
Synthesis of Axially Chiral Boron Compounds



Abstract
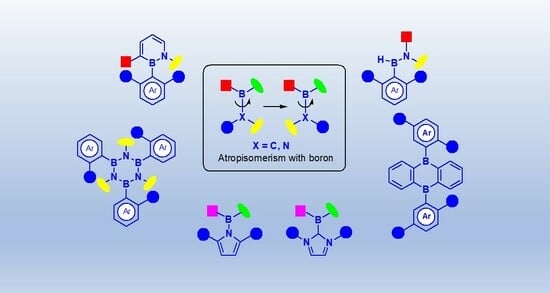
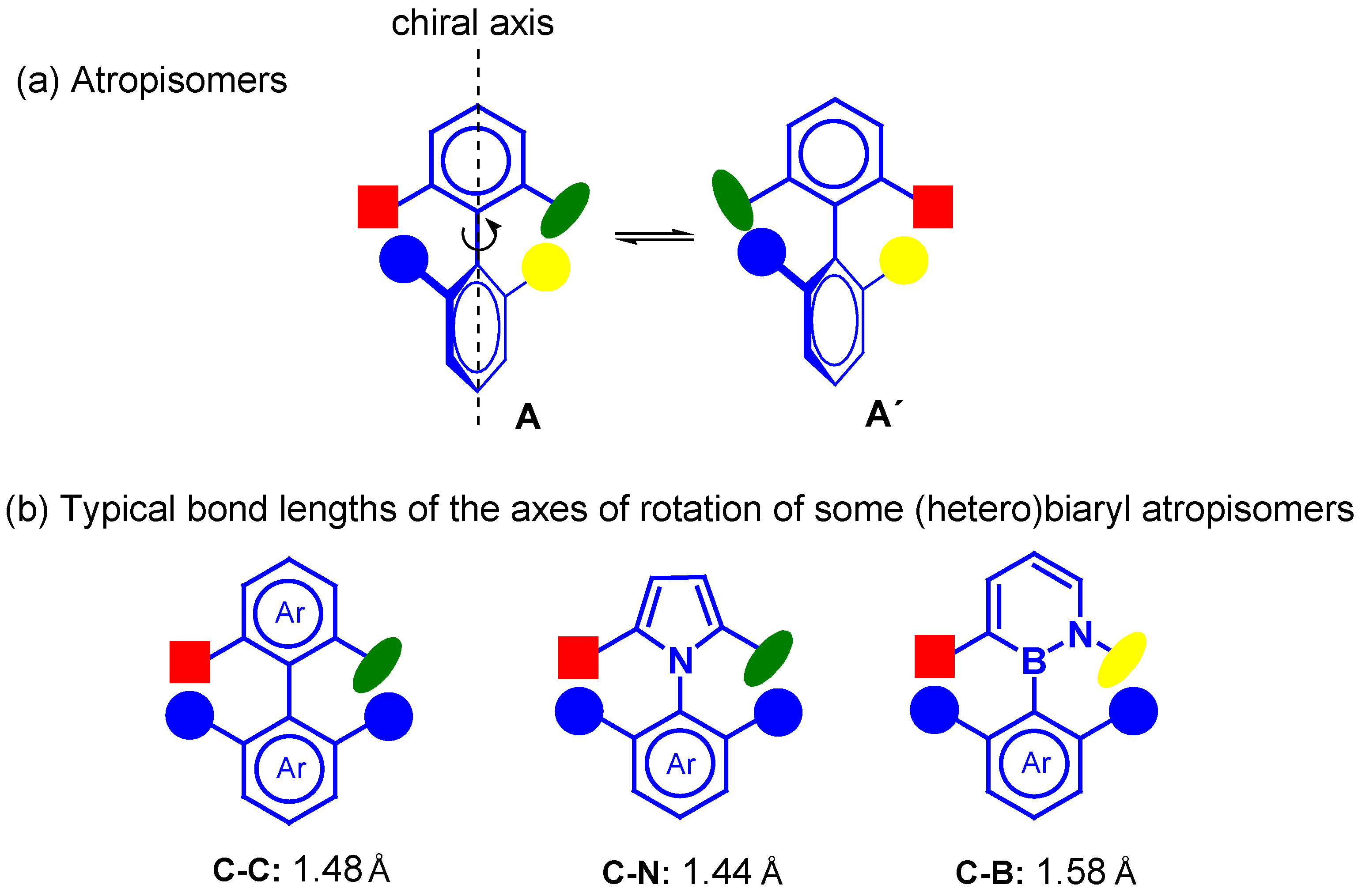
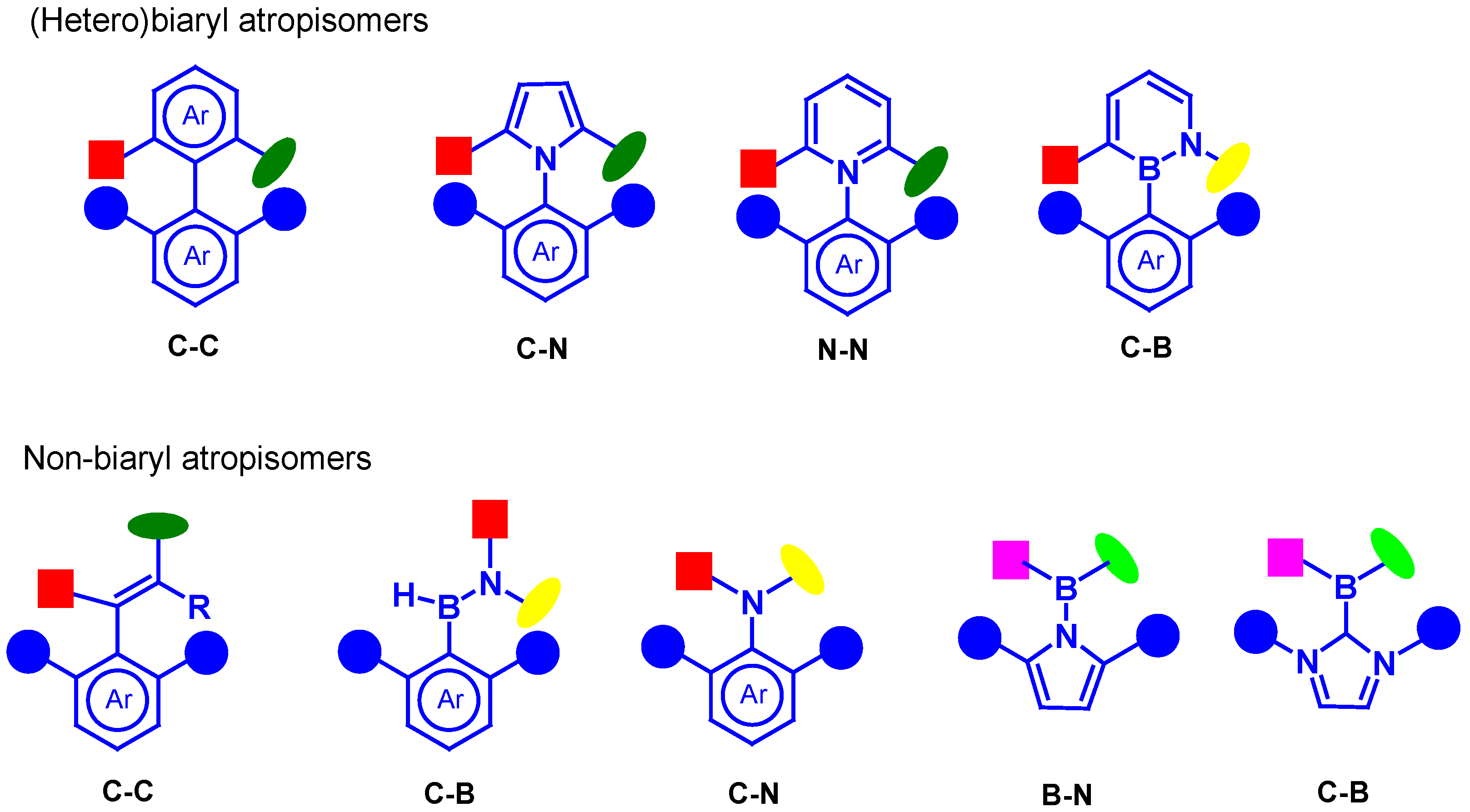
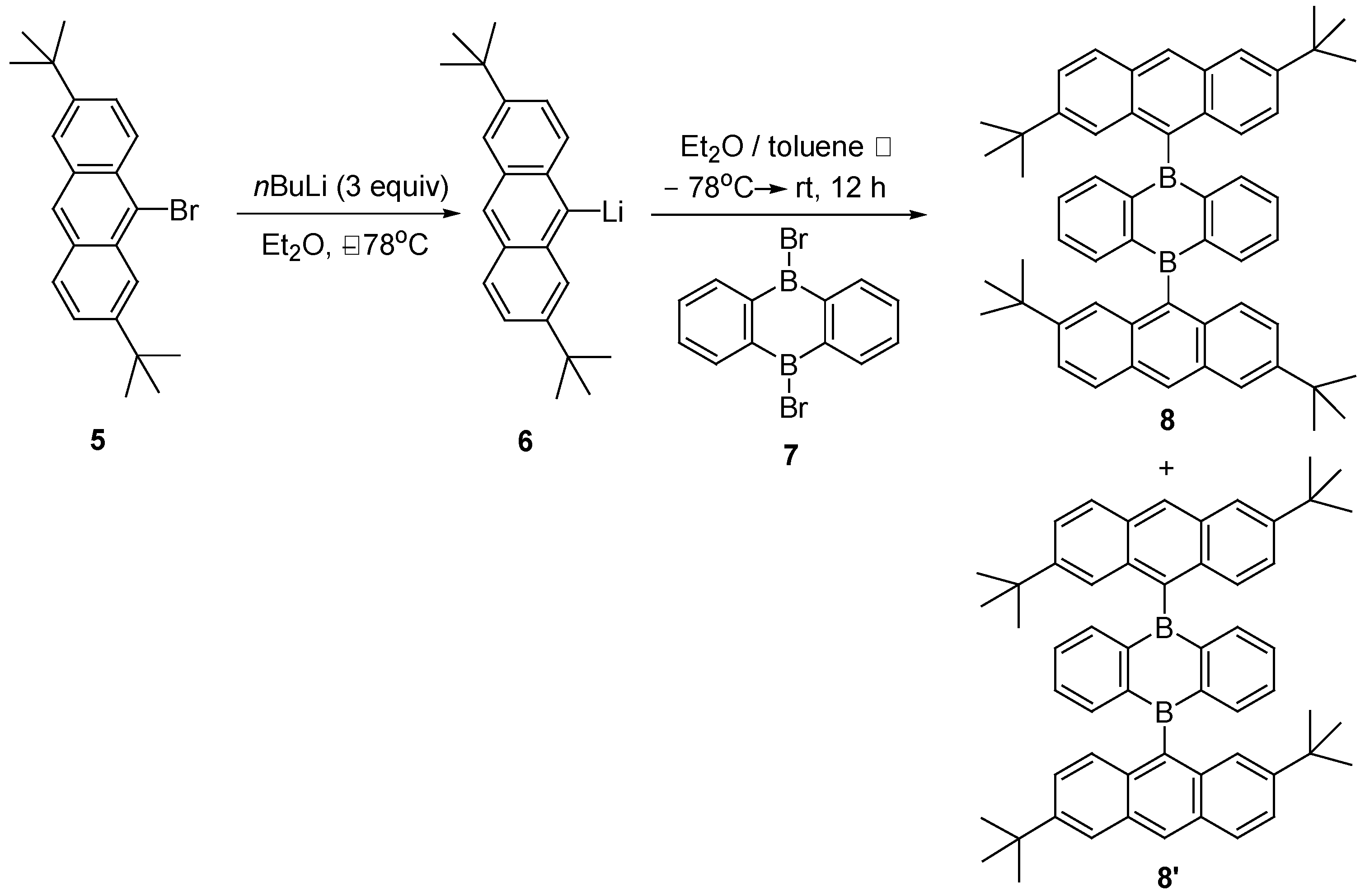
:
1. Introduction
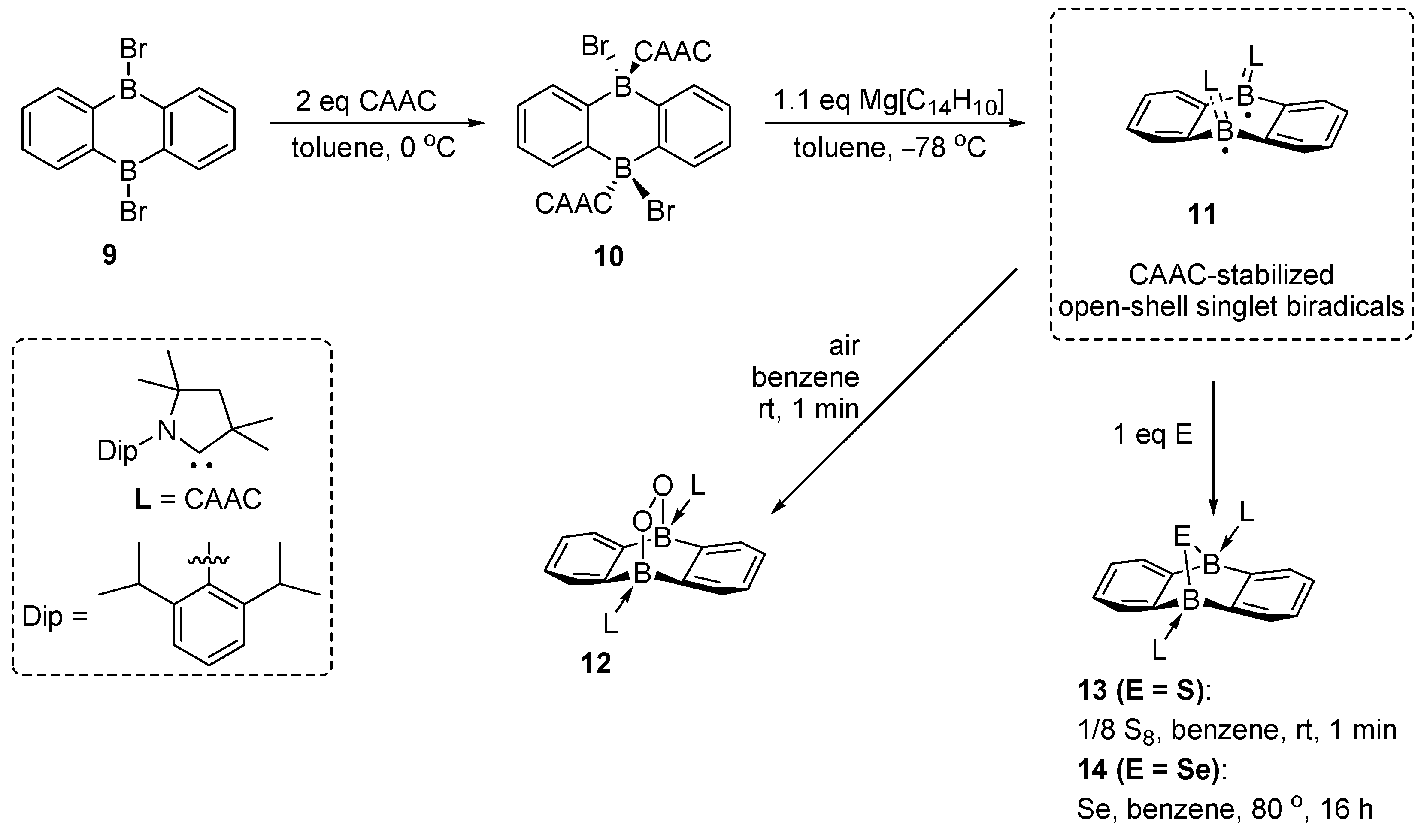
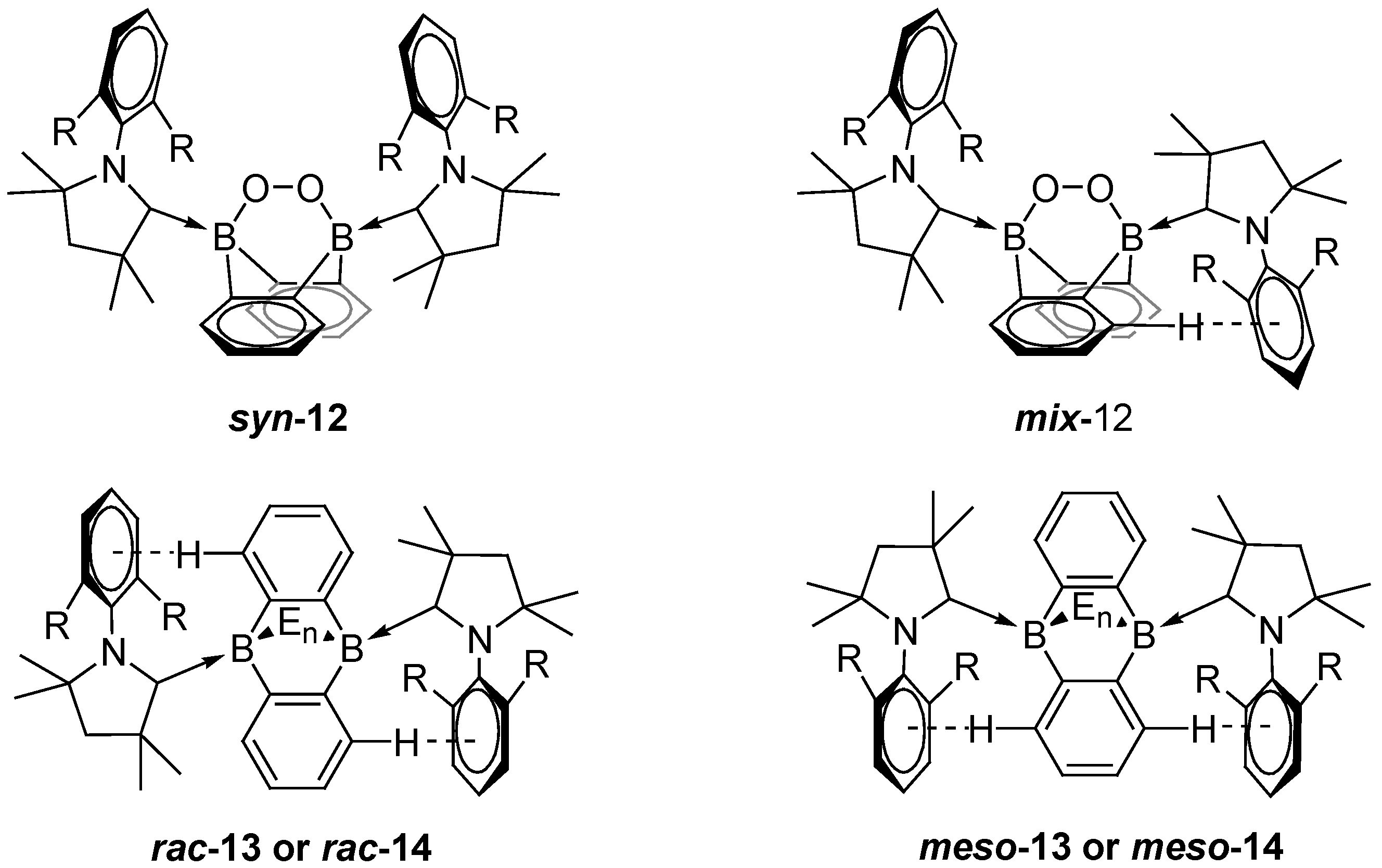
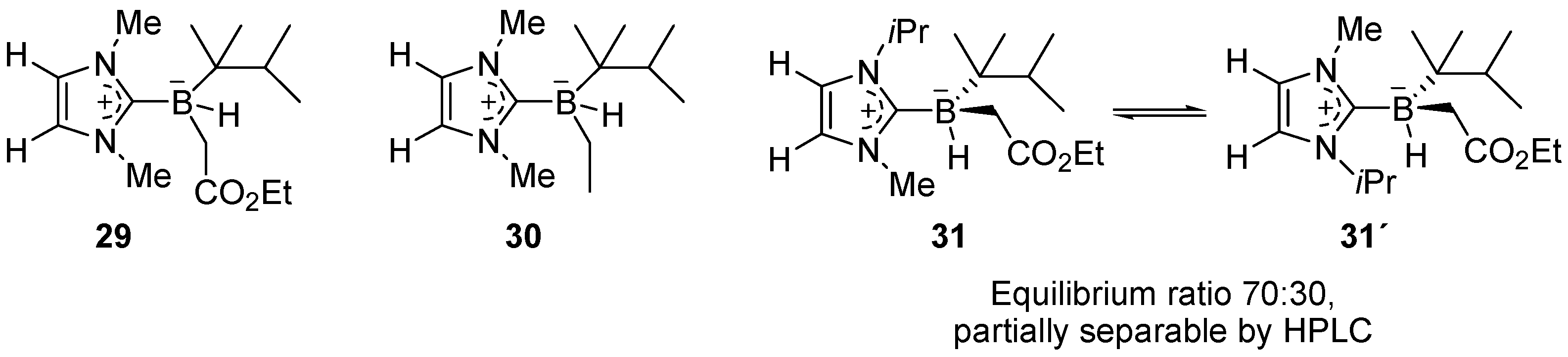
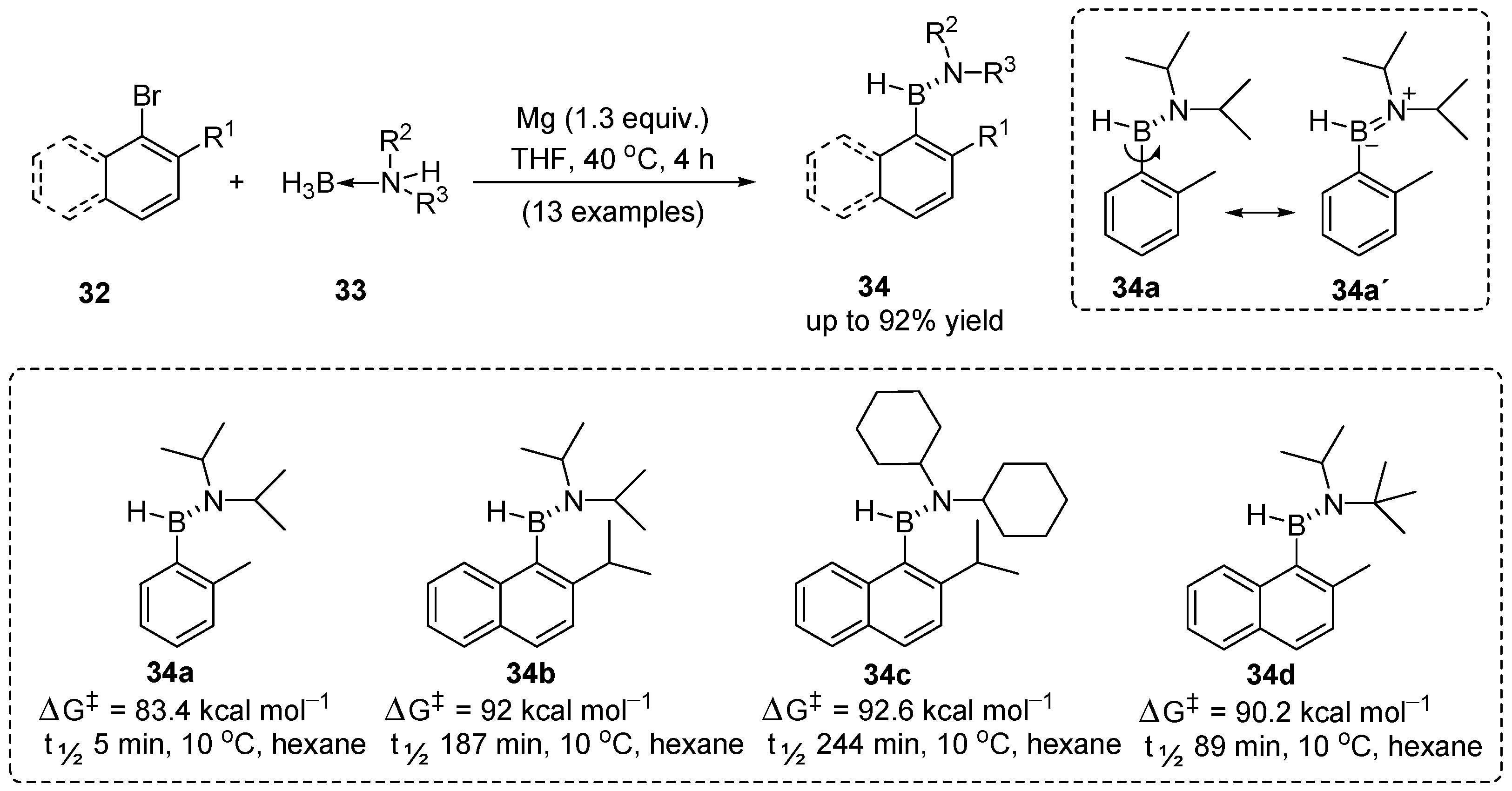
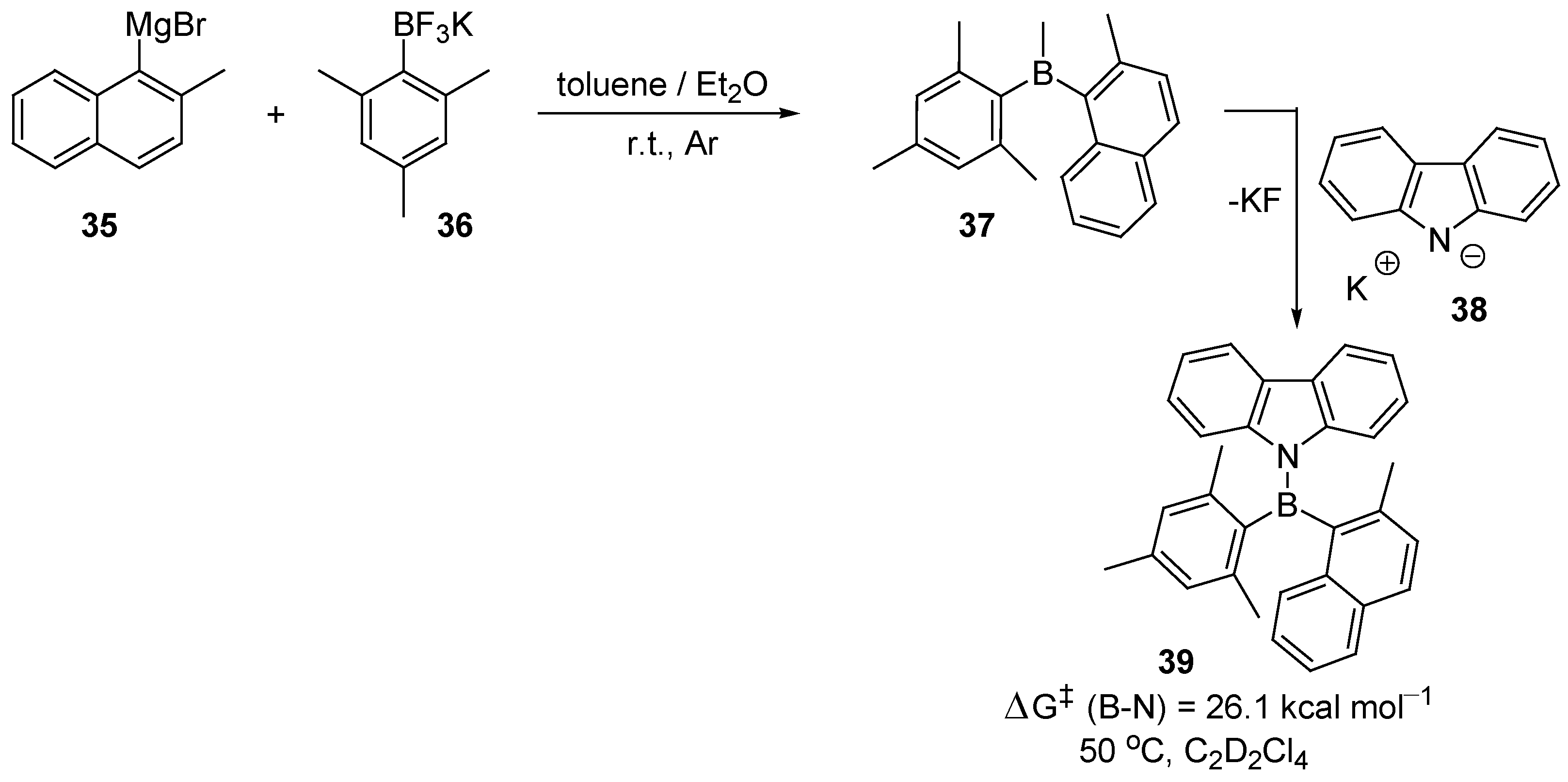
2. Synthesis of Atropisomeric Racemates with Atropisomer Resolution
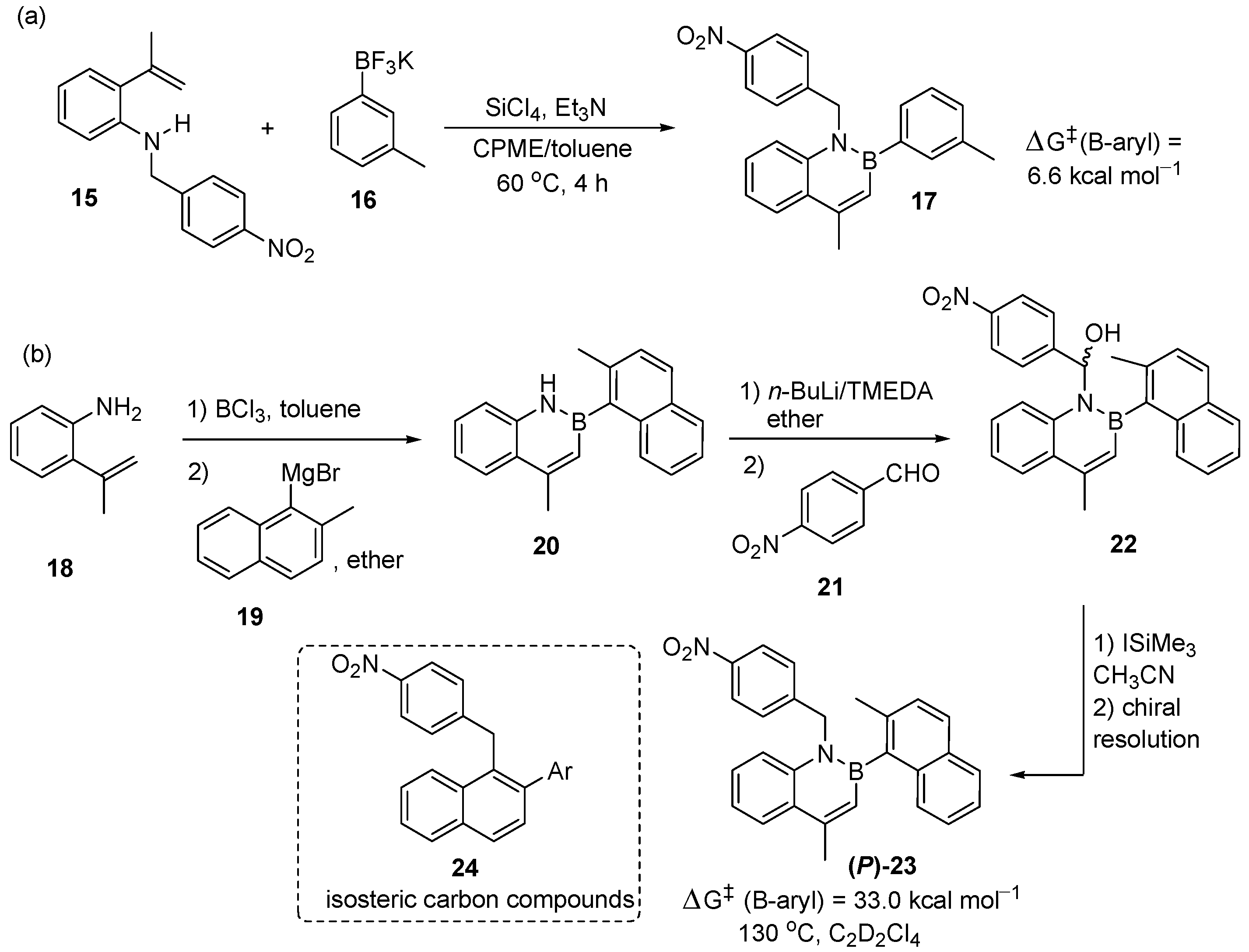
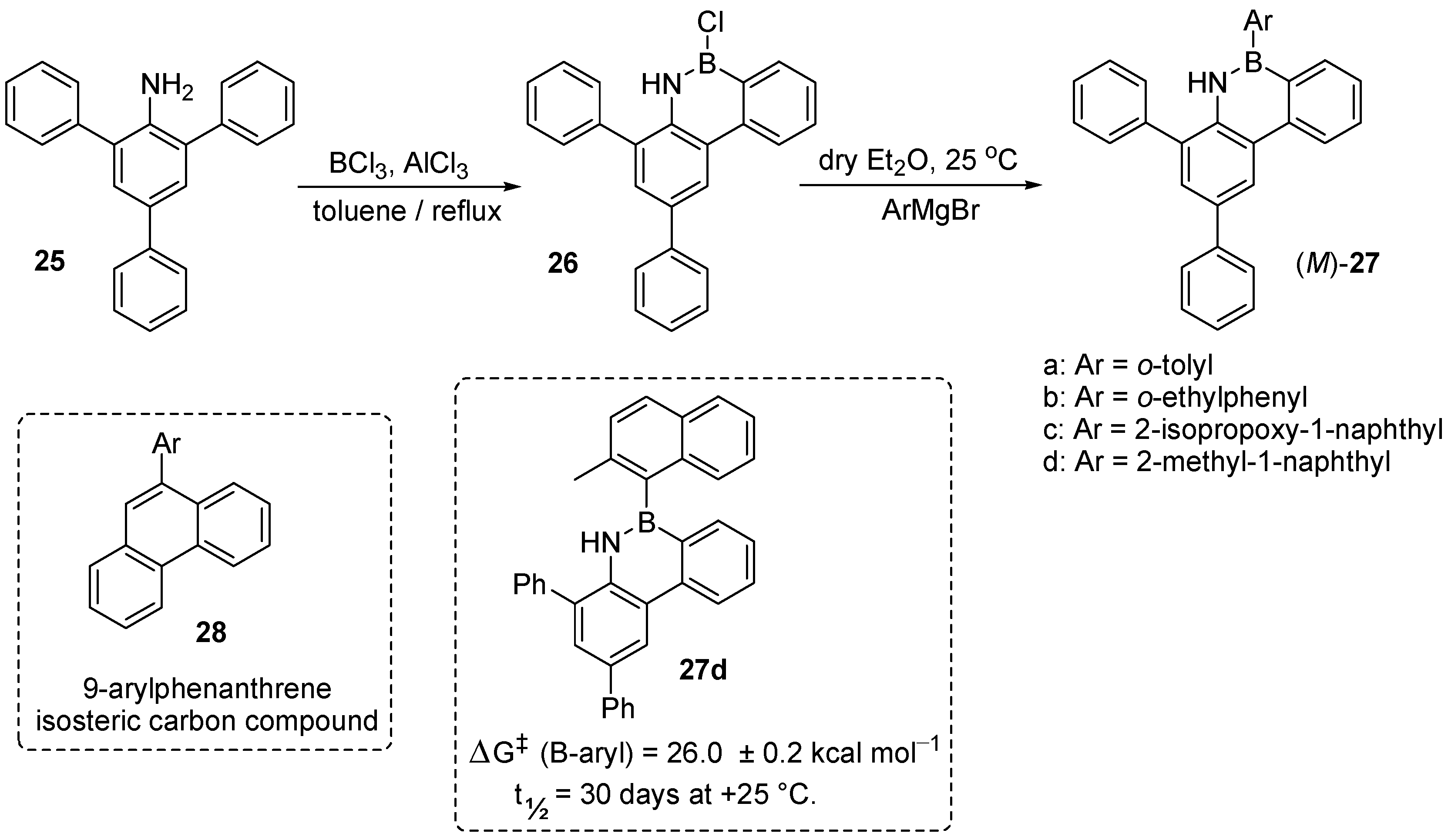
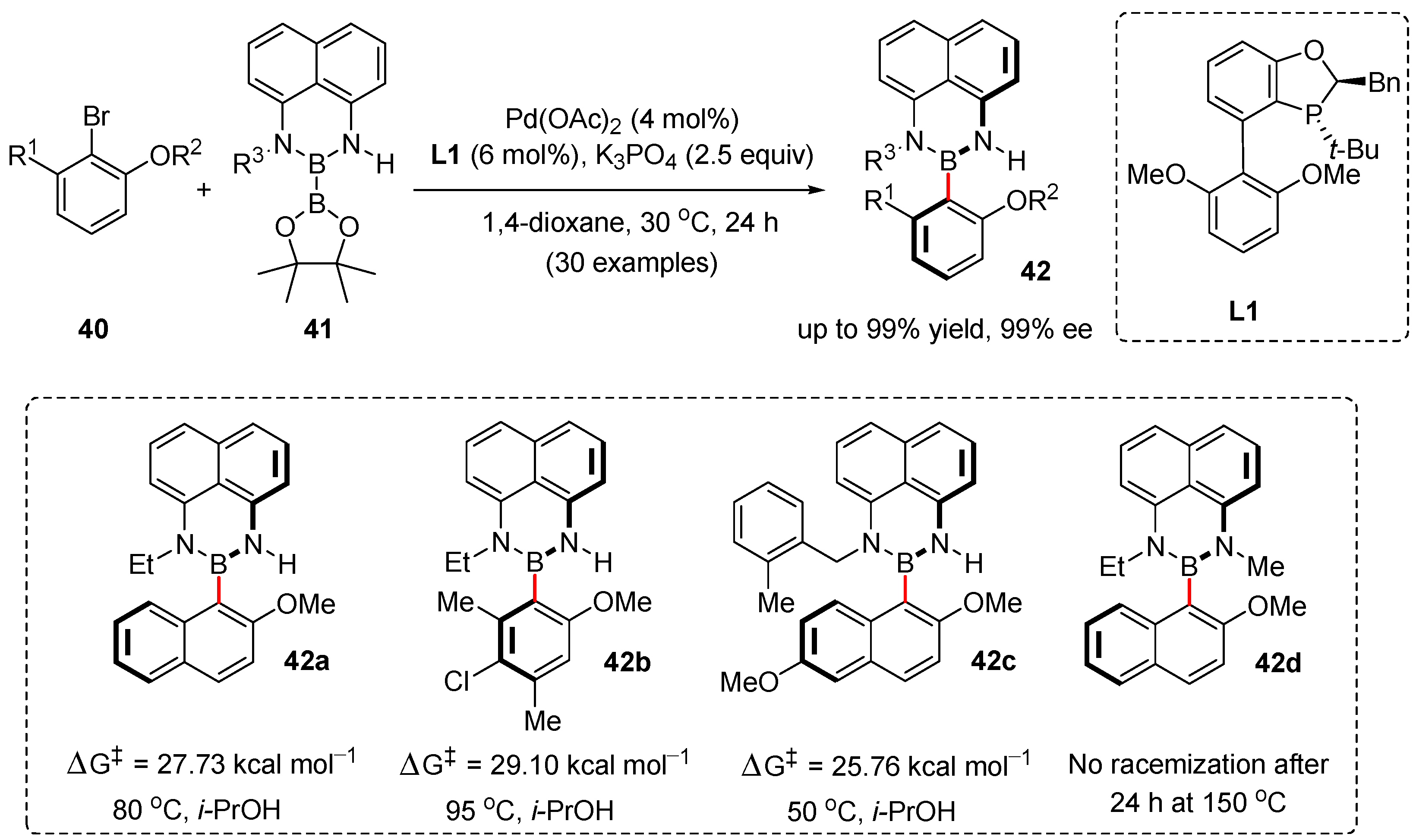
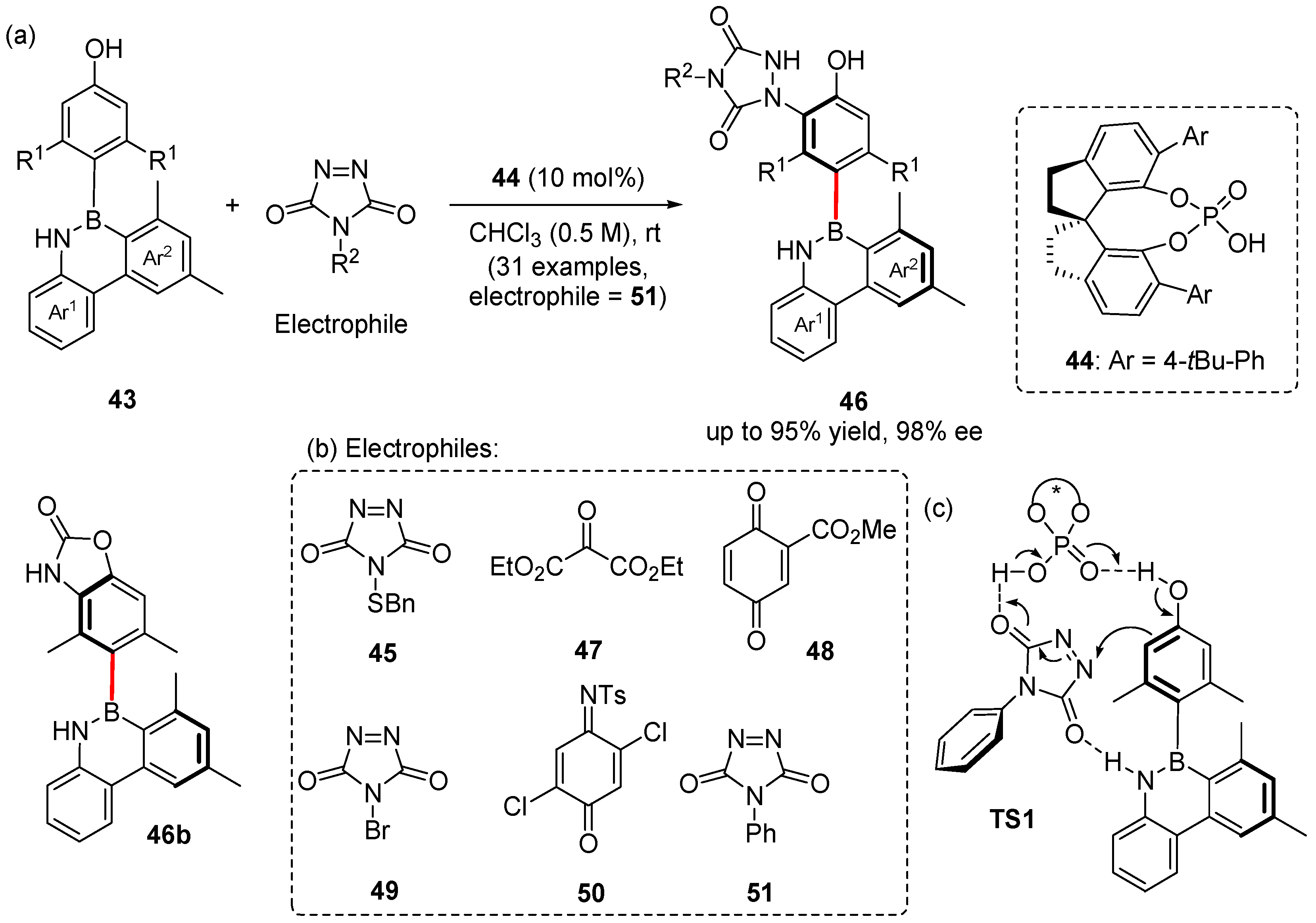
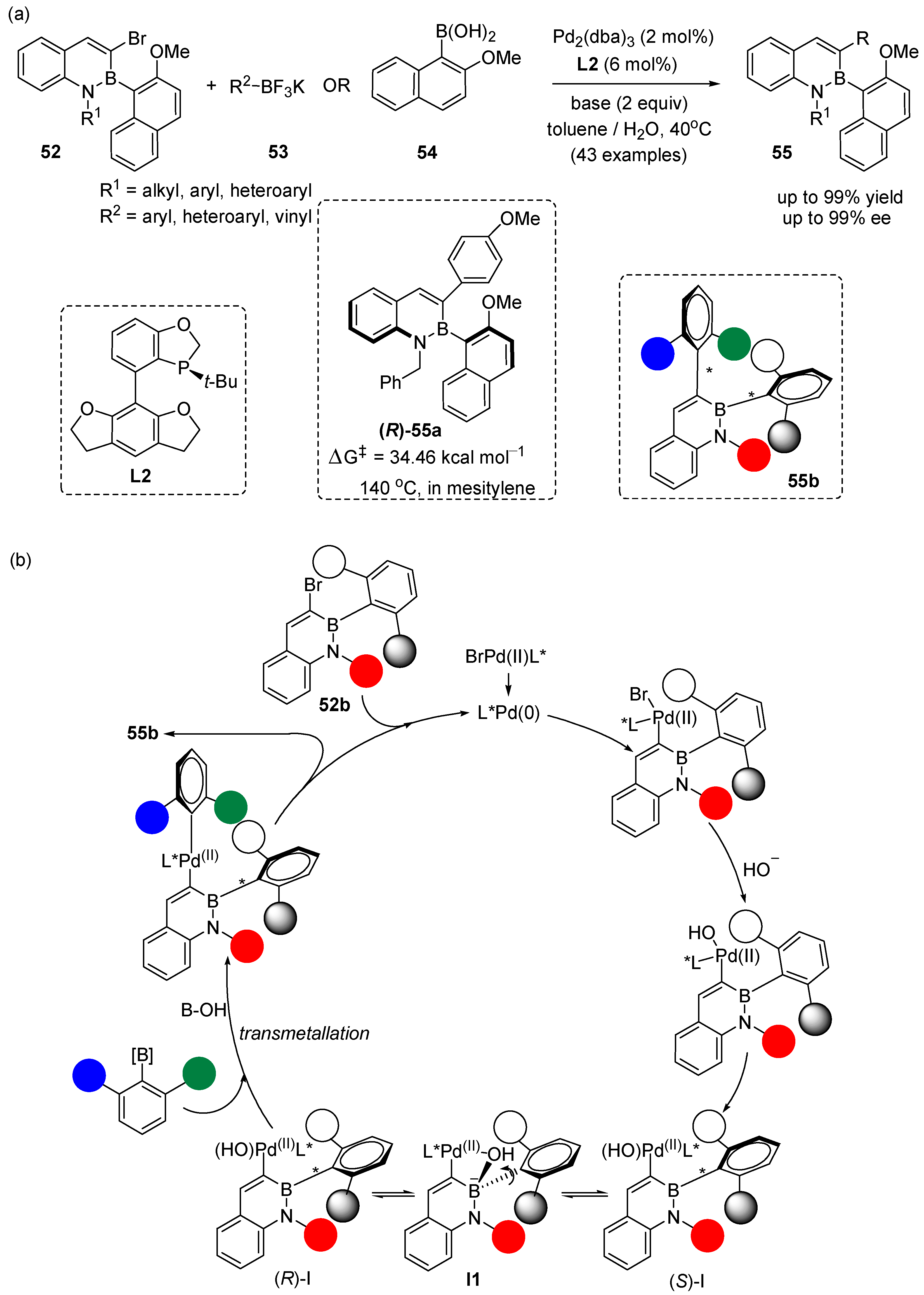
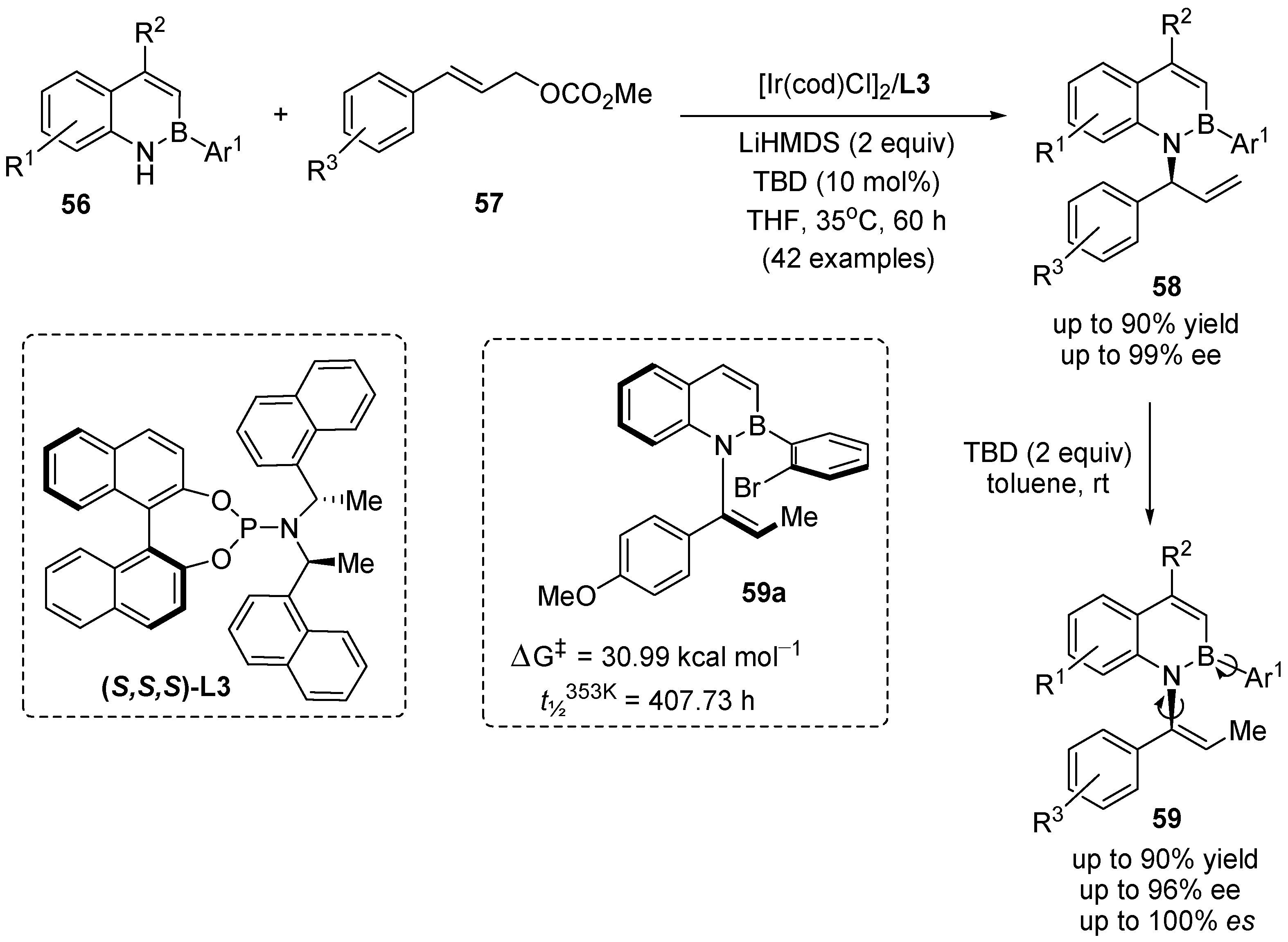
3. Enantioselective Synthesis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baker, S.J.; Ding, C.Z.; Akama, T.; Zhang, Y.-K.; Hernandez, V.; Xia, Y. Therapeutic Potential of Boron-Containing Compounds. Future Med. Chem. 2009, 1, 1275–1288. [Google Scholar] [CrossRef]
- Chatterjee, S.; Tripathi, N.M.; Bandyopadhyay, A. The Modern Role of Boron as a ‘Magic Element’. Chem. Commun. 2021, 57, 13629–13640. [Google Scholar] [CrossRef] [PubMed]
- Jäkle, F. Advances in the Synthesis of Organoborane Polymers for Optical, Electronic, and Sensory Applications. Chem. Rev. 2010, 110, 3985–4022. [Google Scholar] [CrossRef] [PubMed]
- Gapare, R.L.; Thompson, A. Substitution at Boron in BODIPYs. Chem. Commun. 2022, 58, 7351–7359. [Google Scholar] [CrossRef] [PubMed]
- Mantel, M.; Brauns, M.; Pietruszka, J. Boron-Containing Chiral Auxiliaries. Top. Heterocycl. Chem. 2020, 55, 73–112. [Google Scholar]
- Xu, L.; Zhang, S.; Li, P. Boron-Selective Reactions as Powerful Tools for Modular Synthesis of Diverse Complex Molecules. Chem. Soc. Rev. 2015, 44, 8848–8858. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.M. The fifth element. Chemistry World, 8 April 2019. [Google Scholar]
