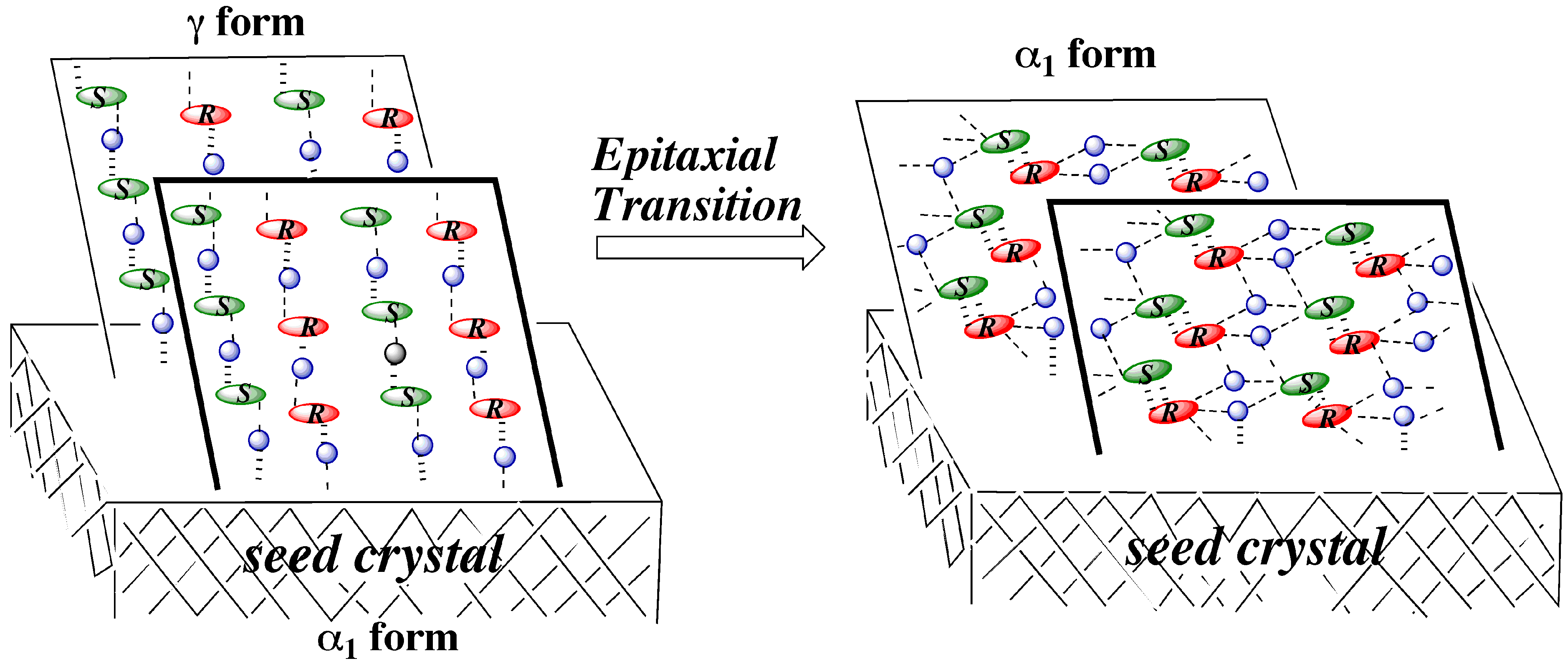
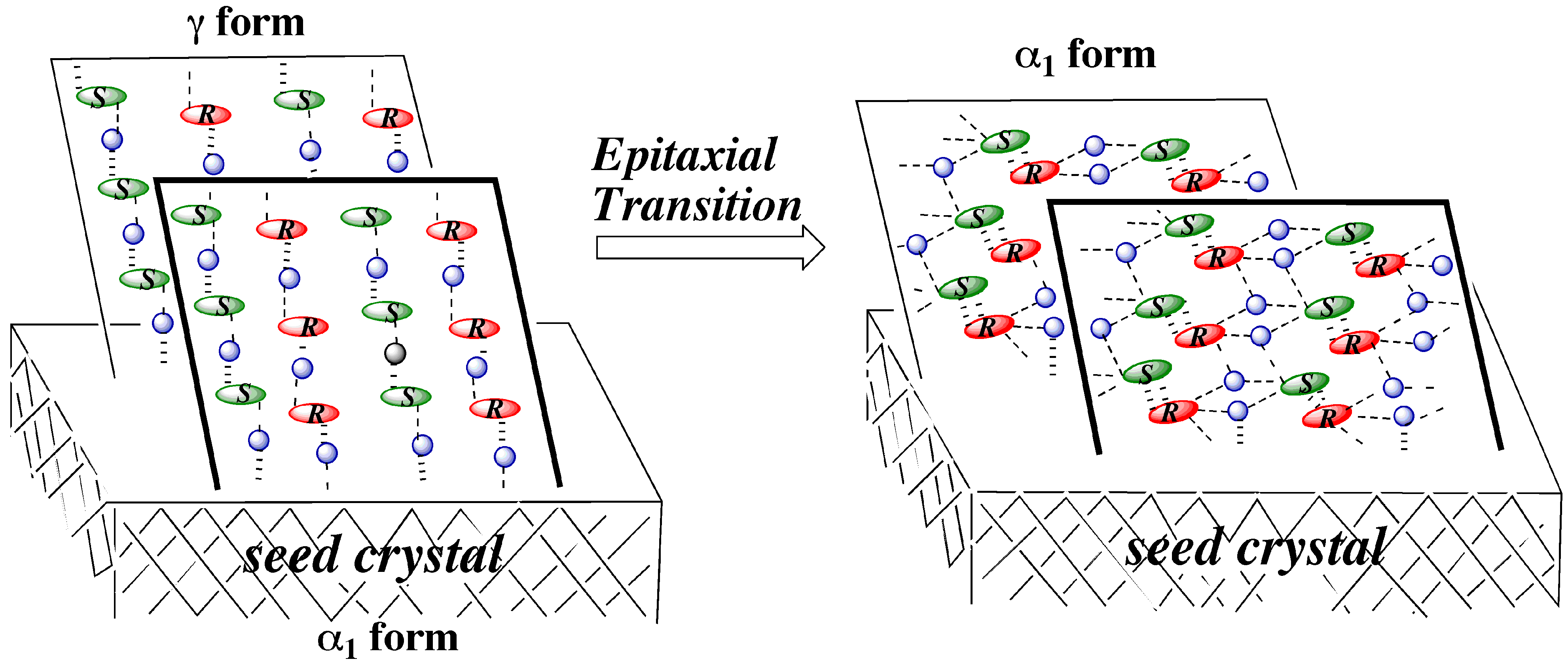
3.4.2. γ to α1 transition
3.5. Overall Mechanism of Preferential Enrichment
3.6. Requirements for the Occurrence of Preferential Enrichment
- 1)
- Polymorphic transition of a metastable polymorphic form into a stable one must occur during crystallization; this process is the essence of chiral symmetry breaking. This polymorphic transition can be monitored by the in situ ATR-IR spectroscopy. If there is a distinct difference in the vibrations of hydrogen bond forming groups between the supersaturated solution before crystallization and the deposited crystals after crystallization, polymorphic transition is most likely to occur.
- 2)
- The solubility of the enantiomerically pure sample is much higher than that of the corresponding racemic sample; this phenomenon is closely related to the preferential formation of homochiral 1D chain even in the racemic solution. The difference in the solubility between the racemic sample and the enantiomerically enriched one substantially affect the ee value reached in the mother liquor after crystallization; the larger the difference is, the higher the reached ee value is.
- 3)
- The deposited crystals must be a mixed crystal composed of fairly random alignment of two enantiomers so that the chiral symmetry breaking can be memorized.
