
Computational Study of H2 Catalytic Combustion on Pd38 Cluster Model and Pd(111) Slab Model

Abstract
:1. Introduction
2. Computing Method
3. Results and Discussion
3.1. Pd Catalyst Model
3.2. Reactants and Intermediates on the Pd38 Cluster Model and Pd(111) Slab Model
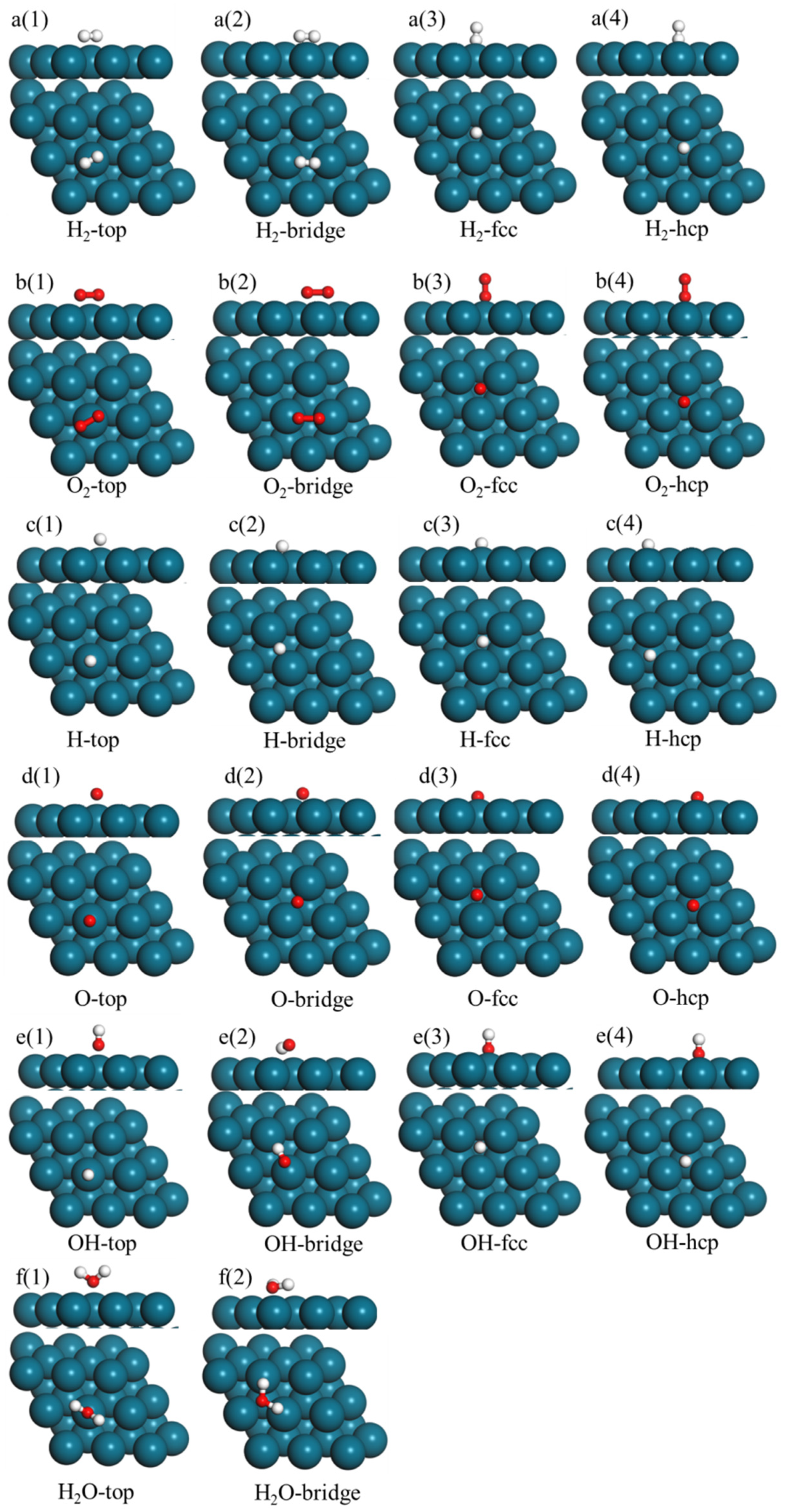
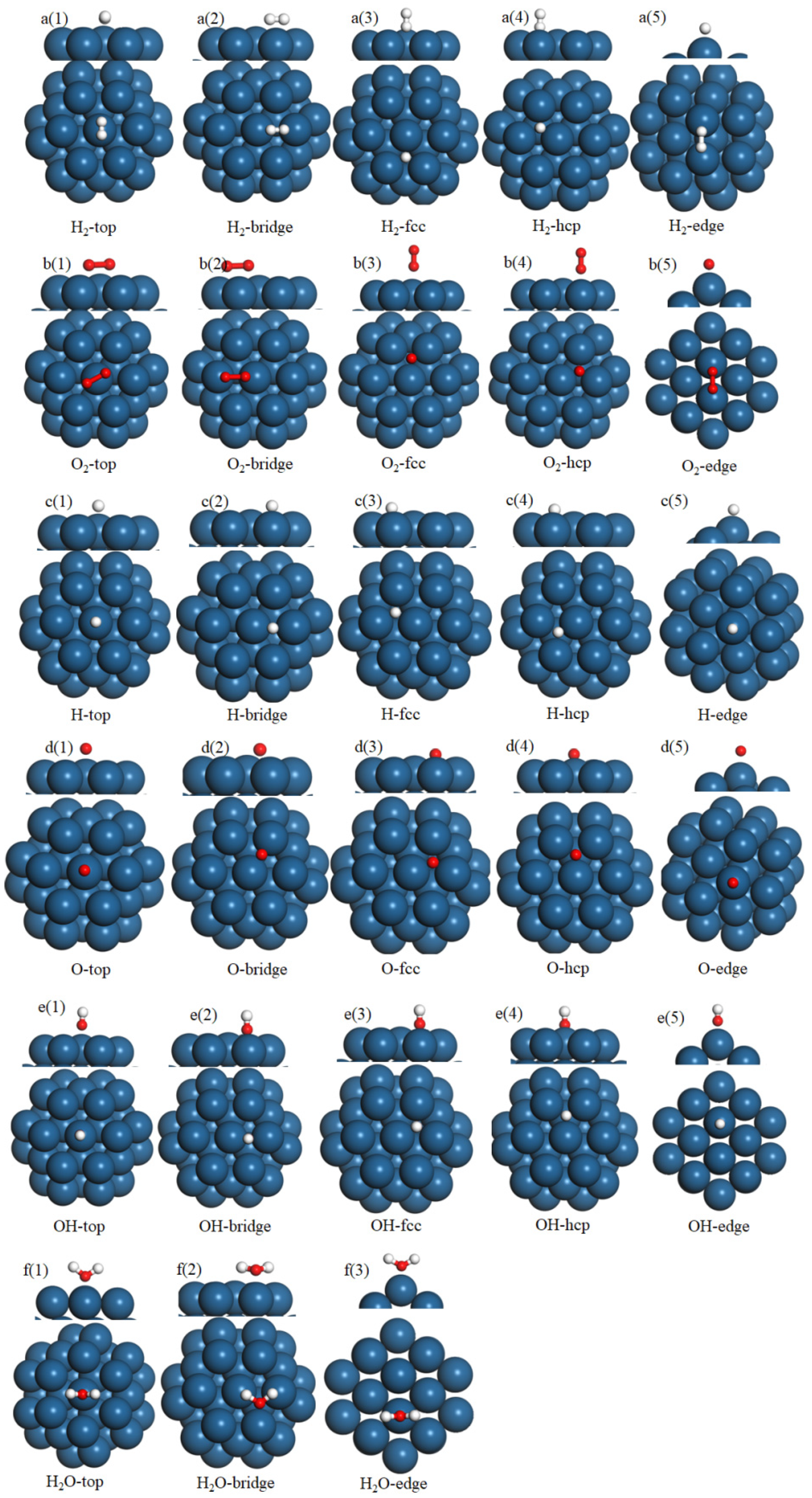

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Figures | Name | Adsorption Energy/eV | Geometric Parameter/Å |
|---|---|---|---|
| Figure 2a(1) | H2-top | −1.31 | Pd1-H1:1.762; Pd1-H2:1.760 |
| Figure 2a(2) | H2-bridge | −1.43 | Pd1-H1:1.788; Pd2-H2:1.792 |
| Figure 2a(3) | H2-fcc | −1.66 | Pd1-H1:1.798; Pd2-H1:1.799; Pd3-H1:1.799 |
| Figure 2a(4) | H2-hcp | −1.62 | Pd1-H1:1.802; Pd2-H1:1.801; Pd3-H1:1.801 |
| Figure 2b(1) | O2-top | −0.68 | Pd1-O1:1.964; Pd1-O 2:1.965 |
| Figure 2b(2) | O2-bridge | −0.77 | Pd1-O1:2.019; Pd2-O 2:2.017 |
| Figure 2b(3) | O2-fcc | −0.98 | Pd1-O 1:1.972; Pd2-O 1:1.970; Pd3-O 1:1.970 |
| Figure 2b(4) | O2-hcp | −0.95 | Pd1-O 1:1.993; Pd2-O1:1.990; Pd3-O1:1.991 |
| Figure 2c(1) | H-top | −2.08 | Pd1-H:1.646 |
| Figure 2c(2) | H-bridge | −2.49 | Pd1-H:1.736; Pd2-H:1.734 |
| Figure 2c(3) | H-fcc | −2.57 | Pd1-H:1.759; Pd2-H:1.760; Pd3-H:1.760 |
| Figure 2c(4) | H-hcp | −2.54 | Pd1-H:1.768; Pd2-H:1.768; Pd3-H:1.769 |
| Figure 2d(1) | O-top | −2.37 | Pd1-O:1.938 |
| Figure 2d(2) | O-bridge | −3.06 | Pd1-O:1.998; Pd2-O:1.998 |
