
Potential Energy Surfaces for Noble Gas (Ar, Kr, Xe, Rn)–Propylene Oxide Systems: Analytical Formulation and Binding


Abstract
1. Introduction
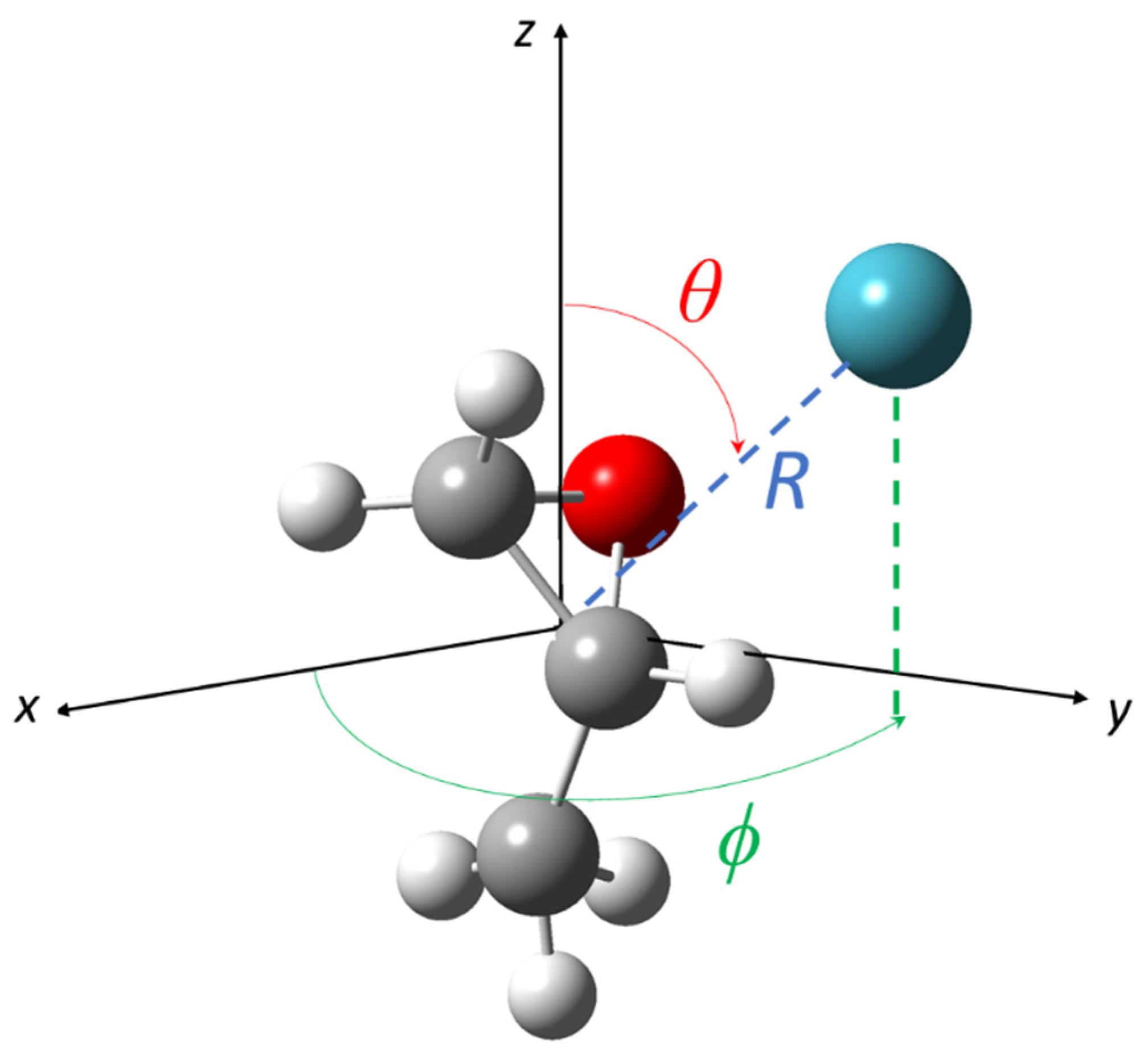
2. Potential Energy Surfaces
2.1. The Improved Lennard-Jones Potential
2.2. Ab Initio Calculations
3. Results and Discussion
4. Final Remarks and Conclusions
- (1)
- The adopted method represents the intermolecular potential by an analytic formulation that makes use of few parameters, all having a well-defined physical meaning. This physically grounded restriction, at variance with most interpolation procedures, ensures the reliability of the interaction in the whole space of the relative configurations;