4. Induction of Preferential Enrichment
5. Scope of Preferential Enrichment
6. Conclusions
Acknowledgements
References and Notes
- Mainzer, K. Symmetry and Complexity: The Sprit and Beauty of Nonlinear Science; World Scientific: Singapore, 2005. [Google Scholar]
- Kauffman, S. Investigations; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Waldrop, M.M. Complexity; Simon &Schuster: New York, NY, USA, 1992. [Google Scholar]
- Pérez-García, L.; Amabilino, D.B. Spontaneous Resolution Under Supramolecular Control. Chem. Soc. Rev. 2002, 31, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Ushio, T.; Tamura, R.; Takahashi, H.; Yamamoto, K. Unusual Enantiomeric Resolution Phenomenon Observed upon Recrystallization of a Racemic Compound. Angew. Chem. Int. Ed. Engl. 1996, 35, 2372–2374. [Google Scholar] [CrossRef]
- Ushio, T.; Tamura, R.; Azuma, N.; Nakamura, K.; Toda, F.; Kobayashi, K. Polymorphism of Compounds Existing as Both Racemic Compounds Crystals and Mixed Crystals of Enantiomers. Mol. Cryst. Liq. Cryst. 1996, 276, 245–252. [Google Scholar] [CrossRef]
- Tamura, R.; Takahashi, H.; Fujimoto, D.; Ushio, T. Mechanism and Scope of Preferential enrichment, a Symmetry-Breaking Enantiomeric Resolution Phenomenon. Top. Curr. Chem. 2007, 269, 53–82. [Google Scholar] [PubMed]
- Tamura, R.; Ushio, T. Preferential Enrichment: A Dynamic Enantiomeric Resolution Phenomenon Caused by Polymorphic Transition During Crystallization. In Enantiomer Separation: Fundamentals and Practical Methods; Toda, F., Ed.; Kluwer: Dordrecht, The Netherlands, 2004; pp. 135–163. [Google Scholar]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates and Resolutions; Krieger: Malabar, FL, USA, 1994. [Google Scholar]
- CRC Handbook of Optical Resolutions via Diastereomeric Salt Formation; Kozma, D. (Ed.) CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Kinbara, K.; Saigo, K. Chiral Discrimination during Crystallization. Top. Stereochem. 2003, 23, 207–265. [Google Scholar]
- Sakai, K.; Sakurai, R.; Nohira, H. New Resolution Technologies Controlled by Chiral Discrimination Mechanisms. Top. Curr. Chem. 2007, 269, 199–231. [Google Scholar] [PubMed]
- Kellogg, R.M.; Kaptein, B.; Vries, T.R. Dutch Rresolution of Racemates and the Roles of Solid Solution Formation and Nucleation Inhibition. Top. Curr. Chem. 2007, 269, 159–197. [Google Scholar] [PubMed]
- Toda, F. Optical Resolutions by Inclusion Complexation with a Chiral Host Compound. In Enantiomer Separation: Fundamentals and Practical Methods; Toda, F., Ed.; Kluwer: Dordrecht, The Netherlands, 2004; pp. 1–47. [Google Scholar]
- Collet, A.; Brienne, M.-J.; Jacques, J. Optical Resolution by Direct Crystallization of Enantiomer Mixtures. Chem. Rev. 1980, 80, 215–230. [Google Scholar] [CrossRef]
- Collet, A. Optical Resolution. In Comprehensive Supramolecular Chemistry; Reinhoudt, D.N., Ed.; Pergamon: Oxford, UK, 1996; Vol. 10, pp. 113–149. [Google Scholar]