| Figure 2d(3) | O-fcc | −3.25 | Pd1-O:2.034; Pd2-H:2.035; Pd3-O:2.035 |
| Figure 2d(4) | O-hcp | −2.96 | Pd1-O:2.079; Pd2-O:2.079; Pd3-O:2.078 |
| Figure 2e(1) | OH-top | −1.03 | Pd1-O:1.957 |
| Figure 2e(2) | OH-bridge | −1.39 | Pd1-O:2.016; Pd2-O:2.016 |
| Figure 2e(3) | OH-fcc | −1.40 | Pd1-O:2.055; Pd2-H:2.056; Pd3-O:2.055 |
| Figure 2e(4) | OH-hcp | −1.33 | Pd1-O:2.092; Pd2-O:2.093; Pd3-O:2.093 |
| Figure 2f(1) | H2O-top | −0.36 | Pd1-O:2.105 |
| Figure 2f(2) | H2O-bridge | −0.59 | Pd1-H1:1.887; Pd2-H2:1.886 |
| Figures | Name | Adsorption Energy/eV | Geometric Parameter/Å |
|---|---|---|---|
| Figure 3a(1) | H2-top | −1.34 | Pd1-H1:1.733; Pd1-H2:1.732 |
| Figure 3a(2) | H2-bridge | −1.55 | Pd1-H1:1.745; Pd2-H2:1.752 |
| Figure 3a(3) | H2-fcc | −1.76 | Pd1-H1:1.756; Pd2-H1:1.768; Pd3-H1:1.769 |
| Figure 3a(4) | H2-hcp | −1.73 | Pd1-H1:1.781; Pd2-H1:1.794; Pd3-H1:1.795 |
| Figure 3a(5) | H2-edge | −0.92 | Pd1-H1:1.881; Pd2-H2:1.882 |
| Figure 3b(1) | O2-top | −0.83 | Pd1-O1:1.968; Pd1-O2:1.968 |
| Figure 3b(2) | O2-bridge | −0.95 | Pd1-O1:2.114; Pd2-O2:2.121 |
| Figure 3b(3) | O2-fcc | −1.06 | Pd1-O1:2.070; Pd2-O1:2.077; Pd3-O1:2.077 |
| Figure 3b(4) | O2-hcp | −1.02 | Pd1-O1:2.059; Pd2-O1:2.066; Pd3-O1:2.067 |
| Figure 3b(5) | O2-edge | −0.89 | Pd1-O1:1.993; Pd2-O2:1.994 |
| Figure 3c(1) | H-top | −1.14 | Pd1-H:1.630 |
| Figure 3c(2) | H-bridge | −1.38 | Pd1-H:1.724; Pd2-H:1.729 |
