4.1. Properties of the Potential Energy Surface
Table 1 compares the parameters of the new PES with the previous one [
28]. Firstly, due to the employment of the new scheme to choose the value of Euclidean distance, the number of sampled data points is decreased from 82,680 to 72,113. Secondly, the sampled data points in this work cover a larger region, in which the separation of reactants (or products) is up to 25 Å, in sharp contrast to 8 Å for the previous PES. Thirdly, the parameter in Morse-like variables,
, is taken as 2.0. A relatively large value of
should be used when the long-range interaction is strong. Finally, the overall RMSE of the new PES is 4.14 meV, 0.58 meV lower than that of the previous PES.
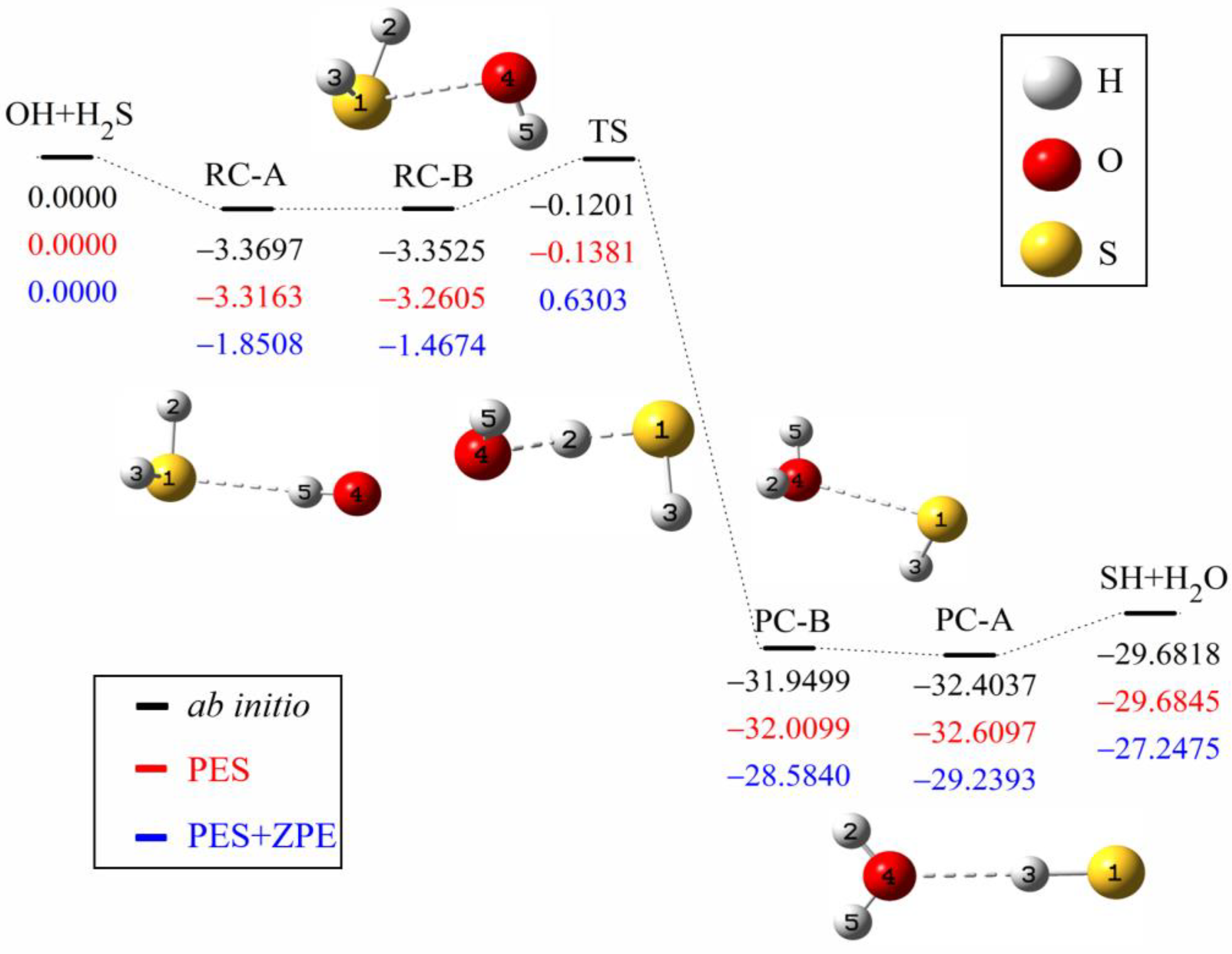
The minimum energy path of the OH + H
2S→H
2O + SH reaction is illustrated in
Figure 1. The ab initio energies are calculated at the level of UCCSD(T)-F12a/aug-cc-pVTZ [
40,
41]. All energies are given in kcal mol
−1 and with respect to the reactant asymptote. Clearly, four minima exist in the entrance and exit valleys, in which RC-A and PC-A are formed by hydrogen bond, while RC-B and PC-B are hemibonded complexes with two center and triple electron S-O bond (2c-3e). The interaction of hydrogen bond between H
2S (H
2O) and OH (SH) is slightly stronger than the corresponding hemibond, consistent with Tang et al.’s results [
27].