- Kinbara, K.; Hashimoto, Y.; Sukegawa, M.; Nohira, H.; Saigo, K. Crystal Structures of the Salts of Chiral Primary Amines with Achiral Carboxylic Acids: Recognition of the Commonly-Occurring Supramolecular Assemblies of hydrogen-Bond Networks and Their Role in the Formation of Conglomerates. J. Am. Chem. Soc. 1996, 118, 3441–3449. [Google Scholar] [CrossRef]
- Coquerel, G. Preferential Crystallization. Top. Curr. Chem. 2007, 269, 1–51. [Google Scholar]
- Eliel, E.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; Wiley: New York, NY, USA, 1994; pp. 297–464. [Google Scholar]
- Tamura, R.; Takahashi, H.; Hirotsu, K.; Nakajima, Y.; Ushio, T.; Toda, F. Unusual Disordered Crystal Structure of a Racemate Exhibiting a Novel Enantiomeric Resolution: Preferential Enrichment. Angew. Chem. Int. Ed. Engl. 1998, 37, 2876–2878. [Google Scholar]
- Tamura, R.; Fujimoto, D.; Lepp, Z.; Misaki, K.; Miura, H.; Takahashi, H.; Ushio, T.; Nakai, T.; Hirotsu, K. Mechanism of Preferential Enrichment, an Unusual Enantiomeric Resolution Phenomenon Caused by Polymorphic Transition during Crystallization of Mixed Crystals Composed of Two Enantiomers. J. Am. Chem. Soc. 2002, 124, 13139–13153. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, D.; Tamura, R.; Lepp, Z.; Takahashi, H.; Ushio, T. Mechanism of a New type of Solvent-Assisted Solid-to-Solid Polymorphic Transition Causing Preferential Enrichment: Prominent Influence of C(sp2)H---O Interaction on the Control of a Crystal Structure. Cryst. Growth Des. 2003, 3, 973–979. [Google Scholar] [CrossRef]
- Tamura, R.; Mizuta, M.; Yabunaka, S.; Fujimoto, D.; Ariga, T.; Okuhara, S.; Ikuma, N.; Takahashi, H.; Tsue, H. Induction and Inhibition of Preferential Enrichment by Controlling the Mode of the Polymorphic Transition with Seed Crystals. Chem. Eur. J. 2006, 12, 3515–3527. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Okuhara, S.; Shimano, E.; Fujimoto, D.; Takahashi, H.; Tsue, H.; Tamura, R. Control of the Mode of Polymorphic Transition Inducing Preferential Enrichment by Modifying the Molecular Structure or Adding Seed Crystals: Significant Influence of CH/F Hydrogen Bonds. Cryst. Growth Des. 2008, 8, 540–548. [Google Scholar] [CrossRef]
- Horiguchi, M.; Okuhara, S.; Shimano, E.; Fujimoto, D.; Takahashi, H.; Tsue, H.; Tamura, R. Mechanistic Flexibility of Solvent-Assisted Solid-to-Solid Polymorphic Transition Causing Preferential Enrichment: Significant Contribution of π/π and CH/π Interactions as Well as Hydrogen Bonds. Cryst. Growth Des. 2007, 7, 1643–1652. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Polymorphism in Pharmaceutical Solids, Drugs and the Pharmaceutical Sciences; Brittain, H.G. (Ed.) Marcel Dekker: New York, NY, USA, 1999; Vol. 95. [Google Scholar]
- Bernstein, J.; Davey, R.J.; Henck, J.-O. Concomitant Polymorphs. Angew. Chem. Int. Ed. Engl. 1999, 38, 3440–3461. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Bernstein, J. Disappearing Polymorphs. Acc. Chem. Res. 1995, 28, 193–200. [Google Scholar] [CrossRef]
- McCrone, W.C. Polymorphism. In Physics and Chemistry of the Organic Solid State; Fox, D., Labes, M.M., Weissberger, A., Eds.; Interscience: New York, NY, USA, 1965; Vol. II, pp. 726–767. [Google Scholar]