| Figure 3c(3) | H-fcc | −1.57 | Pd1-H:1.738; Pd2-H:1.756; Pd3-H:1.754 |
| Figure 3c(4) | H-hcp | −1.54 | Pd1-H:1.749; Pd2-H:1.752; Pd3-H:1.750 |
| Figure 3c(5) | H-edge | −1.11 | Pd1-H:1.688 |
| Figure 3d(1) | O-top | −2.45 | Pd1-O:1.920 |
| Figure 3d(2) | O-bridge | −2.59 | Pd1-O:1.974; Pd2-O:1.979 |
| Figure 3d(3) | O-fcc | −3.62 | Pd1-O:2.011; Pd2-H:2.017; Pd3-O:2.018 |
| Figure 3d(4) | O-hcp | −3.56 | Pd1-O:2.062; Pd2-O:2.074; Pd3-O:2.073 |
| Figure 3d(5) | O-edge | −1.73 | Pd1-O:2.122 |
| Figure 3e(1) | OH-top | −1.57 | Pd1-O:1.924 |
| Figure 3e(2) | OH-bridge | −2.51 | Pd1-O:2.001; Pd2-O:2.009 |
| Figure 3e(3) | OH-fcc | −2.46 | Pd1-O:2.022; Pd2-H:2.028; Pd3-O:2.029 |
| Figure 3e(4) | OH-hcp | −2.41 | Pd1-O:2.069; Pd2-O:2.077; Pd3-O:2.078 |
| Figure 3e(5) | OH-edge | −0.61 | Pd1-O:2.143 |
| Figure 3f(1) | H2O-top | −0.36 | Pd1-O:2.075 |
| Figure 3f(2) | H2O-bridge | −0.59 | Pd1-H1:1.869; Pd2-H2:1.869 |
| Figure 3f(3) | H2O-edge | −0.44 | Pd1-H1:1.869; Pd2-H2:1.869 |
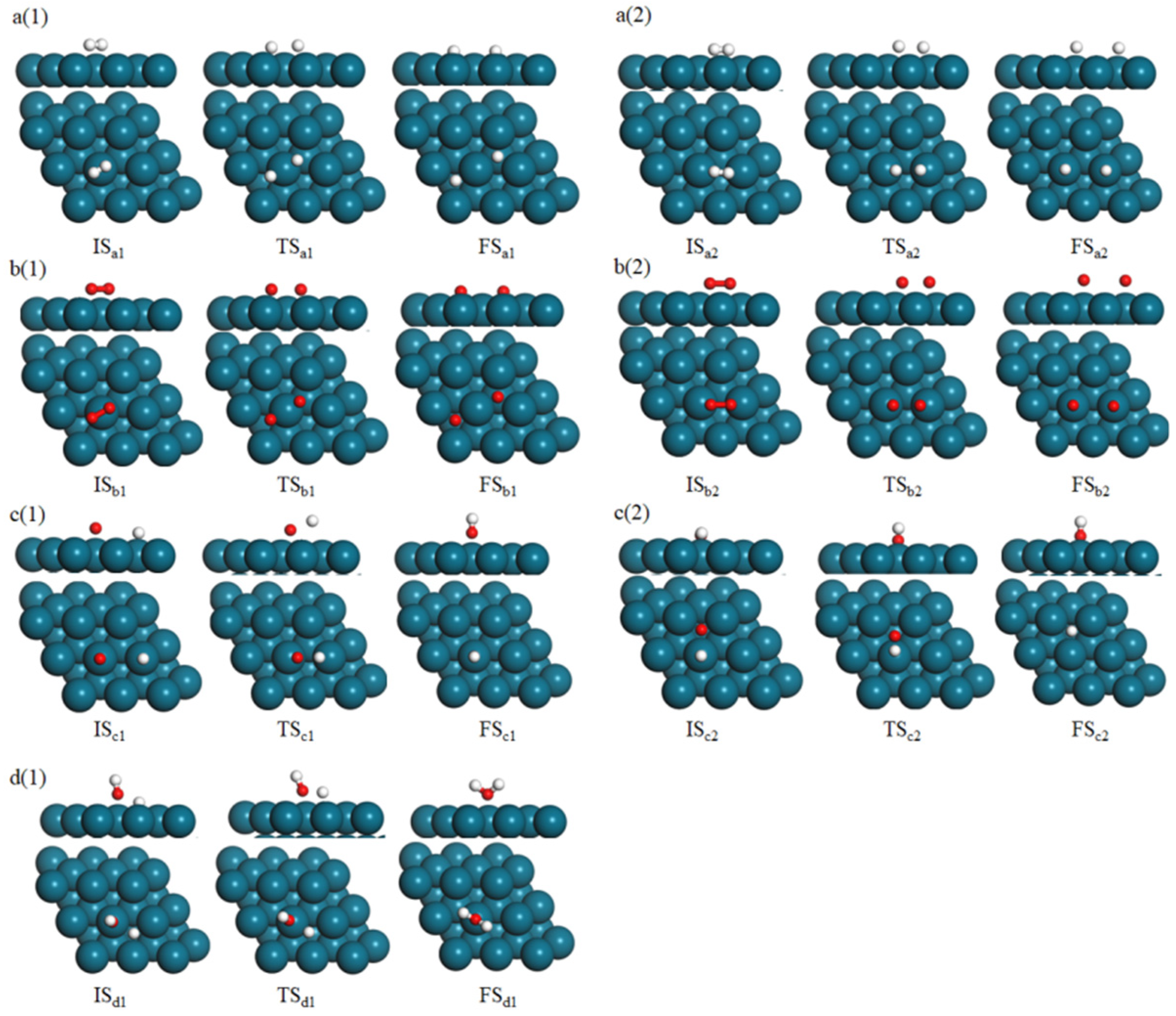
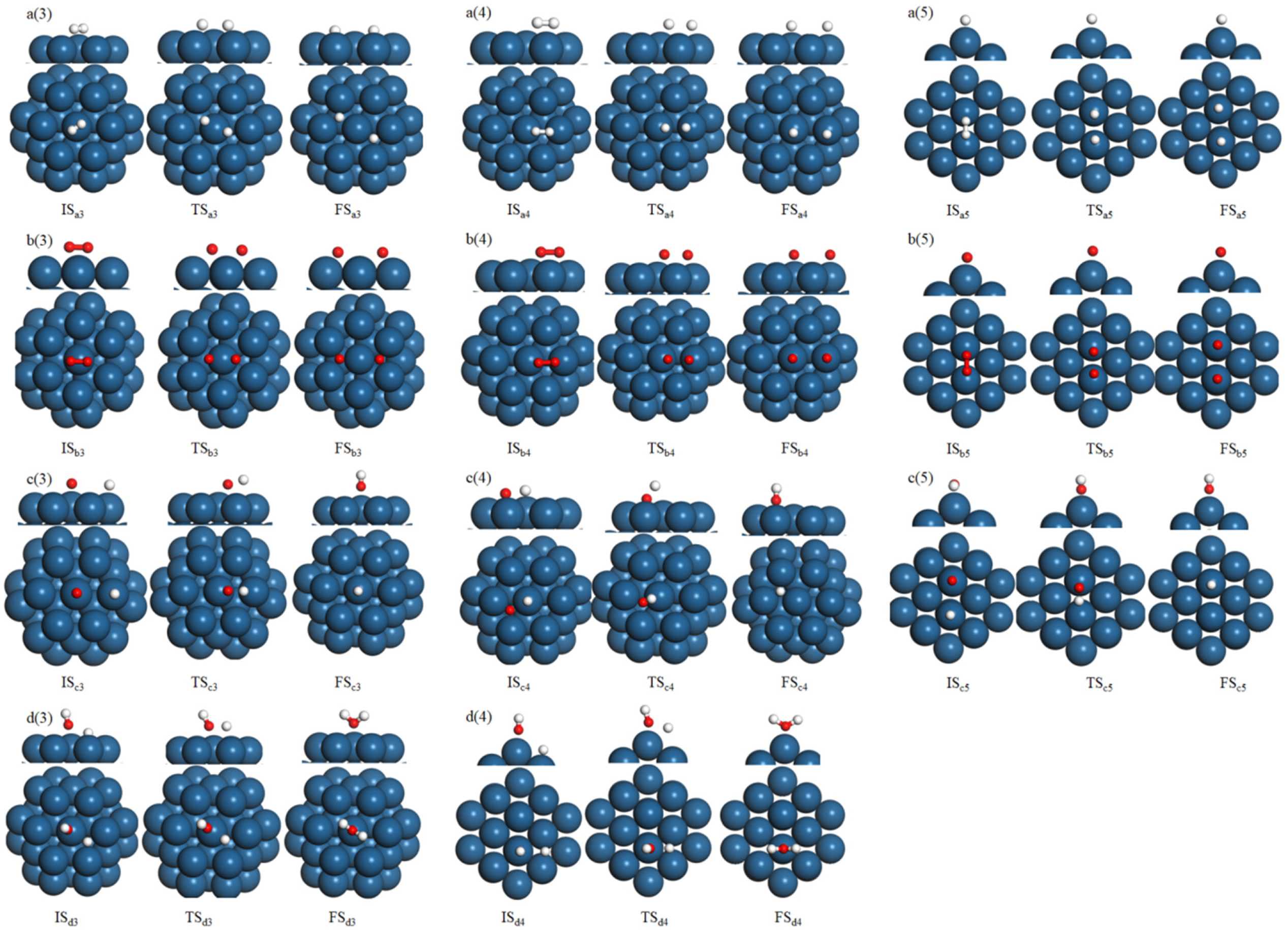
3.3. Reaction Mechanism of H2 Catalytic Combustion on Pd38 Cluster Model and Pd(111) Slab Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pfefferle, W.C. The catalytic combustor—An approach to cleaner combustion (by reaction control in gas turbine engines). J. Energy 2012, 2, 142–146. [Google Scholar]