- Bosdet, M.J.D.; Piers, W.E. B–N as a C–C Substitute in Aromatic Systems. Can. J. Chem. 2009, 87, 8–29. [Google Scholar] [CrossRef]
- Campbell, P.G.; Marwitz, A.J.V.; Liu, S.-Y. Recent Advances in Azaborine Chemistry. Angew. Chem. Int. Ed. 2012, 51, 6074–6092. [Google Scholar] [CrossRef]
- Bélanger-Chabot, G.; Braunschweig, H.; Roy, D.K. Recent Developments in Azaborinine Chemistry. Eur. J. Inorg. Chem. 2017, 2017, 4353–4368. [Google Scholar] [CrossRef]
- Giustra, Z.X.; Liu, S.-Y. The State of the Art in Azaborine Chemistry: New Synthetic Methods and Applications. J. Am. Chem. Soc. 2018, 140, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Abdou-Mohamed, A.; Aupic, C.; Fournet, C.; Parrain, J.-L.; Chouraqui, G.; Chuzel, O. Stereoselective Formation of Boron-Stereogenic Organoboron Derivatives. Chem. Soc. Rev. 2023, 52, 4381–4391. [Google Scholar] [CrossRef] [PubMed]
- Zu, B.; Guo, Y.; He, C. Catalytic Enantioselective Construction of Chiroptical Boron-Stereogenic Compounds. J. Am. Chem. Soc. 2021, 143, 16302–16310. [Google Scholar] [CrossRef] [PubMed]
- Schraff, S.; Sun, Y.; Pammer, F. Tuning of Electronic Properties via Labile N→B-Coordination in Conjugated Organoboranes. J. Mater. Chem. C 2017, 5, 1730–1741. [Google Scholar] [CrossRef]
- Mellerup, S.K.; Wang, S. Boron-Based Stimuli Responsive Materials. Chem. Soc. Rev. 2019, 48, 3537–3549. [Google Scholar] [CrossRef]
- Mei, G.-J.; Koay, W.L.; Guan, C.-Y.; Lu, Y. Atropisomers Beyond the C–C Axial Chirality: Advances in Catalytic Asymmetric Synthesis. Chem 2022, 8, 1855–1893. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, J.W.; Bao, W.; Qiu, S.Q.; Li, S.; Xiang, S.H.; Song, J.; Zhang, J.; Tan, B. Chiral Phosphoric Acid-Catalyzed Remote Control of Axial Chirality at Boron-Carbon Bond. J. Am. Chem. Soc. 2021, 143, 12924–12929. [Google Scholar] [CrossRef]