- (2)
- The present method allows a direct evaluation of the long-range dispersion coefficient C6, that controls the capture character of the long-range forces;
- (3)
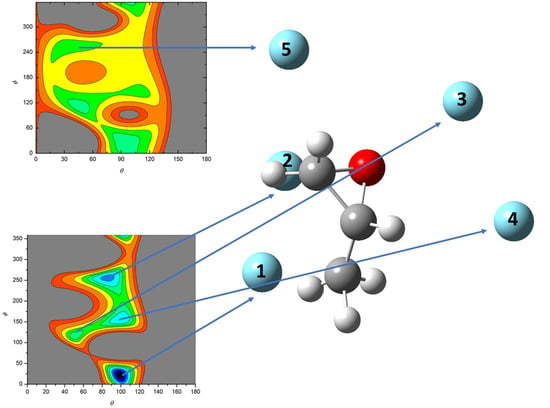
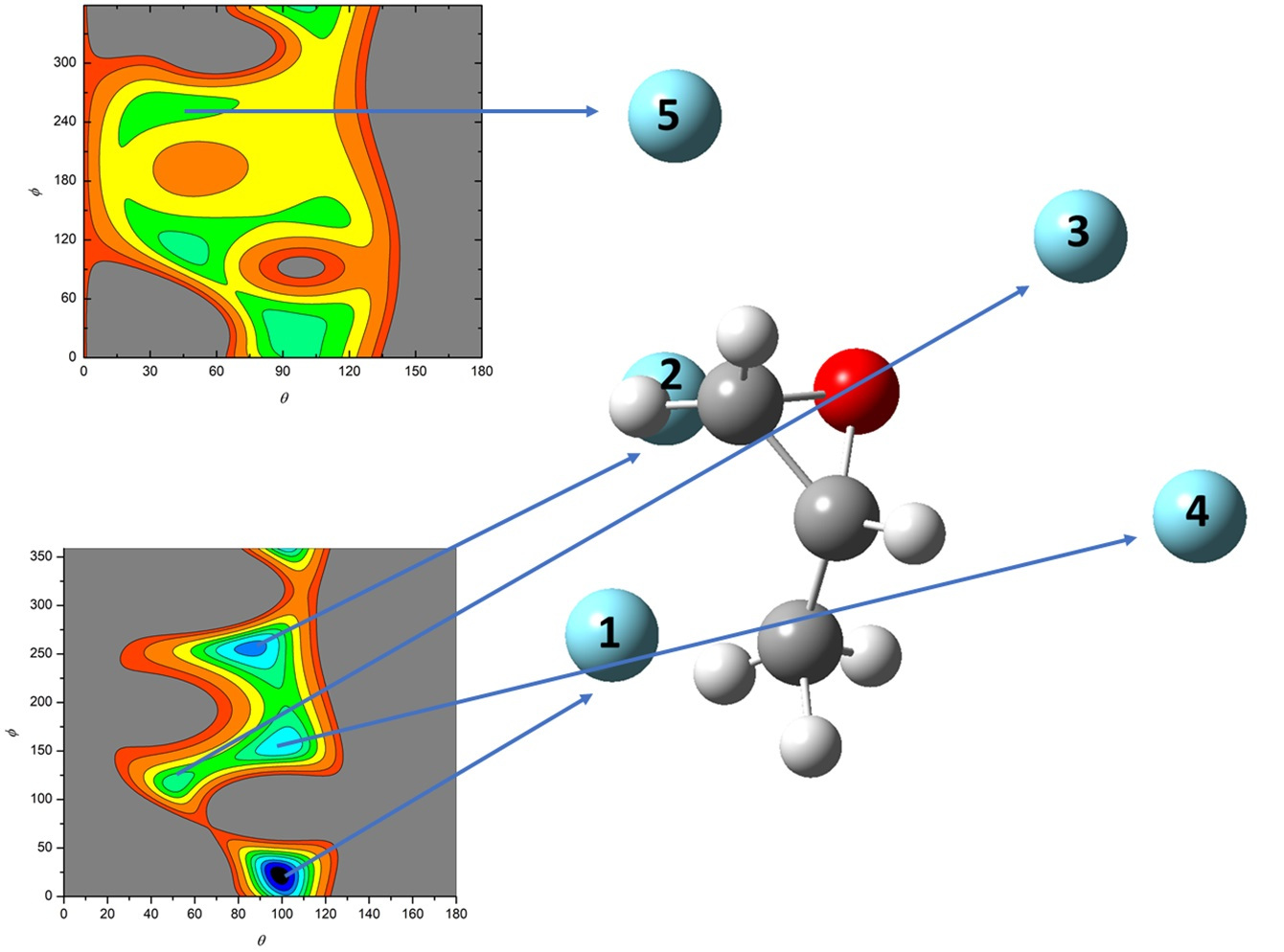
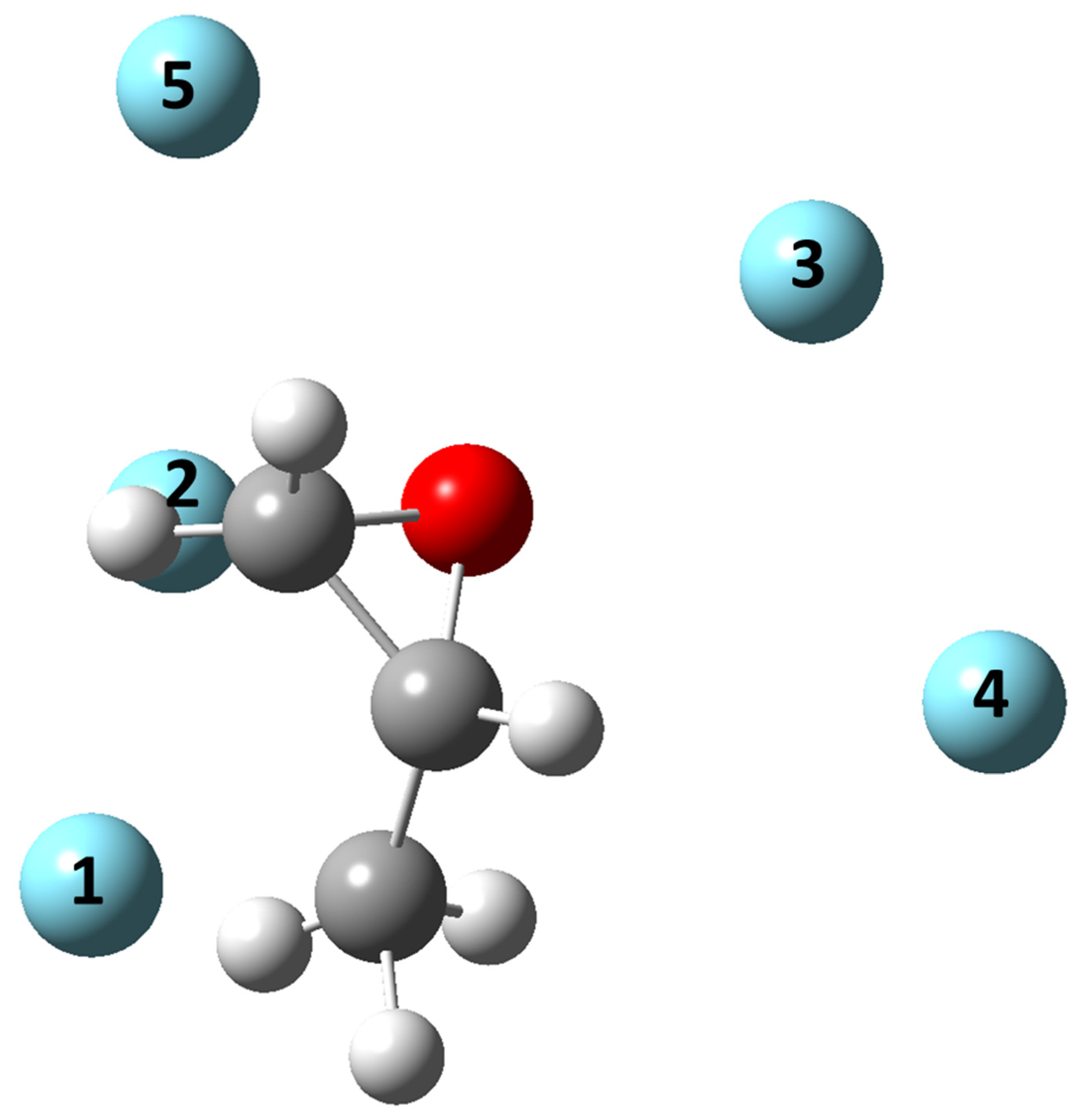
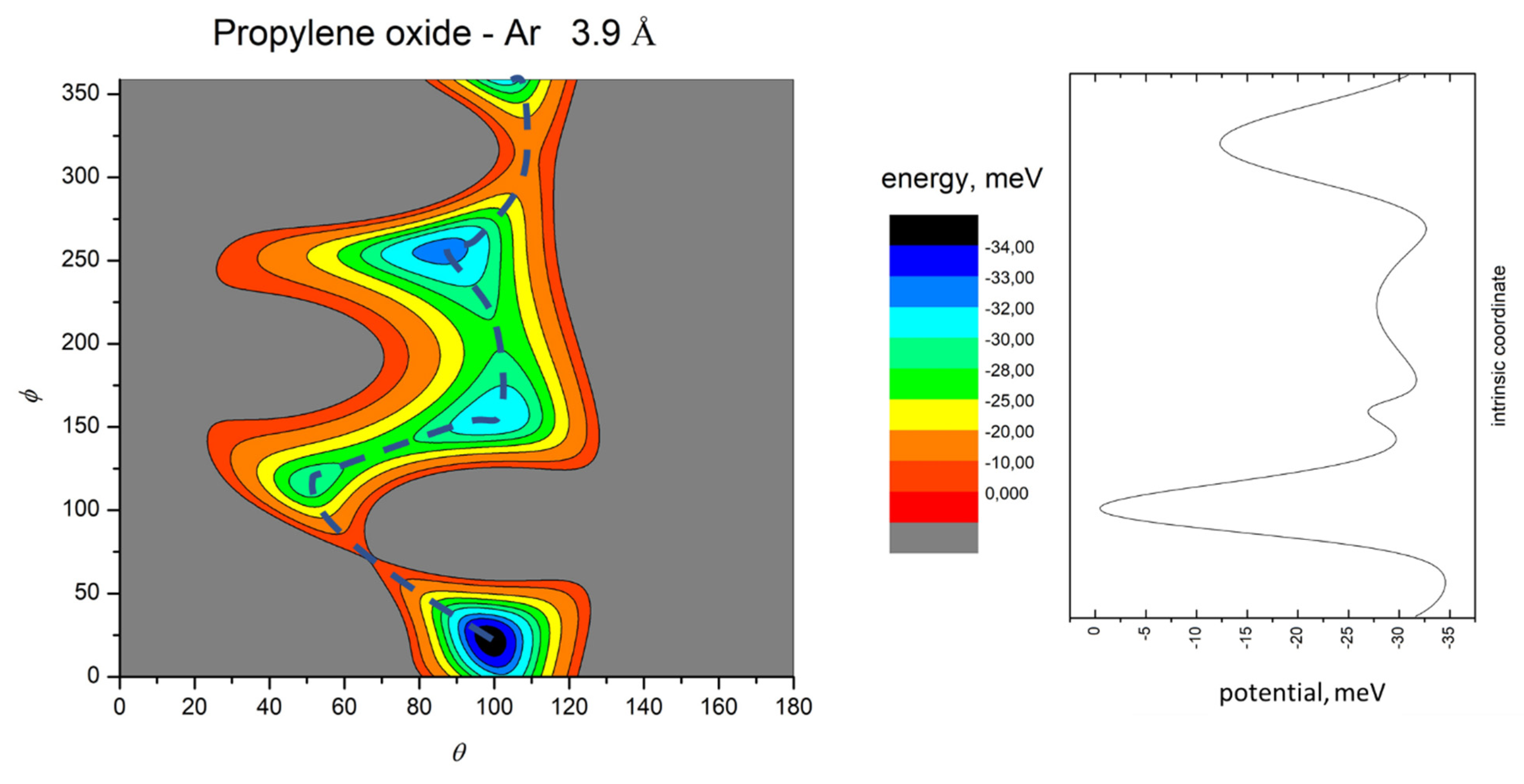
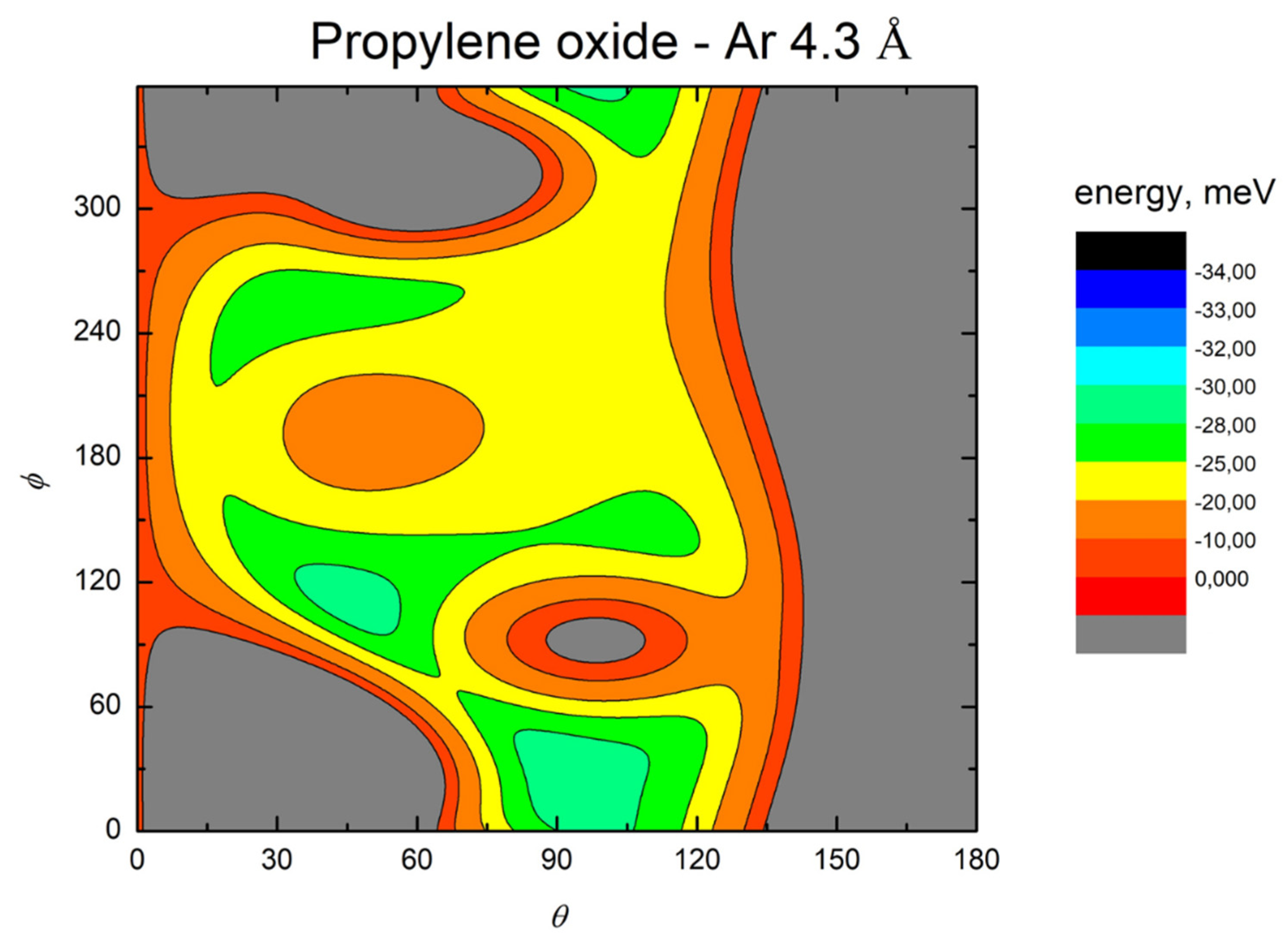
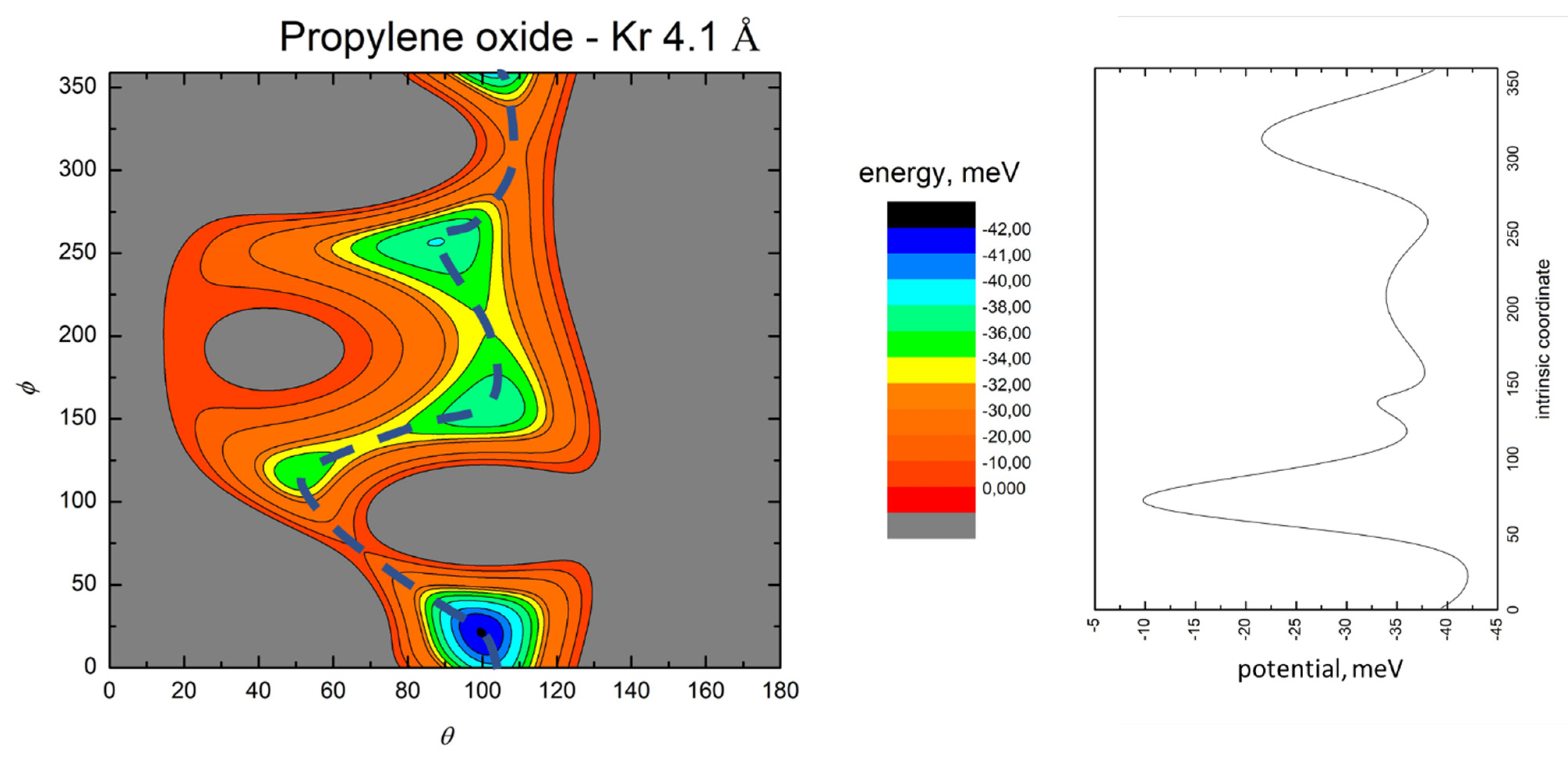
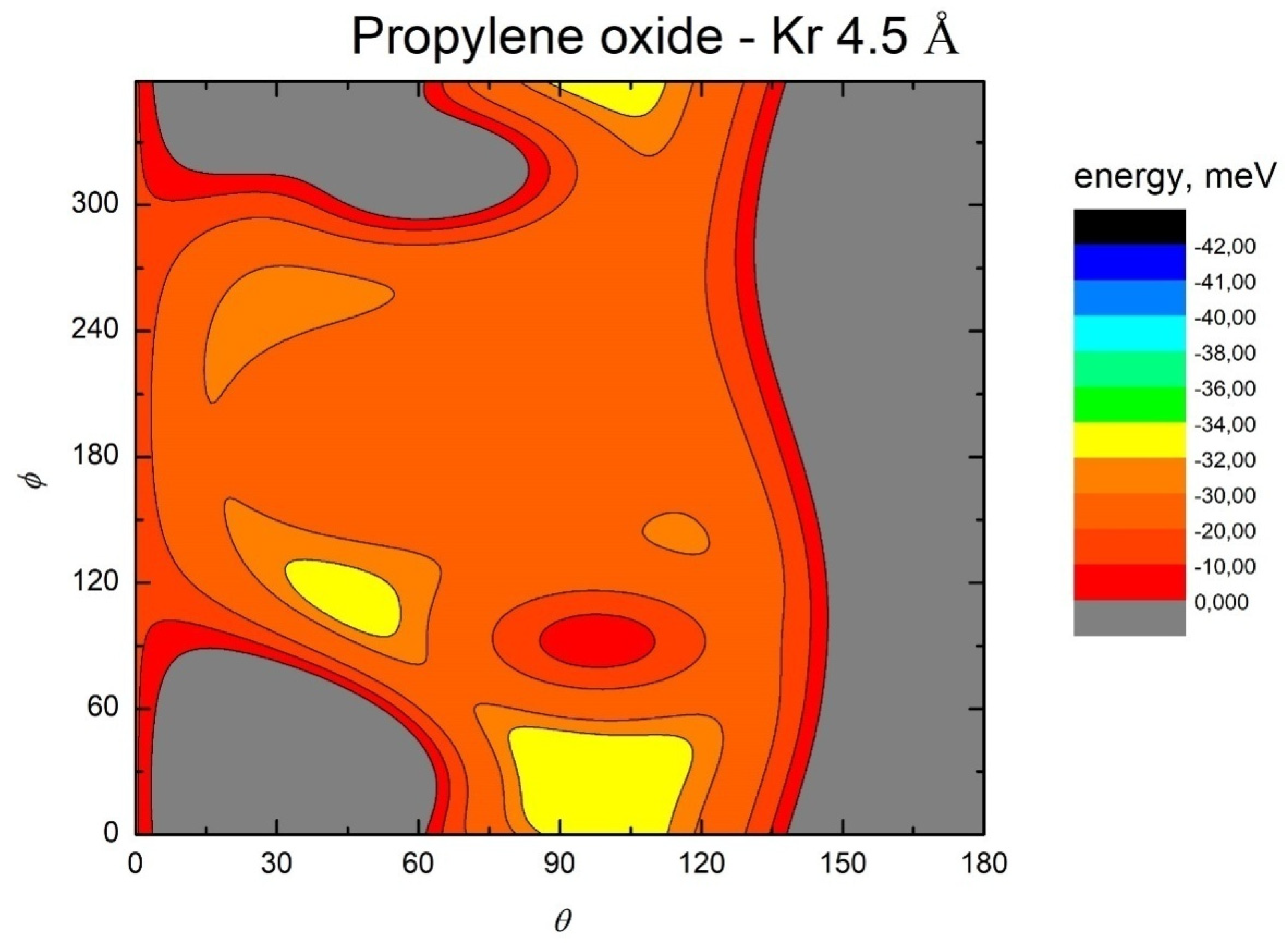
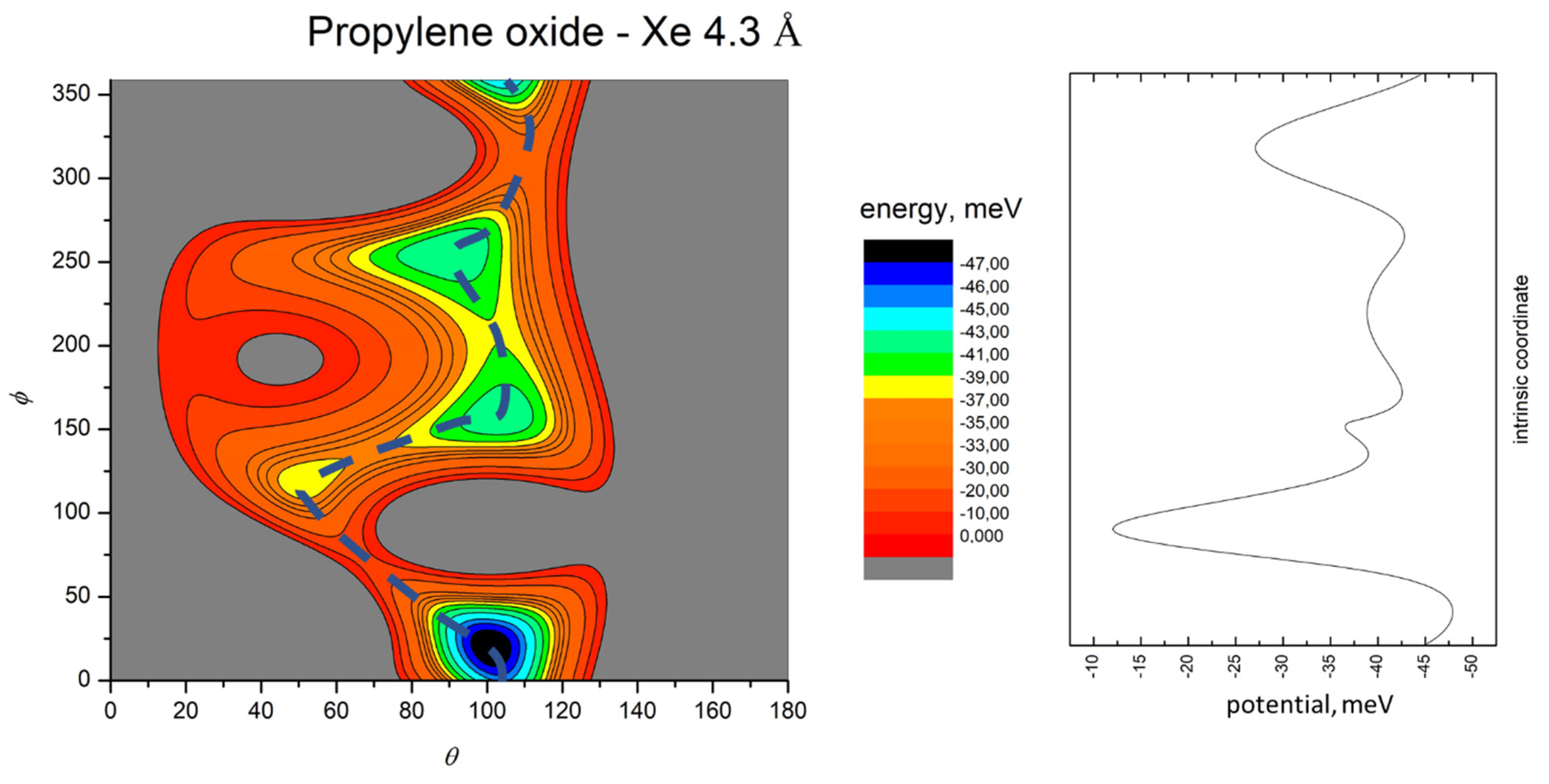
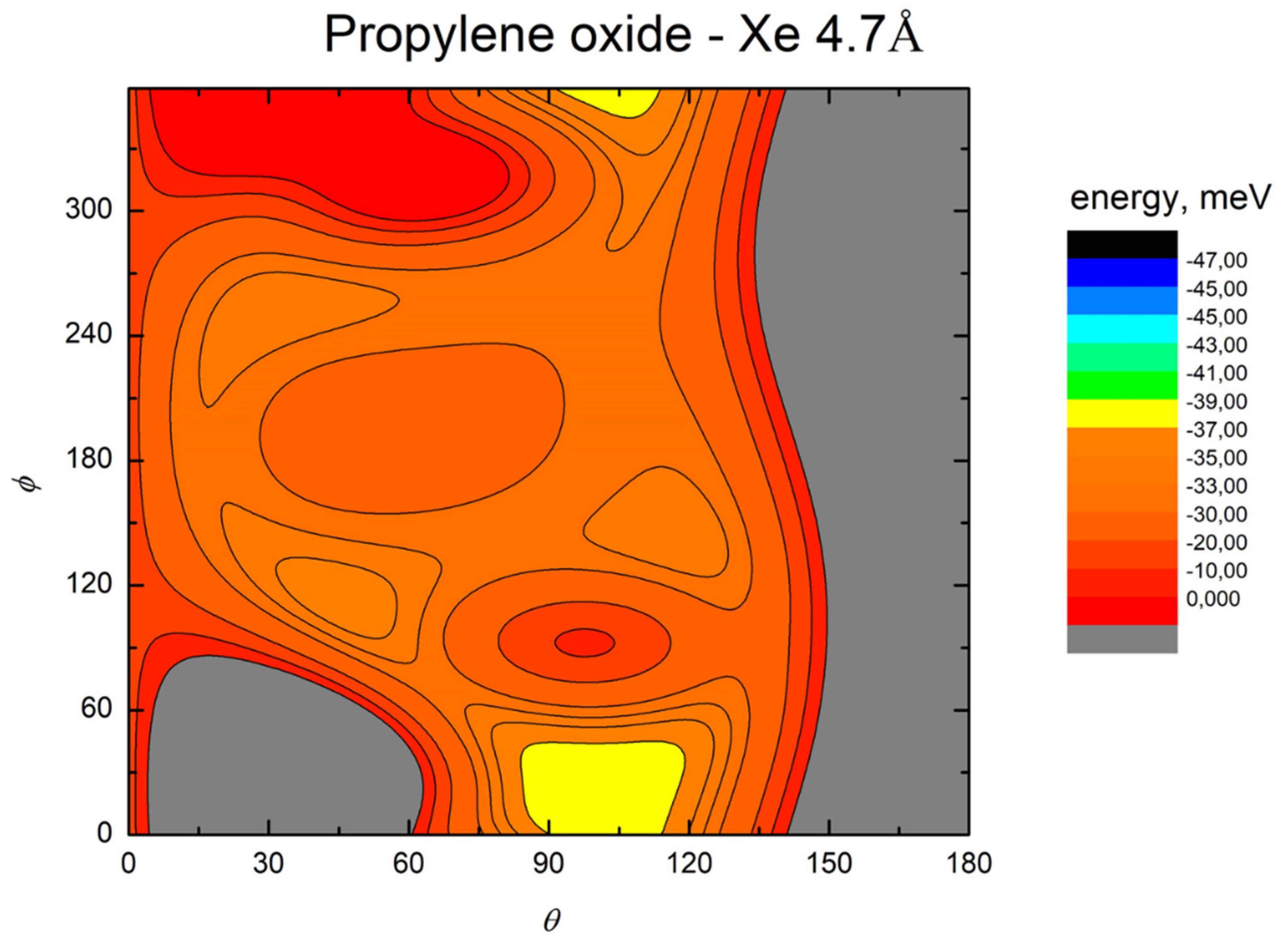
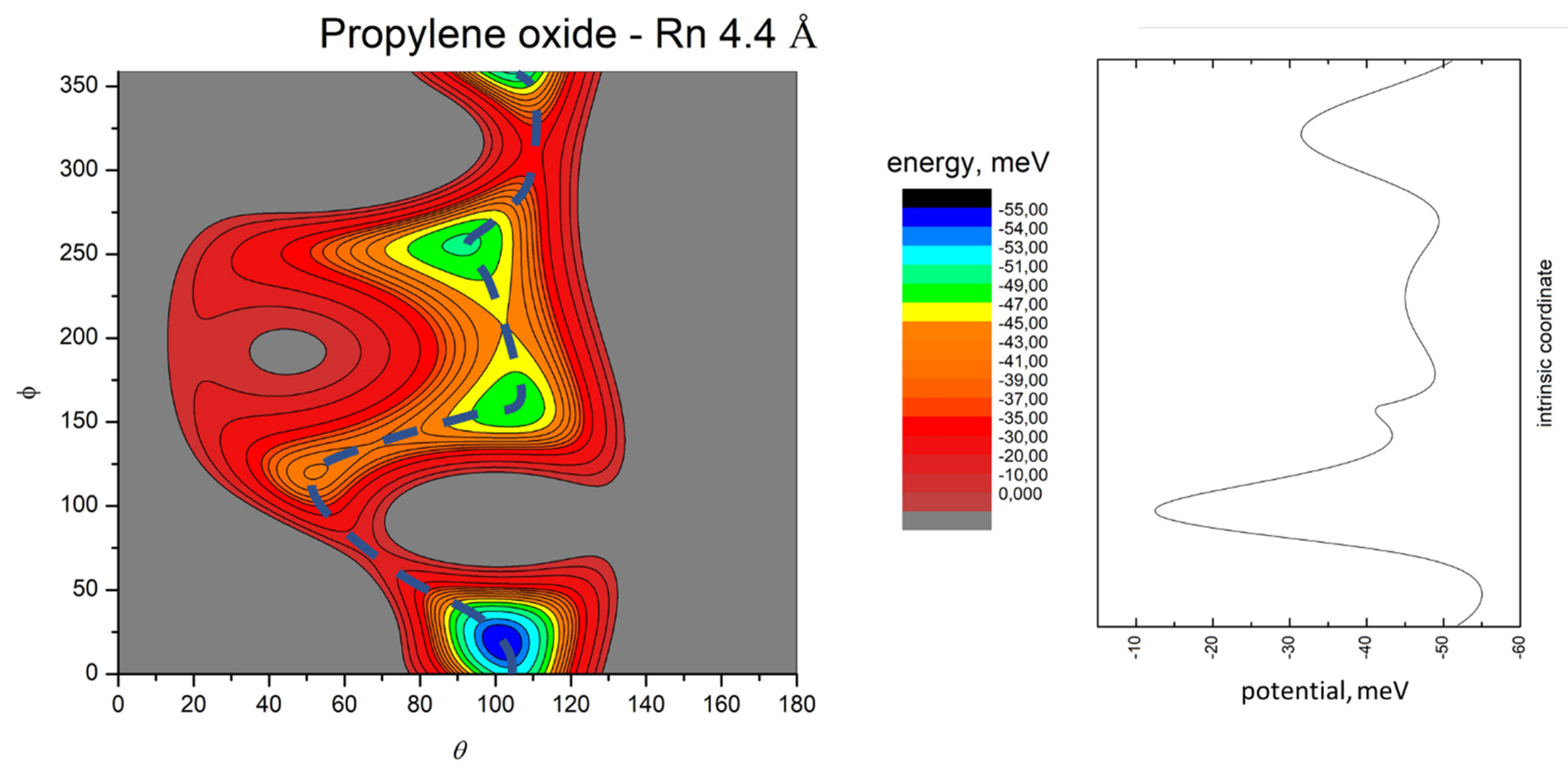
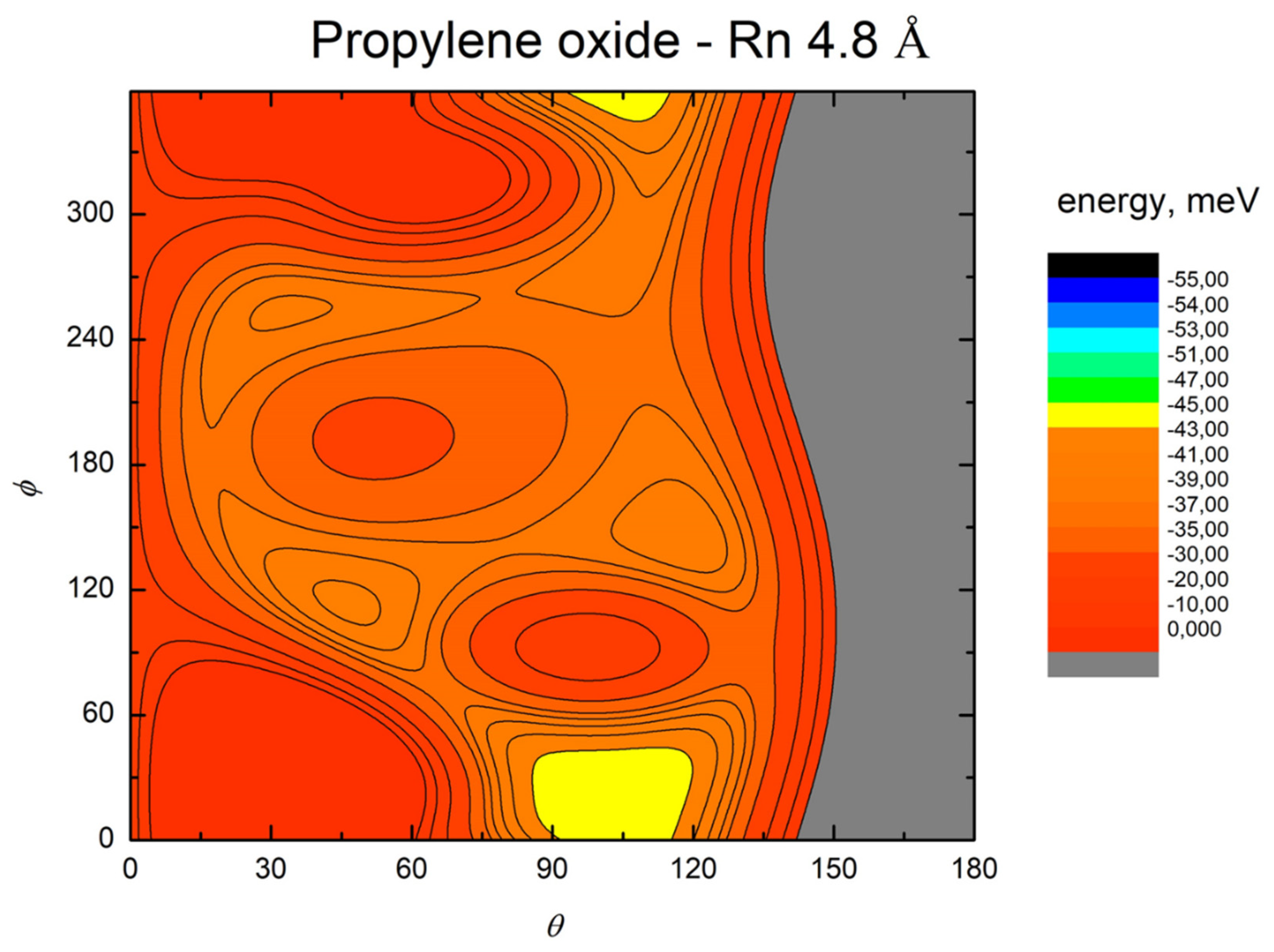