- Pangborn, J.; Scott, M.; Sharer, J. Technical prospects for commercial and residential distribution and utilization of hydrogen. Int. J. Hydrogen Energy 1977, 2, 431–445. [Google Scholar]
- Kramer, J.F.; Reihani, S.A.S.; Jackson, G.S. Low-temperature combustion of hydrogen on supported Pd catalysts. Proc. Combust. Inst. 2002, 29, 989–996. [Google Scholar] [CrossRef]
- Zhang, C.M.; Zhang, J.; Ma, J.X. Hydrogen catalytic combustion over a Pt/Ce0.6Zr0.4O2/MgAl2O4 mesoporous coating monolithic catalyst. Int. J. Hydrogen Energy 2012, 37, 12941–12946. [Google Scholar] [CrossRef]
- Shinde, V.M.; Madras, G. Nanostructured Pd modified Ni/CeO2 catalyst for water gas shift and catalytic hydrogen combustion reaction. Appl. Catal. B 2013, 132, 28–38. [Google Scholar] [CrossRef]
- Singh, S.A.; Vishwanath, K.; Madras, G. Role of Hydrogen and Oxygen Activation over Pt and Pd-Doped Composites for Catalytic Hydrogen Combustion. ACS Appl. Mater. Interfaces 2017, 9, 19380–19388. [Google Scholar]
- Zhou, J.H.; Wang, Y.; Yang, W.J.; Liu, J.Z.; Wang, Z.H.; Cen, K.F. Combustion of hydrogen-air in catalytic micro-combustors made of different material. Int. J. Hydrogen Energy 2009, 34, 3535–3545. [Google Scholar] [CrossRef]
- Du Preez, S.P.; Jones, D.R.; Bessarabov, D.G.; Falch, A.; Quaresma, C.M.D.; Dunnill, C.W. Development of a Pt/stainless steel mesh catalyst and its application in catalytic hydrogen combustion. Int. J. Hydrogen Energy 2019, 44, 27094–27106. [Google Scholar] [CrossRef]