- da Silva, E.M.; Vidal, H.D.A.; Januário, M.A.P.; Corrêa, A.G. Advances in the Asymmetric Synthesis of BINOL Derivatives. Molecules 2023, 28, 12. [Google Scholar] [CrossRef]
- Zhou, Q.L. (Ed.) Priviledged Chiral Ligands and Catalysts; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Börner, A. (Ed.) Phosphorus Ligands in Asymmetric Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Xie, J.-H.; Zhou, Q.-L. Chiral Diphosphine and Monodentate Phosphorus Ligands on a Spiro Scaffold for Transition-Metal-Catalyzed Asymmetric Reactions. Acc. Chem. Res. 2008, 41, 581–593. [Google Scholar] [CrossRef]
- Ding, K.; Li, X.; Ji, B.; Guo, H.; Kitamura, M. Ten Years of Research on NOBIN Chemistry. Curr. Org. Synth. 2005, 2, 499–545. [Google Scholar] [CrossRef]
- Akiyama, T.; Mori, K. Stronger Brønsted Acids: Recent Progress. Chem. Rev. 2015, 115, 9277–9306. [Google Scholar] [CrossRef] [PubMed]
- Faisca Phillips, A.M. Non-covalent Interactions in Asymmetric Reactions Catalyzed by Chiral Phosphoric Acids. In Noncovalent Interactions in Catalysis; Mahmudov, K.T., Kopylovich, M.N., Guedes da Silva, M.F.C., Pombeiro, A.J.L., Eds.; Royal Society of Chemistry Publishing: Cambridge, UK, 2019; pp. 253–281. [Google Scholar]
- James, T.; van Gemmeren, M.; List, B. Development and Applications of Disulfonimides in Enantioselective Organocatalysis. Chem. Rev. 2015, 115, 9388–9409. [Google Scholar] [CrossRef] [PubMed]
- Benda, M.C.; France, S. Chiral Disulfonimides: A Versatile Template for Asymmetric Catalysis. Org. Biomol. Chem. 2020, 18, 7485–7513. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.K.; Xiang, S.-H.; Tan, B. Organocatalytic Enantioselective Synthesis of Axially Chiral Molecules: Development of Strategies and Skeletons. Acc. Chem. Res. 2022, 55, 2920–2937. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, S.R.; Fader, L.D.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.F.; Edwards, P.J. Assessing Atropisomer Axial Chirality in Drug Discovery and Development. J. Med. Chem. 2011, 54, 7005–7022. [Google Scholar] [CrossRef] [PubMed]
- Basilaia, M.; Chen, M.H.; Secka, J.; Gustafson, J.L. Atropisomerism in the Pharmaceutically Relevant Realm. Acc. Chem. Res. 2022, 55, 2904–2919. [Google Scholar] [CrossRef] [PubMed]
- Perreault, S.; Chandrasekhar, J.; Patel, L. Atropisomerism in Drug Discovery: A Medicinal Chemistry Perspective Inspired by Atropisomeric Class I PI3K Inhibitors. Acc. Chem. Res. 2022, 55, 2581–2593. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M.; Pombeiro, A.J.L. Atropselective Organocatalytic Synthesis of Chiral Compounds Containing Nitrogen Along the Axis of Chirality. Symmetry 2023, 15, 1261. [Google Scholar] [CrossRef]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A Twist of Nature—The Significance of Atropisomers in Biological Systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef]
- Mancinelli, M.; Bencivenni, G.; Pecorari, D.; Mazzanti, A. Stereochemistry and Recent Applications of Axially Chiral Organic Molecules. Eur. J. Org. Chem. 2020, 2020, 4070–4086. [Google Scholar] [CrossRef]
- Mondal, A.; Toyoda, R.; Costil, R.; Feringa, B.L. Chemically Driven Rotatory Molecular Machines. Angew. Chem. Int. Ed. 2022, 61, e202206631. [Google Scholar] [CrossRef] [PubMed]
- Oki, M. Recent Advances in Atropisomerism. In Topics in Stereochemistry; Allinger, N.L., Eliel, E.L., Wilen, S.H., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 1983; Volume 14, pp. 1–81. [Google Scholar]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing Atropisomer Axial Chirality in Drug Discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Mercanti, E.; Mancinelli, M. Axial Chirality about Boron−Carbon Bond: Atropisomeric Azaborines. Org. Lett. 2016, 18, 2692–2695. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S.; Dietz, R. 546. New Heteroaromatic Compounds. Part III. 2,1-Borazaro-naphthalene (1,2-dihydro-1-aza-2-boranaphthalene). J. Chem. Soc. 1959, 1959, 2728–2730. [Google Scholar] [CrossRef]
- Islas, R.; Chamorro, E.; Robles, J.; Heine, T.; Santos, J.C.; Merino, G. Borazine: To Be or Not To Be Aromatic. Struct. Chem. 2007, 18, 833–839. [Google Scholar] [CrossRef]
- Johnson, M., Jr.; Mellon, E.K. Atropisomerism in Aryl-Substituted Borazines. Inorg. Chem. 1974, 13, 2769–2772. [Google Scholar] [CrossRef]
- Köster, R.; Seidel, C.; Erschl, S.; Rackmeyer, B. Atropisomerism in Boron-Nitrogen Heterocycles. Z2. Naturforsch. Teil B 1987, 42b, 191. (In German) [Google Scholar] [CrossRef]
- Allaoud, S.; Zair, T.; Karim, A.; Frange, B. Atropisomerism in o-Aryl-Substituted Borazines. Inorg. Chem. 1990, 29, 1447–1449. [Google Scholar] [CrossRef]
- Cornu, D.; Miele, P.; Bonnetot, B.; Guenot, P.; Mongeot, H.; Bouix, J. Synthesis and Characterization of 2,4,6-Tris{[bis(isopropylamino)borylkisopropyl)amino}borazine. Main Group Met. Chem. 1998, 21, 301–302. [Google Scholar] [CrossRef]
- Hoffend, C.; Schödel, F.; Bolte, M.; Lerner, H.W.; Wagner, M. Boron-Doped Tri(9,10-anthrylene)s: Synthesis, Structural Characterization, and Optoelectronic Properties. Chem 2012, 18, 15394–15405. [Google Scholar] [CrossRef]
- Preya, S.E.; Wagner, M. Threat to the Throne: Can Two Cooperating Boron Atoms Rival Transition Metals in Chemical Bond Activation and Catalysis? Adv. Synth. Catal. 2021, 363, 2290–2309. [Google Scholar] [CrossRef]
- Su, Y.; Kinjo, R. Small Molecule Activation by Boron-Containing Heterocycles. Chem. Soc. Rev. 2019, 48, 3613–3659. [Google Scholar] [CrossRef] [PubMed]