To assess the quality of the fitted PES, the optimized geometries by ab initio calculations, the geometries on the fitted PES and available experimental values are listed in
Table 2. On one hand, the geometries of the stationary points at the level of UCCSD(T)-F12a/aug-cc-pVTZ are in accord with the geometries at the level of CCSD(T)/aug-cc-pV(Q+d)Z, where the biggest difference of bond length is 0.255 Å and the biggest difference of angle is 12.7°. On the other hand, the predicted geometries by the PES are also consistent with ab initio results. The deviations from the UCCSD(T)-F12a/aug-cc-pVTZ results are less than 0.171 Å for the bond length and 12.0° for the angle.
Table 3 shows the energies and frequencies of the stationary points. To solve the problem of non-convergence, the basis function extrapolation and energy shift are adopted in the calculation of single-point energy, which causes a small difference from Ping et al.’s results [
28]. The energy difference between the predicted PES and the corresponding ab initio one of UCCSD(T)-F12a/aug-cc-pVTZ is less than 0.1866 kcal mol
−1, and the energy difference between UCCSD(T)-F12a/aug-cc-pVTZ and CCSD(T)/aug-cc-pV(Q+d)Z is less than 0.2425 kcal mol
−1 [
27]. The barrier height on the PES is to be −0.1381 kcal mol
−1, which agrees well with the UCCSD(T)-F12a/aug-cc-pVTZ value of −0.1201 kcal mol