- Fernández, A.; Arzac, G.; Vogt, U.; Hosoglu, F.; Borgschulte, A.; de Haro, M.J.; Montes, O.; Züttel, A. Investigation of a Pt containing washcoat on SiC foam for hydrogen combustion applications. Appl. Catal. B 2016, 180, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, L.; Niu, F.; Chen, D.; Qin, L.; Sun, X.; Huang, Y. Catalytic combustion of hydrogen for residential heat supply application. Int. J. Energy Res. 2016, 40, 1979–1985. [Google Scholar]
- Fumey, B.; Buetler, T.; Vogt, U.F. Ultra-low NO emissions from catalytic hydrogen combustion. Appl. Energy 2018, 213, 334–342. [Google Scholar] [CrossRef]
- Nguyen, V.N.; Deja, R.; Peters, R.; Blum, L.; Stolten, D. Study of the catalytic combustion of lean hydrogen-air mixtures in a monolith reactor. Int. J. Hydrogen Energy 2018, 43, 17520–17530. [Google Scholar] [CrossRef]
- Arzac, G.M.; Montes, O.; Fernandez, A. Pt-impregnated catalysts on powdery SiC and other commercial supports for the combustion of hydrogen under oxidant conditions. Appl. Catal. B 2017, 201, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Mantzaras, J.; Bombach, R. Kinetic interactions between hydrogen and carbon monoxide oxidation over platinum. Combust. Flame 2014, 161, 332–346. [Google Scholar] [CrossRef]