- All systems present five minima very close in energy. Therefore, the collision dynamics is likely to be determined from their overall contribution, as well as from the possibility of adiabatic inter-conversion between them, hindered by the energy barriers that increase with the noble gas mass, and as the R distance decreases;
- (4)
- For each system, the location of the minimum energy changes as the intermolecular distance varies, that is the most stable configuration at long range, can substantially differ from that exhibited at intermediate and short range. An important consequence is that a system, formed by the trapping long-range anisotropic attractive forces, can be channeled during the relaxation of its internal degrees of freedom via fast non-adiabatic cooling, in configurations that can differ from that of the PES global minimum;
- (5)
- The dissociation energy of Ar−propylene oxide and Kr−propylene oxide, estimated by Blanco et al. [25], is consistent with the well depth and geometries of the minima determined in the present work. Moreover, considerations made at the previous point 4 can also applied to this point;
- (6)
- The analytical formulation of the PES allows the calculation of physical, dynamical, and spectroscopic properties of the systems, which selectively depend on the weak anisotropic forces, effective both at long and intermediate range of separation distances;
- (7)
- The same method can be applied, after parameters scaling for the change in polarizability, to the description of simple diatomic molecule-propylene oxide systems, when the diatom (as H2, N2 and O2), with rotational levels following a Boltzmann distribution at room and higher temperature, rotates sufficiently faster than propylene oxide. Therefore, under such conditions, the diatom, which during the collision behaves as a pseudo-atom, interacts with propylene oxide with intermolecular forces basically of van der Waals nature. In particular, N2 and O2 would behave like Ar, as they show a similar isotropic polarizability component, while the polarizability of H2 suggests for this molecule an intermediate behavior between Ne and Ar. However, the anisotropic character of the intermolecular forces emerges at low temperature, which is when only slowly rotating diatoms are interacting with the propylene oxide. In this case, the complete potential formulation must also include the anisotropic contribution of the electrostatic components, as those arising from permanent molecular electric multipole interactions. The method can also be extended to more complex systems of interest for their chiral properties, like propylene oxide dimers.