−1, while it is slightly lower than the CCSD(T)/aug-cc-pV(Q+d)Z value of 0.0600 kcal mol
−1. The frequency difference is less than 53 cm
−1 compared with the UCCSD(T)-F12a/aug-cc-pVTZ results and the imaginary frequency of the transition state is the same.
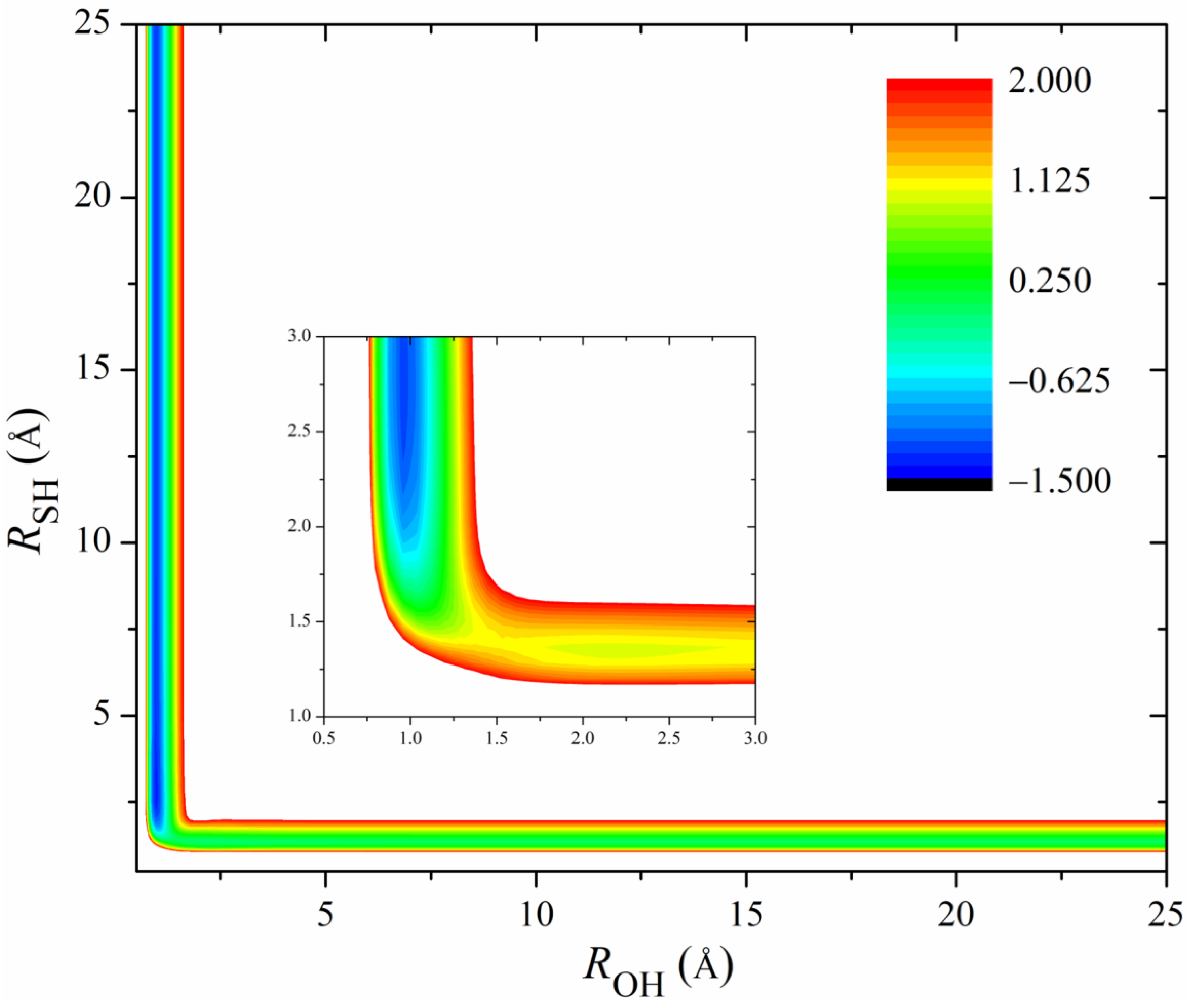
Figure 2 shows the 2D contours of the PES as a function of the breaking HS and the forming OH with the other coordinates fixed at the geometry of the transition state. The energy gap of the fitted PES is from −1.5 eV to 2.0 eV. Clearly, the fitted PES is globally smooth. The insert zooms in the contours near the transition state. The pre- and post-reaction wells are nicely reproduced by the PES. The fitting errors of the sampled points are plotted in
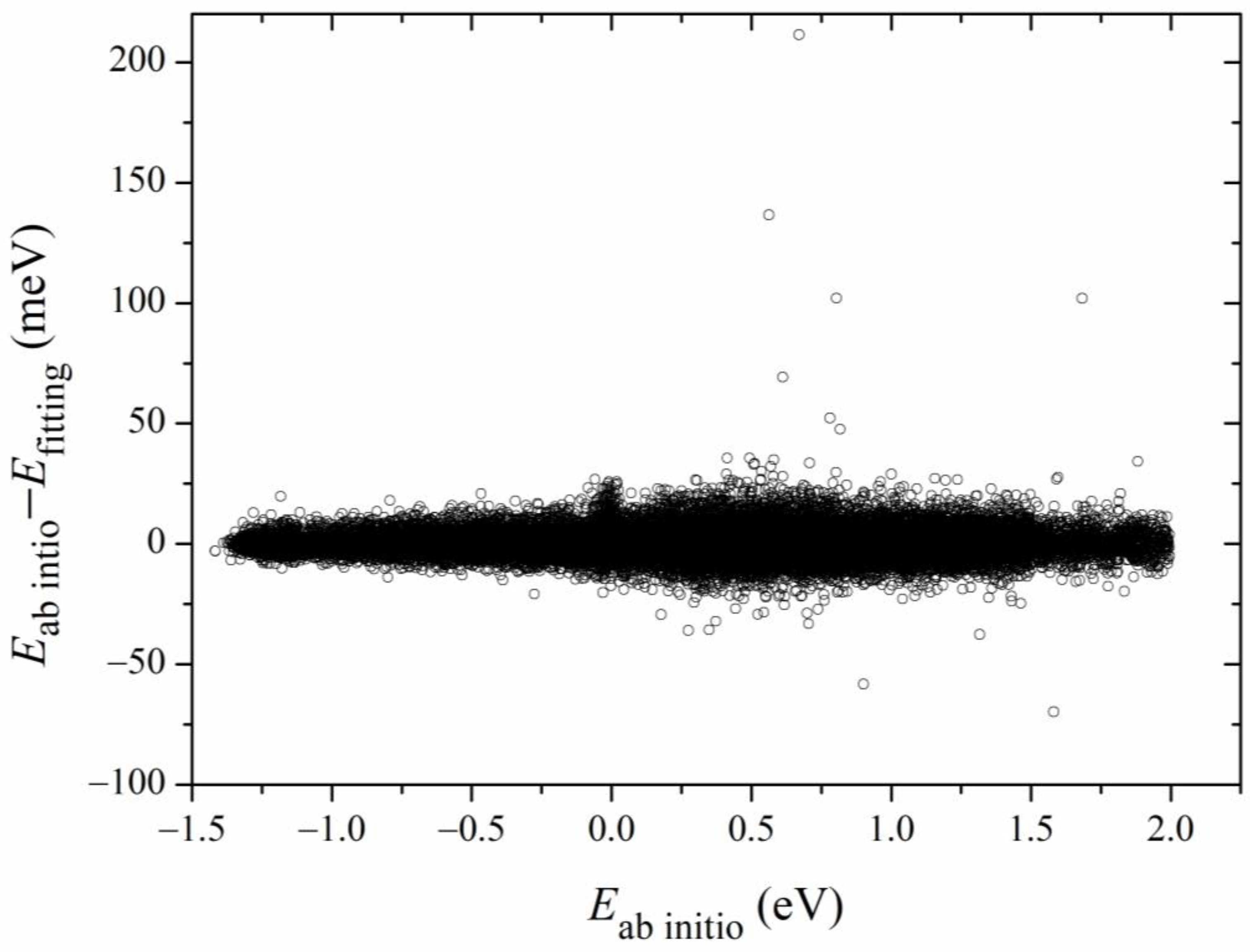
Figure 3. The errors for most of the sampled points are distributed in the range of −20 to 20 meV, with several scattered points as large as 200 meV. Overall, the new PES is globally accurate and satisfies the requirement of dynamics calculations.
4.2. Product Energy Disposal
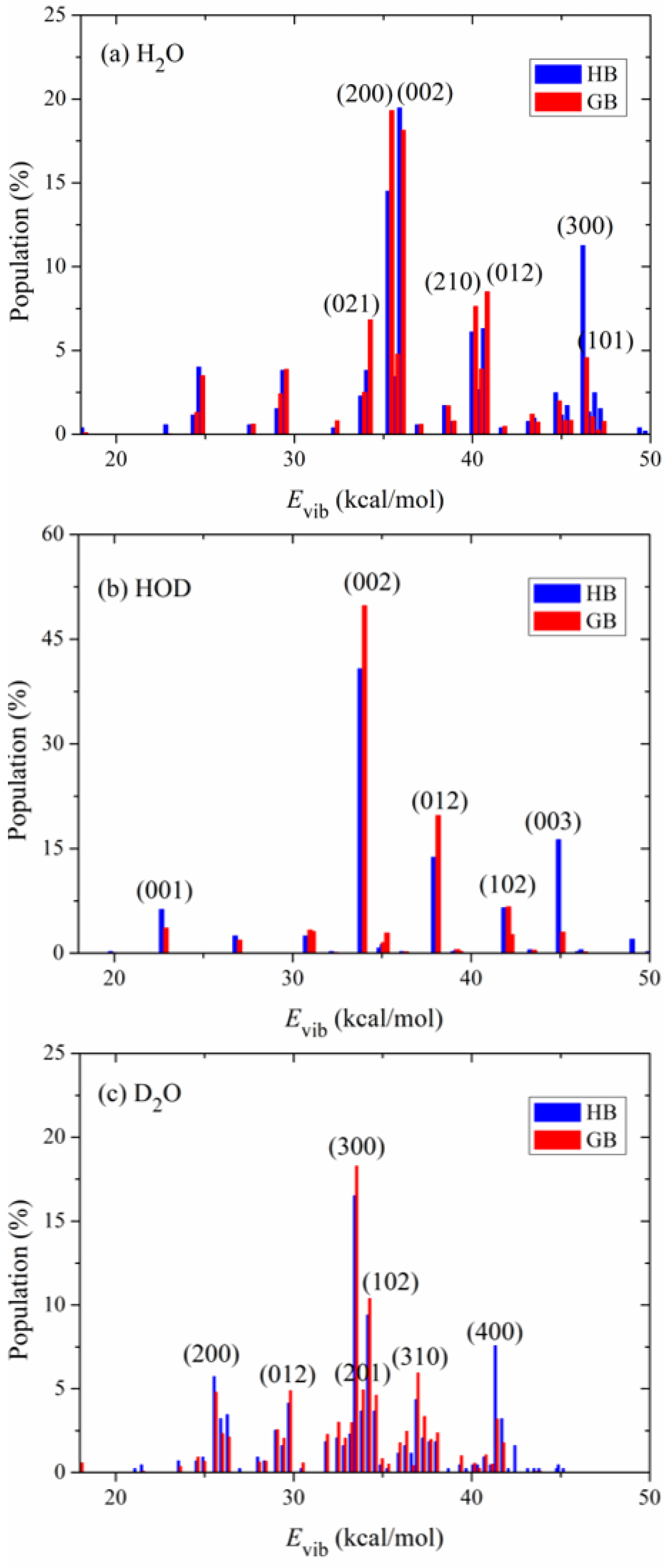
Figure 4 shows the calculated product vibrational state distributions. The reactant ro-vibrational states are sampled at 298 K. The three quantum numbers in the parentheses, (