- Légaré, M.-A.; Pranckevicius, C.; Braunschweig, H. Metallomimetic Chemistry at Boron. Chem. Rev. 2019, 119, 8231–8261. [Google Scholar] [CrossRef] [PubMed]
- Dietz, M.; Arrowsmith, M.; Gärtner, A.; Radacki, K.; Bertermann, R.; Braunschweig, H. Harnessing the Electronic Differences Between CAAC-Stabilised 1,4-Diborabenzene and 9,10-Diboraanthracene for Synthesis. Chem. Commun. 2021, 57, 13526–13529. [Google Scholar] [CrossRef] [PubMed]
- Bosdet, M.J.D.; Jaska, C.A.; Piers, W.E.; Sorensen, T.S.; Parvez, M. Blue Fluorescent 4a-Aza-4b-boraphenanthrenes. Org. Lett. 2007, 9, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Y.; Lin, H.-R.; Lei, T.; Yang, D.-C.; Zhuang, F.-D.; Wang, J.-Y.; Yuan, S.-C.; Pei, J. Azaborine Compounds for Organic Field-Effect Transistors: Efficient Synthesis, Remarkable Stability, and BN Dipole Interactions. Angew. Chem. Int. Ed. 2013, 52, 3117–3120. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Boffa, M.; Marotta, E.; Mancinelli, M. Axial Chirality at the Boron−Carbon Bond: Synthesis, Stereodynamic Analysis, and Atropisomeric Resolution of 6-Aryl-5,6-dihydrodibenzo[c,e][1,2]azaborinines. J. Org. Chem. 2019, 84, 12253–12258. [Google Scholar] [CrossRef]
- Curran, D.P.; Solovyev, A.; Makhlouf Brahmi, M.; Fensterbank, L.; Malacria, M.; Lacôte, E. Synthesis and Reactions of N-Heterocyclic Carbene Boranes. Angew. Chem. Int. Ed. 2011, 50, 10294–10317. [Google Scholar] [CrossRef]
- Damodaran, K.; Li, X.; Pan, X.; Curran, D.P. Dynamic Behavior of N-Heterocyclic Carbene Boranes: Boron−Carbene Bonds in B,B-Disubstituted N,N-Dimethylimidazol-2-ylidene Boranes Have Substantial Rotation Barriers. J. Org. Chem. 2015, 80, 4465–4469. [Google Scholar] [CrossRef]