- (8)
- These potential energy surfaces can be used to evaluate and investigate possible collision alignment processes, basic for chiral discrimination in gaseous streams and vortices occurring in no equilibrium atmosphere environments [40];
- (9)
- Finally, it is also worth pointing out that this methodology can be profitably used to preliminarily address the search of ab-initio stationary points in multidimensional surfaces for weakly bound systems. This might be a difficult task, as they require a higher level of theory than bound systems, and may present many minima, particularly when some of them are shallow and can easily escape a grid search. The ab-initio calculation of transition states may be even more elusive. A preliminary detailed search on the present PES is reliable and much faster and allows identification of confined regions where minima or transition state points are located, strongly reducing the number of points to be evaluated quantum mechanically. Moreover, its formulation permits both forces and force constants to be obtained analytically, speeding up their calculation in molecular dynamics simulations, where these quantities play a crucial role, therefore alleviating the related computational burden.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Falcinelli, S.; Cappelletti, D. Special issue “Long-Range Intermolecular Interactions in chemistry and Physics”. Chem. Phys. Lett. 2021, 786, 139190. [Google Scholar]
- Hong, Q.; Bartolomei, M.; Esposito, F.; Coletti, C.; Sun, Q.; Pirani, F. Reconciling experimental and theoretical vibrational deactivation in low energy O + N2 collisions. Phys. Chem. Chem. Phys. 2021, 23, 15475–15479. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Sun, Q.; Pirani, F.; Valentín-Rodríguez, M.A.; Hernández-Lamoneda, R.; Coletti, C.; Hernández, M.I.; Bartolomei, M. Energy exchange rate coefficients from vibrational inelastic O2 ( + O2( collisions on a new spin-averaged potential energy surface. J. Chem. Phys. 2021, 154, 064304. [Google Scholar] [CrossRef] [PubMed]
- McGuire, B.A.; Carroll, P.B.; Loomis, R.A.; Finneran, I.A.; Jewell, P.R.; Remijan, A.J.; Blake, G.A. Discovery of the interstellar chiral molecule propylene oxide (CH3CHCH2O). Science 2016, 352, 1449–1452. [Google Scholar] [CrossRef]
- Palazzetti, F.; Maciel, G.S.; Lombardi, A.; Grossi, G.; Aquilanti, V. The astrochemical observatory: Molecules in the laboratory and in the cosmos. J. Chin. Chem. Soc. 2013, 59, 1045–1052. [Google Scholar] [CrossRef]
- Aquilanti, V.; Grossi, G.; Lombardi, A.; Maciel, G.S.; Palazzetti, F. The origin of chiral discrimination: Supersonic molecular beam experiments and molecular dynamics simulations of collisional mechanisms. Phys. Scr. 2008, 78, 058119. [Google Scholar] [CrossRef]
- Quack, M. How important is parity violation for molecular and biomolecular chirality? Angew. Chem. Int. Ed. Engl. 2002, 41, 4618–4630. [Google Scholar] [CrossRef]
- Lombardi, A.; Palazzetti, F.; Maciel, G.S.; Aquilanti, V.; Sevryuk, M.B. Simulation of oriented collision dynamics of simple chiral molecules. Int. J. Quant. Chem. 2011, 111, 1651–1658. [Google Scholar] [CrossRef]
- Swalen, J.D.; Herschbach, D.R. Internal Barrier of Propylene Oxide from the Microwave Spectrum. I. J. Chem. Phys. 1957, 27, 100. [Google Scholar] [CrossRef]
- Herschbach, D.R.; Swalen, J.D. Internal Barrier of Propylene Oxide from the Microwave Spectrum. II. J. Chem. Phys. 1958, 29, 761. [Google Scholar] [CrossRef]
- Kawiecki, R.W.; Devlin, F.; Stephens, P.J.; Amos, R.D.; Handy, N.C. Vibrational circular dichroism of propylene oxide. Chem. Phys. Lett. 1988, 145, 411–417. [Google Scholar] [CrossRef]
- Šebestìk, J.; Bouř, P. Raman Optical Activity of Methyloxirane Gas and Liquid. J. Phys. Chem. Lett. 2011, 2, 498–502. [Google Scholar] [CrossRef]
- Merten, C.; Bloino, J.; Barone, V.; Xu, Y. Anharmonicity effects in the vibrational CD spectra of propylene oxide. J. Phys. Chem. Lett. 2013, 4, 3424–3428. [Google Scholar] [CrossRef]
- Che, D.-C.; Palazzetti, F.; Okuno, Y.; Aquilanti, V.; Kasai, T. Electrostatic hexapole state-selection of the asymmetric-top molecule propylene oxide. J. Phys. Chem. A 2010, 114, 3280. [Google Scholar] [CrossRef] [PubMed]
- Che, D.-C.; Kanda, K.; Palazzetti, F.; Aquilanti, V.; Kasai, T. Electrostatic Hexapole State-Selection of the Asymmetric-Top Molecule Propylene Oxide: Rotational and Orientational Distributions. Chem. Phys. 2012, 399, 180–192. [Google Scholar] [CrossRef]
- Falcinelli, S.; Vecchiocattivi, F.; Alagia, M.; Schio, L.; Richter, R.; Stranges, S.; Catone, D.; Arruda, M.S.; Mendes, L.A.V.; Palazzetti, F.; et al. Double photoionization of propylene oxide: A coincidence study of the ejection of a pair of valence-shell electrons. J. Chem. Phys. 2018, 148, 114302. [Google Scholar] [CrossRef]
- Falcinelli, S.; Rosi, M.; Pirani, F.; Bassi, D.; Alagia, M.; Schio, L.; Richter, R.; Stranges, S.; Balucani, N.; Lorent, V.; et al. Angular distribution of ion products in the double photoionization of propylene oxide. Front. Chem. 2019, 7, 621. [Google Scholar] [CrossRef]
- Dubnikova, F.; Lifshitz, A. Isomerization of Propylene Oxide. Quantum Chemical Calculations and Kinetic Modeling. J. Phys. Chem. A 2000, 104, 4489–4496. [Google Scholar] [CrossRef]
- Elango, M.; Maciel, G.S.; Palazzetti, F.; Lombardi, A.; Aquilanti, V. Quantum Chemistry of C3H6O Molecules: Structure and Stability, Isomerization Pathways, and Chirality Changing Mechanisms. J. Phys. Chem. A 2010, 114, 9864–9874. [Google Scholar] [CrossRef]
- Witt, A.N. The Chemical Composition of the Interstellar Medium. Phil. Trans. Math Phys. Eng. Sci. 2001, 359, 1949–1959. [Google Scholar] [CrossRef][Green Version]
- McGuire, B.A. 2018 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. Astrophys. J. 2018, 239, 17. [Google Scholar] [CrossRef]
- Orek, C.; Kłos, J.; Lique, F.; Bulut, N. Ab initio studies of the Rg-NO+ (X1Σ+) van der Waals complexes (Rg = He, Ne, Ar, Kr, and Xe). J. Chem. Phys. 2016, 144, 204303. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Maris, A.; Melandri, S.; Caminati, W. Rotational spectrum of propylene oxide-neon. Mol. Phys. 2002, 100, 3245–3249. [Google Scholar] [CrossRef]
- Blanco, S.; Maris, A.; Millemaggi, A.; Caminati, W. The most stable conformer of the propylene oxide-argon complex. J. Mol. Struct. 2002, 612, 309–313. [Google Scholar] [CrossRef]
- Blanco, S.; Melandri, S.; Maris, A.; Caminati, W.; Velino, B.; Kisiel, Z. Free jet rotational spectrum of propylene oxide–krypton and modelling and ab initio calculations for propylene oxide–rare gas dimers. Phys. Chem. Chem. Phys. 2003, 5, 1359–1364. [Google Scholar] [CrossRef]