), denote excitations in the symmetric stretching, bending and asymmetric stretching modes of the products H
2O and D
2O. As to the product HOD,
refers to excitation in the O-D stretching mode and
in the O-H stretching mode. Two binning methods (HB and GB) are applied in the calculations. Since the two methods give similar distributions, the following discussions are based on the GB results. For the product molecule H
2O in the OH + H
2S reaction, as shown in
Figure 4a, it is mainly populated in the states (200), (002), (012), (210) and (021). The significant excitation of the stretching modes of H
2O results from the visible change of the OH bond among the minimum energy path. The bond length of OH from the transition state to the product H
2O is shortened from 1.501 Å to 0.959 Å. Compared with the stretching modes, the bending mode of H
2O is less excited and mainly populated in the ground and fundamental states.
To study the effect of isotopic substitution on the dynamics, the vibrational state distributions of the products HOD in the reaction OD + H
2S and D
2O in the reaction OD + D
2S are presented in
Figure 4b,c, respectively. In sharp contrast to H
2O, the product HOD is dominantly formed in the state (002), i.e., the first overtone of the OH stretching mode, followed by visible excitation in the state (012). The OD bond in the product HOD is nearly unexcited. According to the analysis of the reaction coordinate at the transition state, the stretching motion of the O-H bond is to a great extent along the reaction coordinate. Therefore, the initial available energy favors to flow into the O-H stretching mode of HOD. However, due to the large frequency difference between the OH and OD stretching modes, the intramolecular vibrational redistribution (IVR) is infeasible in the reaction process. In other words, the OD bond is more like a spectator bond in the OD + H
2S reaction. When both reactants OH and H
2S are isotopically substituted, as shown in
Figure 4c, the product D
2O is mainly populated in the state (300), followed by (102), (400) and (200). Interestingly, it appears that the IVR in D
2O is not as strong as in H
2O. The reaction OD + D
2S prefers to produce D
2O in the vibrational excited states of the symmetric stretching mode and the excitation energy in the symmetric stretching mode cannot efficiently flow into the asymmetric stretching mode. In addition, compared with the products H
2O and HOD, D
2O is formed in higher excited states due to its higher density of states. Actually, the products H
2O, HOD and D
2O are all largely populated in the vibrational states whose energies are around 35 kcal mol
−1, close to the total available energy.