- Han, S.M.; Mullins, C.B. Catalytic Reactions on Pd-Au metallic Model Catalysts. Accounts Chem. Res. 2021, 54, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Morfin, F.; Sabroux, J.C.; Renouprez, A. Catalytic combustion of hydrogen for mitigating hydrogen risk in case of a severe accident in a nuclear power plant: Study of catalysts poisoning in a representative atmosphere. Appl. Catal. B 2004, 47, 47–58. [Google Scholar] [CrossRef]
- Sandeep, K.C.; Bhattacharyya, R.; Warghat, C.; Bhanja, K.; Mohan, S. Experimental investigation on the kinetics of catalytic recombination of hydrogen with oxygen in air. Int. J. Hydrogen Energy 2014, 39, 17906–17912. [Google Scholar] [CrossRef]
- Qi, W.J.; Ran, J.Y.; Wang, R.R.; Du, X.S.; Shi, J.; Ran, M.C. Kinetic mechanism of effects of hydrogen addition on methane catalytic combustion over Pt(111) surface: A DFT study with cluster modeling. Comput. Mater. Sci. 2016, 111, 430–442. [Google Scholar] [CrossRef]
- Sui, R.; Liang, W.K.; Zhang, L.; Mantzaras, J.; Law, C.K. Kinetic interactions between H-2 and CO in catalytic oxidation over PdO. Combust. Flame 2020, 211, 270–280. [Google Scholar] [CrossRef]
- Tsamis, C.; Tsoura, L.; Nassiopoulou, A.; Travlos, A.; Salmas, C.; Hatzilyberis, K.; Androutsopoulos, G. Hydrogen catalytic oxidation reaction on Pd-doped porous silicon. IEEE Sens. J. 2002, 2, 89–95. [Google Scholar] [CrossRef]