- Birepinte, M.; Robert, F.; Pinet, S.; Chabaud, L.; Pucheault, M. Non-biaryl Atropisomerism at the C–B Bond in Sterically Hindered Aminoarylboranes. Org. Biomol. Chem. 2020, 18, 3007–3011. [Google Scholar] [CrossRef]
- Pecorari, D.; Giuliani, E.; Mazzanti, A.; Stagni, S.; Fiorini, V.; Vigarani, G.; Zinna, F.; Pescitelli, G.; Mancinelli, M. Synthesis and Stereodynamic and Emission Properties of Dissymmetric Bis-Aryl Carbazole Boranes and Identification of a CPL-Active B−C Atropisomeric Compound. J. Org. Chem. 2023, 88, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Pecorari, D.; Mazzanti, A.; Gianvittorio, S.; Foschi, S.; Stagni, S.; Fiorini, V.; Mancinelli, M. Highly Twisted Carbazole-Borane Derivates: B-N Stereodynamic Analysis and Consequence on their Emission Properties. Org. Chem. Front. 2021, 8, 4496. [Google Scholar] [CrossRef]
- Carmona, J.A.; Rodríguez-Franco, C.; Fernández, R.; Hornillos, V.; Lassaletta, J.M. Atroposelective transformation of axially chiral (hetero)biaryls. From Desymmetrization to Modern Resolution Strategies. Chem. Soc. Rev. 2021, 50, 2968–2983. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.K.; Xiang, S.-H.; Li, S.; Ye, L.; Tan, B. Recent Advances in Catalytic Asymmetric Construction of Atropisomers. Chem. Rev. 2021, 121, 4805–4902. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Mao, Y.; Xu, J.; Wang, H.; He, Y.; Li, W.; Song, Q. Construction of Axially Chiral Arylborons via Atroposelective Miyaura Borylation. J. Am. Chem. Soc. 2021, 143, 10048–10053. [Google Scholar] [CrossRef]
- Farhang, M.; Akbarzadeh, A.R.; Rabbani, M.; Ghadiri, A.M. A Retrospective-Prospective Review of Suzuki–Miyaura Reaction: From Cross-Coupling Reaction to Pharmaceutical Industry Applications. Polyhedron 2022, 227, 116124. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Phillips, A.M.F.; Pombeiro, A.J.L. C-C bond formation in the sustainable synthesis of pharmaceuticals. In Sustainable Synthesis of Pharmaceuticals: Using Transition Metals as Catalysts; Pereira, M.M., Calvete, M.J.F., Eds.; Green Chemistry Series; Royal Society of Chemistry: Oxford, UK, 2018; pp. 193–229. [Google Scholar]