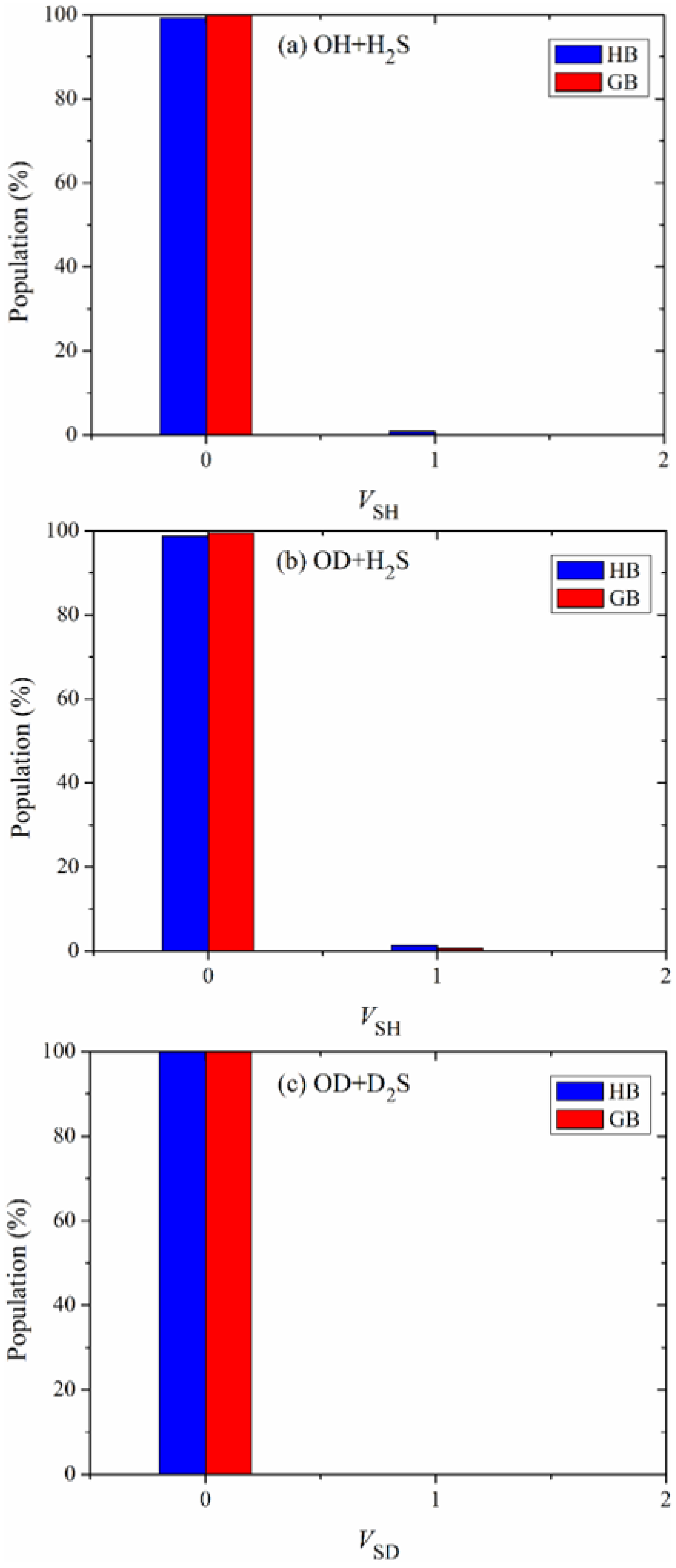
The vibrational state distributions of the products SH in the reactions OH + H
2S and OD + H
2S and SD in the reaction OD + D
2S are illustrated in
Figure 5a–c. Obviously, these products are almost exclusively formed in their ground vibrational states, indicating the nature of a spectator in the reaction OH + H
2S and its isotopic variants.