- Ostwald, W. Grundriss der Allgemeinen Chemie; W. Engelmann: Leipzig, 1899. [Google Scholar]
- Parkinson, G.M.; Thomas, J.M.; Williams, J.O.; Goringe, M.J.; Hobbs, L.W. The Suggested Role of Partial Dislocations in the Single Crystal–Single Crystal Phase Transition of a Cyclo-octane Molecular Cationic Salt. J. Chem. Soc. Perkin 2 1976, 836–838. [Google Scholar] [CrossRef]
- Tamura, R.; Ushio, T.; Nakamura, K.; Takahashi, H.; Azuma, N.; Toda, F. Ideal Enantiomeric Resolution by Recrystallization of a Racemic Compound. Enantiomer 1997, 2, 277–280. [Google Scholar]
- Tamura, R.; Ushio, T.; Takahashi, H.; Nakamura, K.; Azuma, N.; Toda, F.; Endo, K. Ideal Enantiomeric Resolution of a Racemic Compound (Part 4): Relationship Between Enantiomeric Resolution Phenomenon and Crystal Properties. Chirality 1997, 9, 220–224. [Google Scholar] [CrossRef]
- Takahashi, H.; Tamura, R.; Ushio, T.; Nakajima, Y.; Hirotsu, K. Ideal Enantiomeric Resolution (Preferential Enrichment) by Recrystallization of a Racemic Compound. V: Relationship Between Preferential Enrichment and Crystal Structures. Chirality 1998, 10, 705–710. [Google Scholar] [CrossRef]
- Tamura, R.; Takahashi, H.; Hirotsu, K.; Nakajima, Y.; Ushio, T. Preferential Enrichment and Crystal Structure. Mol. Cryst. Liq. Cryst. 2001, 356, 185–194. [Google Scholar] [CrossRef]
- Tamura, R.; Takahashi, H.; Ushio, T.; Nakajima, Y.; Hirotsu, K.; Toda, F. Ideal Enantiomeric Resolution (Preferential Enrichment) by Recrystallization of a Racemic Compound (Part 6): Hydrogen Bonding Mode in the Crystal Structure. Enantiomer 1998, 3, 149–157. [Google Scholar]
- Tamura, R.; Takahashi, H.; Miura, H.; Lepp, Z.; Nakajima, Y.; Hirotsu, K.; Ushio, T. Comparison of Crystal Structures of New Racemic Chiral Compounds Showing and Not Showing the Phenomenon of Preferential Enrichment. Supramol. Chem. 2001, 13, 71–78. [Google Scholar] [CrossRef]
- Takahashi, H.; Tamura, R.; Lepp, Z.; Kobayashi, K.; Ushio, T. Preferential Enrichment: An Essential Crystal Structure. Enantiomer 2001, 6, 57–66. [Google Scholar] [PubMed]
- Takahashi, H.; Tamura, R.; Fujimoto, D.; Lepp, Z.; Kobayashi, K.; Ushio, T. Preferential Enrichment: Full Crystallographic Analysis of the Unusual Phenomenon in the Mixed Crystals’ Version. Chirality 2002, 14, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Tamura, R.; Yabunaka, S.; Ushio, T. Crystal Structure of a New Racemate Showing Preferential Enrichment: Evidence for the Existence as a Racemic Mixed Crystal Composed of the Two Enantiomers. Mendeleev Commun. 2003, 119–121. [Google Scholar] [CrossRef]
- Miura, H.; Ushio, T.; Nagai, K.; Fujimoto, D.; Lepp, Z.; Takahashi, H.; Tamura, R. Crystallization of a Desired Metastable Polymorph by Pseudoseeding, Crystal Structure Solution from Its Powder X-ray Diffraction Data, and Confirmation of Polymorphic Transition. Cryst. Growth Des. 2003, 3, 959–965. [Google Scholar] [CrossRef]
- Takahashi, H.; Tamura, R.; Yabunaka, S.; Mizuta, M.; Ikums, N.; Tsue, H.; Ushio, T. Significant Contribution of Phenyl Centroid---I–C(sp2) Coulombic Donor-Acceptor Attraction to the Buildup of a Crystal Structure. Mendeleev Commun. 2004, 239–241. [Google Scholar] [CrossRef]