- Abate, S.; Barbera, K.; Centi, G.; Giorgianni, G.; Perathoner, S. Role of size and pretreatment of Pd particles on their behaviour in the direct synthesis of H2O2. J. Energy Chem. 2016, 25, 9. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Z.; Tan, J. New insights into catalysis for Heck reactions with fine supported Pd particles. React. Chem. Eng. 2020, 5, 921–934. [Google Scholar] [CrossRef]
- Chen, H.; Wu, Y.L.; Qi, S.T.; Chen, Y.; Yang, M.D. Deoxygenation of octanoic acid catalyzed by hollow spherical Ni/ZrO2. Appl. Catal. A 2017, 529, 79–90. [Google Scholar] [CrossRef]
- Wang, B.J.; Song, L.Z.; Zhang, R.G. The dehydrogenation of CH4 on Rh(111), Rh(110) and Rh(100) surfaces: A density functional theory study. Appl. Surf. Sci. 2012, 258, 3714–3722. [Google Scholar] [CrossRef]
- Zhang, R.G.; Song, L.Z.; Wang, Y.H. Insight into the adsorption and dissociation of CH4 on Pt(h k l) surfaces: A theoretical study. Appl. Surf. Sci. 2012, 258, 7154–7160. [Google Scholar] [CrossRef]
- Qi, D.; Luo, X.; Yao, J.; Yao, Y.; Lu, X. Computational study of CO catalytic oxidation on Pd38 cluster model and Pd slab model. J. Fuel Chem. Technol. 2020, 48, 9. [Google Scholar]
- Wang, D.S.; Li, Y.D. Bimetallic Nanocrystals: Liquid-Phase Synthesis and Catalytic Applications. Adv. Mater. 2011, 23, 1044–1060. [Google Scholar] [CrossRef] [PubMed]

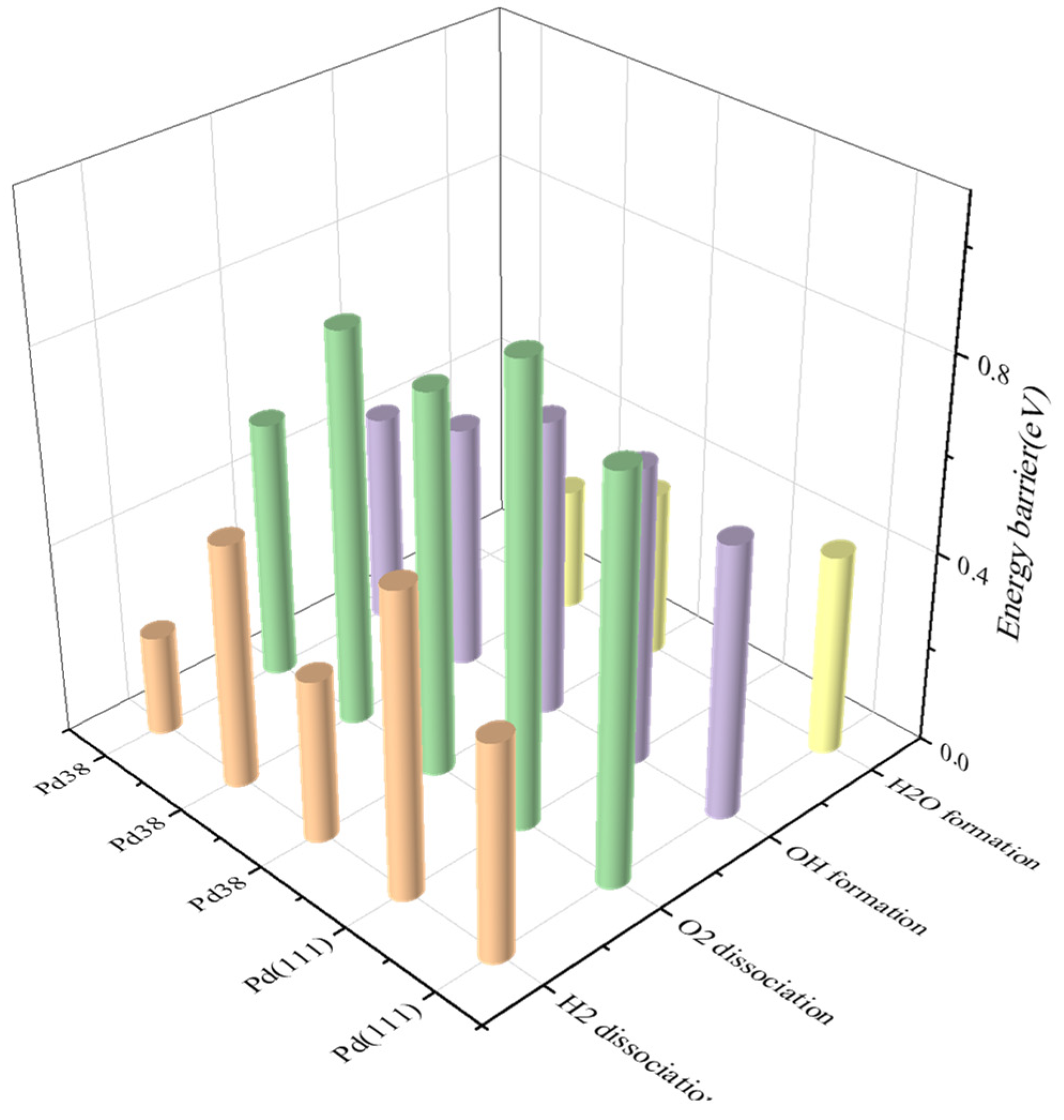
| Elementary Step | No. | Pd(111)/(eV) | Pd38/(eV) | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| H2 → 2H | a | Ea: 0.45 Erxn: −0.12 | Ea: 0.63 Erxn: −0.22 | Ea: 0.34 Erxn: −0.23 | Ea: 0.51 Erxn: −0.29 | Ea: 0.21 Erxn: −0.32 |