Table 4 compares the calculated vibrational state distribution of the product H
2O in the OH + H
2S reaction with the experimental result from Seter’s group [
19,
21]. In the experiment, the symmetric (
) and asymmetric (
) stretching modes are difficult to distinguish. Thus,
in the table represents the sum of
and
. Theoretical calculations show that the product is largely populated in the states
for
and in the states
for
. They are consistent with experimental observations, although visible quantitative difference exists. The theoretical proportion of the states
for
is higher than the corresponding experimental values, while the theoretical proportion of the states
for
is slightly lower. For
, the experimental distributions are somewhat scattered, and one experiment gave the highest proportion for the states
[
19], consistent with theoretical results; the other experiment reported the highest proportion for the states
[
21]. The reactions in the experiment were carried out under the pressure of 0.5 Torr. However, this condition is not considered in theoretical calculations. Therefore, the quantitative difference between experimental and theoretical results is possibly caused by the pressure.
The vibrational state distribution of the product HOD in the OD + H
2S reaction is listed in
Table 5. The quantum numbers
and
represent excitations in the OD stretching, bending and OH stretching modes. Since the frequency of OD stretching mode (2831 cm
−1) is very close the to the double frequency of the bending mode (2888 cm
−1), it is hard for the experiment to distinguish them as well. Hence,
in the table is defined as the maximum value of
and
. For
, theoretical calculations predict that the product HOD is largely distributed in the state
, in accord with the experimental results. On the other hand, both experiment and theory show that the product HOD has larger distributions in
and
for
and 2, respectively. In addition, visible difference between experimental and theoretical values also exists.
Table 6 lists the vibrational state distribution of the product D
2O in the OD + D
2S reaction. Unfortunately, there are no experimental results for comparison. Like
Table 4,
in the table represents the sum of
and
. The product D
2O is largely populated in states
for
and 1. For
D
2O is largely distributed in the states
.
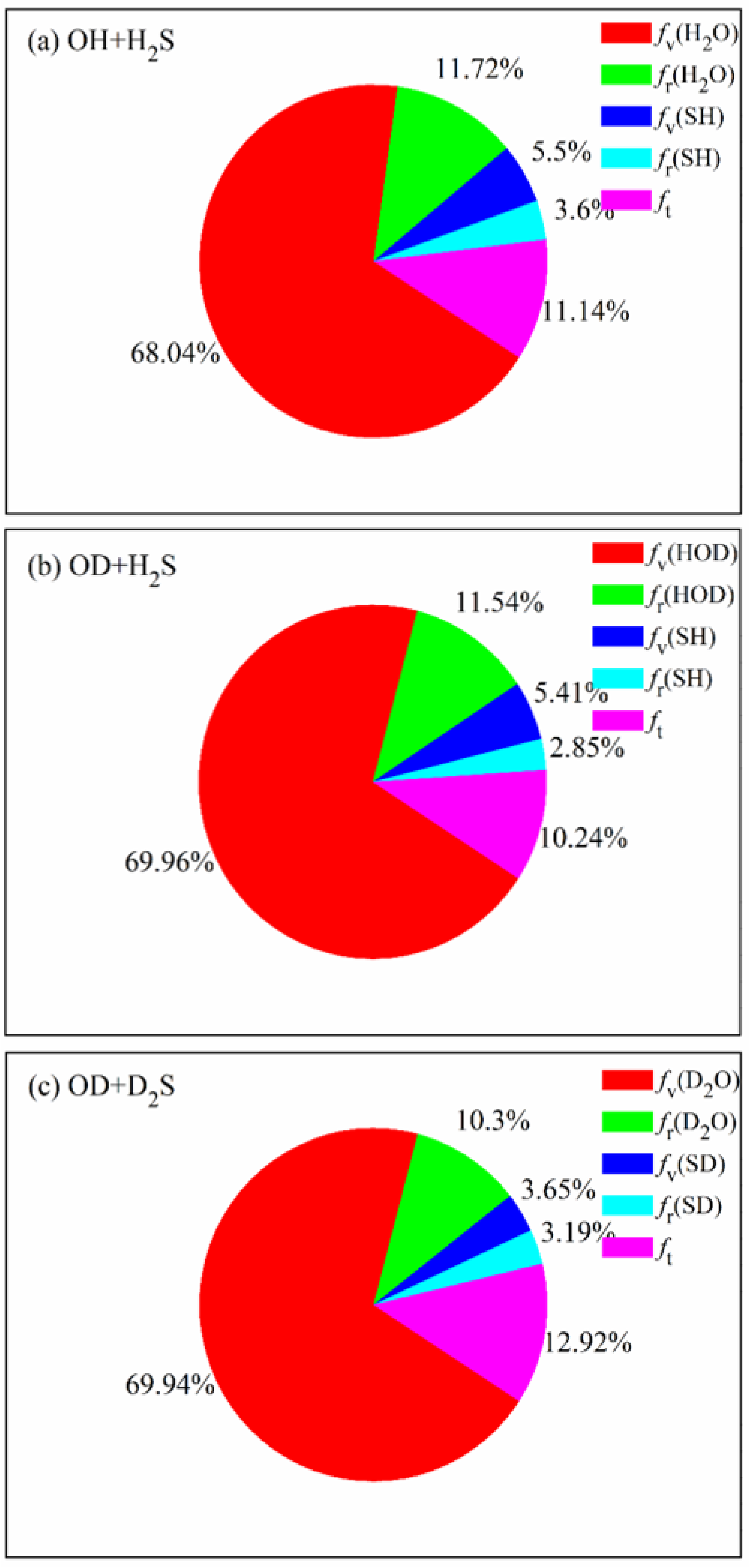
The product energy disposals of the three reactions are shown in
Figure 6.
fv,
fr and
ft denote the fractions of available energy released as the product vibrational energy, the rotational energy and the translation energy, respectively. The product zero-point energy is subtracted from the corresponding vibrational energy. It can be seen that 68.04% of available energy is released as the vibrational energy of H
2O in the OH + H
2S reaction, followed by the rotational energy of H