- Fujimoto, D.; Takahashi, H.; Ariga, T.; Tamura, R. Preferential Enrichment: Significant Influence of Minor Molecular Modification on the Mode of Polymorphic Transition during Crystallization. Chiraliry 2006, 18, 188–195. [Google Scholar] [CrossRef]
- Horiguchi, M.; Yanunaka, S.; Iwama, S.; Shimano, E.; Lepp, Z.; Takahashi, H.; Tsue, H.; Tamura, R. Case Study on the Effects of Molecular Structure on the Mode of Polymorphic Transition Inducing Preferential Enrichment. Eur. J. Org. Chem. 2008, 3496–3505. [Google Scholar] [CrossRef]
- Maruyama, S.; Ooshima, H. Crystallization Behavior of Taltirelin Polymorphs in a Mixture of Water and Methanol. J. Crystal Growth 2000, 212, 239–245. [Google Scholar] [CrossRef]
- Kitamura, M.; Ueno, S.; Sato, K. Molecular Aspects of the Polymorphic Crystallization of Amino Acids and Lipids. In Crystallization Processes; Ohtaki, H., Ed.; Wiley: Chichester, UK, 1998; pp. 99–129. [Google Scholar]
- Kitamura, M. Polymorphism in the Crystallization of L-Glutamic Acid. J. Crystal Growth 1989, 96, 541–546. [Google Scholar] [CrossRef]
- Dobashi, A.; Saito, N.; Motoyama, Y.; Hara, S. Self-Induced Nonequivalence in the Association of D- and L-Amino Acid Derivatives. J. Am. Chem. Soc. 1986, 108, 307–308. [Google Scholar] [CrossRef]
- Jursic, B.S.; Goldberg, S.I. Enantiomer Discrimination Arising from Solute-Solute Interactions in Partially Resolved Chloroform Solutions of Chiral Carboxamides. J. Org. Chem. 1992, 57, 7172–7174. [Google Scholar] [CrossRef]
- Harger, M.J.P. Proton Magnetic Resonance Non-equivalence of the Enantiomers of Alkylphenylphosphinic Amides. J. Chem. Soc. Perkin Trans. 2 1977, 1882–1887. [Google Scholar] [CrossRef]
- Harger, M.J.P. Chemical Shift Non-equivalence of Enantiomers in the Proton Magnetic Resonance Spectra of Partly Resolved Phosphinothioic Acids. J. Chem. Soc. Perkin Trans. 2 1978, 326–331. [Google Scholar] [CrossRef]
- Iwama, S.; Horiguchi, M.; Uchida, Y.; Sato, H.; Takahashi, H.; Tsue, H.; Tamura, R. Observation of the Preferential Enrichment Phenomenon for Essential Amino Acids. Manuscript submitted for publication. This work was presented in part at the 19th International Conference on the Chemistry of Organic Solid State, Sestri Levante, Italy, 14–19 June 2009. [Google Scholar]


© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tamura, R.; Iwama, S.; Takahashi, H. Chiral Symmetry Breaking Phenomenon Caused by a Phase Transition. Symmetry 2010, 2, 112-135. https://doi.org/10.3390/sym2010112
Tamura R, Iwama S, Takahashi H. Chiral Symmetry Breaking Phenomenon Caused by a Phase Transition. Symmetry. 2010; 2(1):112-135. https://doi.org/10.3390/sym2010112
Chicago/Turabian StyleTamura, Rui, Sekai Iwama, and Hiroki Takahashi. 2010. "Chiral Symmetry Breaking Phenomenon Caused by a Phase Transition" Symmetry 2, no. 1: 112-135. https://doi.org/10.3390/sym2010112
APA StyleTamura, R., Iwama, S., & Takahashi, H. (2010). Chiral Symmetry Breaking Phenomenon Caused by a Phase Transition. Symmetry, 2(1), 112-135. https://doi.org/10.3390/sym2010112