| O2 → 2O | b | Ea: 0.83 Erxn: −0.47 | Ea: 0.94 Erxn: −0.32 | Ea: 0.79 Erxn: −0.42 | Ea: 0.82 Erxn: −0.35 | Ea: 0.54 Erxn: −0.50 |
| H + O → OH | c | Ea: 0.57 Erxn: −0.31 | Ea: 0.62 Erxn: −0.19 | Ea: 0.62 Erxn: −0.33 | Ea: 0.51 Erxn: −0.24 | Ea: 0.44 Erxn: −0.32 |
| OH + H → H2O | d | Ea: 0.42 Erxn: −0.44 | Ea: 0.62 Erxn: −0.33 | Ea: 0.51 Erxn: −0.24 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, D.; Luo, X.; Yao, Y.; Qi, N.; Lu, X.; Chen, H.; Shi, H. Computational Study of H2 Catalytic Combustion on Pd38 Cluster Model and Pd(111) Slab Model. Symmetry 2022, 14, 1544. https://doi.org/10.3390/sym14081544
Qi D, Luo X, Yao Y, Qi N, Lu X, Chen H, Shi H. Computational Study of H2 Catalytic Combustion on Pd38 Cluster Model and Pd(111) Slab Model. Symmetry. 2022; 14(8):1544. https://doi.org/10.3390/sym14081544
Chicago/Turabian StyleQi, Dabin, Xudong Luo, Yulong Yao, Na Qi, Xiaojun Lu, Hao Chen, and Hongqi Shi. 2022. "Computational Study of H2 Catalytic Combustion on Pd38 Cluster Model and Pd(111) Slab Model" Symmetry 14, no. 8: 1544. https://doi.org/10.3390/sym14081544
APA StyleQi, D., Luo, X., Yao, Y., Qi, N., Lu, X., Chen, H., & Shi, H. (2022). Computational Study of H2 Catalytic Combustion on Pd38 Cluster Model and Pd(111) Slab Model. Symmetry, 14(8), 1544. https://doi.org/10.3390/sym14081544