2O and the relative translational energy. The small proportion of vibrational and rotational energy of SH indicates that it is hard for the energy to flow into the product SH, no matter the vibrational mode and the rotational mode. When the reactants are isotopically substituted, the product energy disposal is nearly unchanged. Most of available energy flows into the vibrational and rotational modes of HOD in the OD + H
2S reaction or D
2O in the OD + D
2S reaction. It appears that, compared with the former two reactions, a little more energy (12.92%) is transferred as the translational energy in the OD + D
2S reaction. The fraction of
fv(H
2O) (
fv(HOD)) is estimated to be 56% in the experiment [
20,
21], lower than the calculated value of 69.96% (69.94%). As mentioned, the difference is possibly caused by the pressure of 0.5 Torr in experiment, which is not considered in the simulation.
Although this reaction is likely to be barrierless, the submerged barrier could exert some control on the reactivity, especially for the reactive trajectories by the direct mechanism. The sudden vector projection (SVP) model [
56,
57,
58,
59] has been widely used to predict reactant mode selectivity and product energy disposal in many gas phase and gas surface reactions by calculating the coupling between the interested reactant or product normal mode vector (
) and the reaction coordinate vector (
) at the transition state:
. The calculated SVP values of different product modes for the three reactions are listed in
Table 7. For the OH + H
2S reaction, the symmetric and asymmetric stretching modes of H
2O have the largest projection on the reaction coordinate, followed by the product translational mode and the bending mode of H
2O. The vibrational mode of the product SH has a negligible coupling with the reaction coordinate. This is consistent with QCT calculations, in which the formed H
2O is largely populated in the excited states of the two stretching modes of H
2O, while its bending mode is less excited. Most of available energy flows into the vibrational modes of H
2O, followed by the translational mode, and a very small percentage of the energy is released as the vibrational and rotational energy of SH. For the OD + H
2S reaction, the stretching mode of OH in HOD has the largest projection value, followed by the translational mode and the bending mode of HOD. The stretching mode of OD in HOD has a weak coupling with the reaction coordinate and the vibrational mode of SH has a negligible value. According to QCT calculations, the HOD is dominantly formed in the first overtone state of the OH moiety, well predicted by the SVP model. The product energy disposal of HOD can be explained by the SVP model as well. Furthermore, the SVP values of the product D
2O are close to those of H
2O, indicating similar energy disposal of D
2O to that of H
2O. This is also confirmed by QCT calculations.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}