- Su, Z.; Borho, N.; Xu, Y. Chiral Self-Recognition: Direct Spectroscopic Detection of the Homochiral and Heterochiral Dimers of Propylene Oxide in the Gas Phase. J. Am. Chem. Soc. 2006, 128, 17126–17131. [Google Scholar] [CrossRef]
- Faure, A.; Dagdigian, P.J.; Rist, C.; Dawes, C.; Qintas-Sànchez, E.; Lique, F.; Hochlaf, M. Interaction of Chiral Propylene Oxide (CH3CHCH2O) with Helium: Potential Energy Surface and Scattering Calculations. ACS Earth Space Chem. 2019, 3, 964–972. [Google Scholar] [CrossRef]
- Palazzetti, F.; Cappelletti, D.; Coletti, C.; Falcinelli, S.; Pirani, F. Molecular Beam Scattering Experiments on Noble Gas–Propylene Oxide: Total Integral Cross Sections and Potential Energy Surfaces of He–and Ne–C3H6O. J. Chem. Phys. 2021, 155, 234301. [Google Scholar] [CrossRef]
- Lombardi, A.; Palazzetti, F. Chirality in molecular collision dynamics. J. Phys. Cond. Matt. 2018, 30, 063003. [Google Scholar] [CrossRef]
- Kiss, B.; Szőri, M.; Jedlovszky, P. Adsorption of Propylene Oxide on Amorphous Ice under Interstellar Conditions. A Grand Canonical Monte Carlo Simulation Study. J. Phys. Chem. C 2020, 124, 16402–16414. [Google Scholar] [CrossRef]
- Capitelli, M.; Cappelletti, D.; Colonna, G.; Gorse, C.; Laricchiuta, A.; Liuti, G.; Longo, S.; Pirani, F. On the possibility of using model potentials for collision integral calculations of interest for planetary atmospheres. Chem. Phys. 2007, 338, 62–68. [Google Scholar] [CrossRef]
- Cambi, R.; Cappelletti, D.; Pirani, F.; Liuti, G. Generalized correlations in terms of polarizability for van der Waals interaction potential parameter calculations. J. Chem. Phys. 1991, 95, 1852–1861. [Google Scholar] [CrossRef]
- Pirani, F.; Cappelletti, D.; Liuti, G. Range strength and anisotropy of intermolecular forces in atom-molecule systems: An atom-bond pairwise additivity approach. Chem. Phys. Lett. 2001, 350, 286–296. [Google Scholar] [CrossRef]
- Pirani, F.; Maciel, G.S.; Cappelletti, D.; Aquilanti, V. Experimental benchmarks and phenomenology of interatomic forces: Open shell and electronic anisotropy effects. Int. Rev. Phys. Chem. 2006, 25, 165–199. [Google Scholar] [CrossRef]
- Olney, T.N.; Cann, C.G.; Cooper, G.; Brion, C.E. Absolute scale determination for photoabsorpion spectra and the calculation of molecular properties using dipole sum-rules. Chem. Phys. 1997, 223, 59–98. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peterson, K.A.; Woon, D.E.; Dunning, T.H., Jr. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H + H2 → H2 + H reaction. J. Chem. Phys. 1994, 100, 7410. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, R. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553. [Google Scholar] [CrossRef]
- Coletti, C.; Re, N. High Level Theoretical Study of Benzene−Halide Adducts: The Importance of C–H−Anion Hydrogen Bonding. J. Phys. Chem. A 2009, 113, 1578–1585. [Google Scholar] [CrossRef]
- Lee, H.-N.; Chang, L.-C.; Su, T.-M. Optical rotamers of substituted simple alkanes induced by macroscopic translation-rotational motions. Chem. Phys. Lett. 2011, 507, 63–68. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ε/meV | rm/Å | β | |
|---|---|---|---|
| CH3-Ar | 9.21 | 4.02 | 7.0 |
| C-Ar | 7.19 | 3.79 | 7.0 |
| O-Ar | 8.51 | 3.61 | 7.0 |
| H-Ar | 3.27 | 3.51 | 8.0 |
| ε/meV | rm/Å | β | |
|---|---|---|---|
| CH3-Kr | 11.78 | 4.11 | 7.0 |
| C-Kr | 8.73 | 3.91 | 7.0 |
| O-Kr | 9.72 | 3.76 | 7.0 |
| H-Kr | 3.59 | 3.69 | 8.0 |
| ε/meV | rm/Å | β | |
|---|---|---|---|
| CH3-Xe | 14.25 | 4.26 | 7.0 |
| C-Xe | 9.93 | 4.09 | 7.0 |
| O-Xe | 10.19 | 3.97 | 7.0 |
| H-Xe | 3.67 | 3.93 | 8.0 |
| ε/meV | rm/Å | β | |
|---|---|---|---|
| CH3-Rn | 16.80 | 4.34 | 7.0 |
| C-Rn | 11.37 | 4.19 | 7.0 |
| O-Rn | 11.39 | 4.10 | 7.0 |
| H-Rn | 3.96 | 4.07 | 8.0 |
| Minimum 1 | Minimum 2 | Minimum 3 | Minimum 4 | Minimum 5 | |
|---|---|---|---|---|---|
| θ | 98.8 (99.8) | 87.6 (81.3) | 49.7 (50.0) | 100.4 (99.4) | 43.1 (30.5) |
| ϕ | 22.6 (10.5) | 254.7 (259.1) | 114.8 (106.7) | 156.5 (66.1) | 255.6 (247.4) |
| R (Å) | 3.93 (3.83) | 3.74 (3.75) | 4.06 (3.87) | 3.88 (3.67) | 4.24 (4.15) |
| V (meV) | −34.6 (−36.8) | −34.1 (−36.0) | −31.8 (−40.2) | −31.7 (−40.3) | −26.9 (−33.0) |
| Minimum 1 | Minimum 2 | Minimum 3 | Minimum 4 | Minimum 5 | |
|---|---|---|---|---|---|
| θ | 99.9 (99.3) | 88.8 (80.8) | 49.9 (49.9) | 101.7 (98.8) | 43.2 (31.5) |
| ϕ | 21.2 (12.0) | 254.1 (257.7) | 115.9 (108.7) | 157.9 (157.0) | 254.9 (246.4) |
| R (Å) | 4.06 (3.93) | 3.88 (3.81) | 4.21 (3.97) | 4.02 (3.77) | 4.38 (4.28) |
| V (meV) | −42.2 (−44.2) | −40.9 (−44.6) | −37.0 (−49.2) | −38.2 (−49.0) | −31.7 (−39.8) |
| Minimum 1 | Minimum 2 | Minimum 3 | Minimum 4 | Minimum 5 | |
|---|---|---|---|---|---|
| θ | 101.2 (98.7) | 90.1 (79.0) | 50.2 (49.9) | 103.1 (97.5) | 41.9 (33.0) |
| ϕ | 20.0 (13.2) | 253.4 (256.2) | 117.3 (110.8) | 159.6 (157.0) | 254.0 (244.6) |
| R (Å) | 4.25 (4.10) | 4.07 (3.97) | 4.42 (4.12) | 4.21 (3.94) | 4.59 (4.39) |
| V (meV) | −48.1 (−53.2) | −46.0 (−55.2) | −40.2 (−60.2) | −43.1 (−59.7) | −34.9 (−49.2) |
| Minimum 1 | Minimum 2 | Minimum 3 | Minimum 4 | Minimum 5 | |
|---|---|---|---|---|---|
| θ | 102.8 (98.6) | 90.8 (79.7) | 50.5 (49.8) | 103.8 (96.9) | 40.8 (33.8) |
| ϕ | 19.4 (13.8 | 253.1 (255.1) | 118.1 (112.0) | 160.4 (156.5) | 253.6 (243.2) |
| R (Å) | 4.36 (4.15) | 4.18 (4.01) | 4.54 (4.17) | 4.32 (3.99) | 4.72 (4.43) |
| V (meV) | −55.1 (−59.9) | −52.6 (−63.7) | −45.1 (−67.6) | −49.4 (−67.1) | −39.5 (−55.2) |
| System | |
|---|---|
| He-C3H6O | 1.65 |
| Ne-C3H6O | 3.65 |
| Ar-C3H6O | 11.80 |
| Kr-C3H6O | 17.43 |
| Xe-C3H6O | 25.78 |
| Rn-C3H6O | 34.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palazzetti, F.; Coletti, C.; Marrone, A.; Pirani, F. Potential Energy Surfaces for Noble Gas (Ar, Kr, Xe, Rn)–Propylene Oxide Systems: Analytical Formulation and Binding. Symmetry 2022, 14, 249. https://doi.org/10.3390/sym14020249
Palazzetti F, Coletti C, Marrone A, Pirani F. Potential Energy Surfaces for Noble Gas (Ar, Kr, Xe, Rn)–Propylene Oxide Systems: Analytical Formulation and Binding. Symmetry. 2022; 14(2):249. https://doi.org/10.3390/sym14020249
Chicago/Turabian StylePalazzetti, Federico, Cecilia Coletti, Alessandro Marrone, and Fernando Pirani. 2022. "Potential Energy Surfaces for Noble Gas (Ar, Kr, Xe, Rn)–Propylene Oxide Systems: Analytical Formulation and Binding" Symmetry 14, no. 2: 249. https://doi.org/10.3390/sym14020249
APA StylePalazzetti, F., Coletti, C., Marrone, A., & Pirani, F. (2022). Potential Energy Surfaces for Noble Gas (Ar, Kr, Xe, Rn)–Propylene Oxide Systems: Analytical Formulation and Binding. Symmetry, 14(2), 249. https://doi.org/10.3390/sym14020249