- Yang, K.; Mao, Y.; Zhang, Z.; Xu, J.; Wang, H.; He, Y.; Yu, P.; Song, Q. Construction of C-B Axial Chirality Via Dynamic Kinetic Asymmetric Cross-Coupling Mediated by Tetracoordinate Boron. Nat. Commun. 2023, 14, 4438. [Google Scholar] [CrossRef]
- Bao, X.; Rodriguez, J.; Bonne, D. Enantioselective Synthesis of Atropisomerswith Multiple Stereogenic Axes. Angew.Chem. Int. Ed. 2020, 59, 12623–12634. [Google Scholar] [CrossRef]
- Zhang, X.L.; Gu, J.; Cui, W.H.; Ye, Z.; Yi, W.; Zhang, Q.; He, Y. Stepwise Asymmetric Allylic Substitution-Isomerization Enabled Mimetic Synthesis of Axially Chiral B,N-Heterocycles. Ang. Chem. Int. Ed. 2022, 61, e202210456. [Google Scholar] [CrossRef]
- Wu, Y.-X.; Liu, Q.; Zhang, Q.; Ye, Z.; He, Y. Asymmetric Allylic Substitution-Isomerization for Accessing Axially Chiral Vinylindoles bby Intramolecular π-π Stacking Interactions. Cell Rep. Phys. Sci. 2022, 3, 101005. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faisca Phillips, A.M.; Pombeiro, A.J.L. Synthesis of Axially Chiral Boron Compounds. Symmetry 2024, 16, 11. https://doi.org/10.3390/sym16010011
Faisca Phillips AM, Pombeiro AJL. Synthesis of Axially Chiral Boron Compounds. Symmetry. 2024; 16(1):11. https://doi.org/10.3390/sym16010011
Chicago/Turabian StyleFaisca Phillips, Ana Maria, and Armando J. L. Pombeiro. 2024. "Synthesis of Axially Chiral Boron Compounds" Symmetry 16, no. 1: 11. https://doi.org/10.3390/sym16010011
APA StyleFaisca Phillips, A. M., & Pombeiro, A. J. L. (2024). Synthesis of Axially Chiral Boron Compounds. Symmetry, 16(1), 11. https://doi.org/10.3390/sym16010011