1. Introduction
In the case of the pharmaceutical industry, the development of continuous technologies can be beneficial to ensure constant product quality. The development of such procedures requires a special approach, and the demand for enantiomeric separation is increasing. In such cases, if the separation is to be accomplished by crystallization, the separation of conglomerate-forming enantiomeric mixtures can be conducted directly, but the separation of the more common (in the majority of cases) racemate-forming enantiomers [
1] cannot be resolved directly (with good efficiency) only by fractional crystallization of diastereomeric mixtures formed with so-called chiral resolving agents (foreign chiral compounds) [
2].
Our group has regularly reported on the investigation of the separation of enantiomers of various structures, during which we studied the dependence of the separation efficiency (F = Y*ee
Dia) [
3] on the choice of the resolving agent, the solvent, the reaction conditions, and the crystallization time [
4,
5,
6,
7,
8,
9,
10,
11,
12].
We found that the F parameter, which is suitable for comparison of the separations, is the function of the eutectic composition of the enantiomeric mixtures, of the racemic compound, or of the enantiomeric mixtures of the resolving agent, namely, because the eeDia separable from the crystalline diastereomer is approximately the same as the eeRac (the eutectic composition of the racemic compound) or the eeResAg (the eutectic composition of the resolving agent). Elucidating which one is the determinant in a given case depends on the solvent (i.e., the possible presence of achiral compounds) and, in the case of a given solvent (at the same temperature), on the time of crystallization (the time between mixing and filtration). Thus, it is advantageous if the determining eeEu is as close as possible to 100% of the eutectic composition of the racemic compound or the resolving agent.
2. Applied Methods for Exploiting the Kinetic Control—Distribution between Two Phases
2.1. Continuous Industrial Processes: Liquid1 + Solid1 to Liguid2 + Solid2
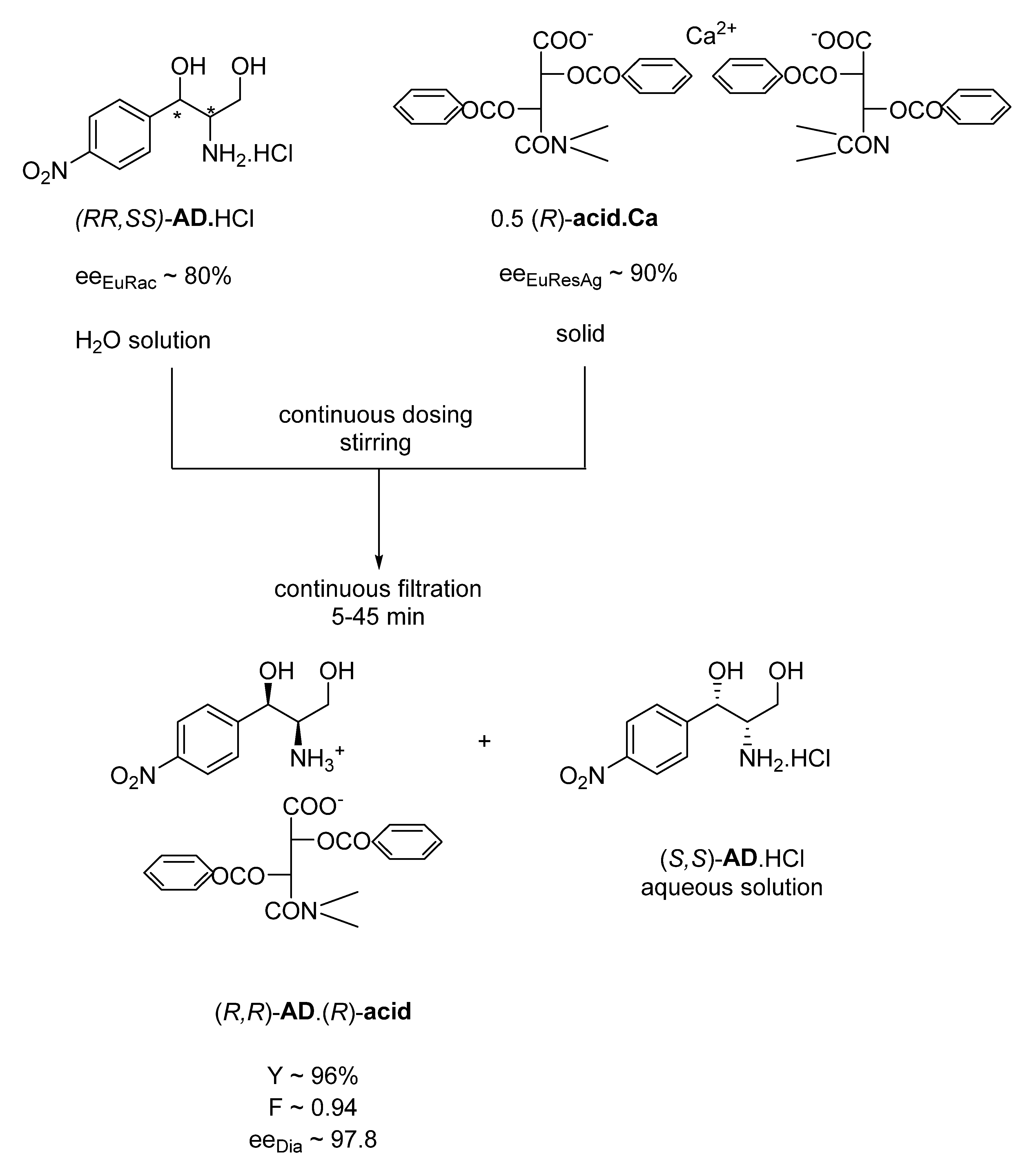
Previously, an industrial process was developed in 1975 under the leadership of Elemér Fogassy, currently working in our group, to produce the crystalline diastereomeric salt of one of the racemic intermediates of chloramphenicol ((
R,R)-
AD)) via a continuous process (
Figure 1) [
13].
In the process, a 0.5 equivalent of solid (R)-acid.Ca1/2 is continuously added to the aqueous solution of the racemic AD.HCl in a stirred apparatus from which the suspension of the salt of the immediately crystallizing (R,R)-AD.(R)-acid is fed continuously to a filter. Under laboratory conditions, complete crystallization is immediate, while, in an industrial apparatus, mixing and filtration takes approximately 45 min (which can also be considered immediate crystallization under these conditions).
If the same reaction is carried out from an aqueous solution of the ammonium salt of the (R)-acid instead of the calcium salt (by a single-phase salt–salt reaction), the crystallization takes 6–8 h.
Thus, a rapid salt–salt reaction between the liquid–solid phases occurs here, during which another solid–liquid equilibrium is formed in the reaction mixture. This equilibrium has shifted almost quantitatively toward the new solid phase, although the (R)-acid.Ca1/2 salt alone precipitates almost quantitatively from water. The eeEu compositions of both the racemic AD and (R)-acid provides high eeDia values. Clearly, kinetic control has prevailed. It appears that if a sufficiently poorly soluble salt of the resolving agent is chosen, kinetic control can be expected.
2.2. The Application of a Quasi-Racemic Resolving Agent with a Related Structure
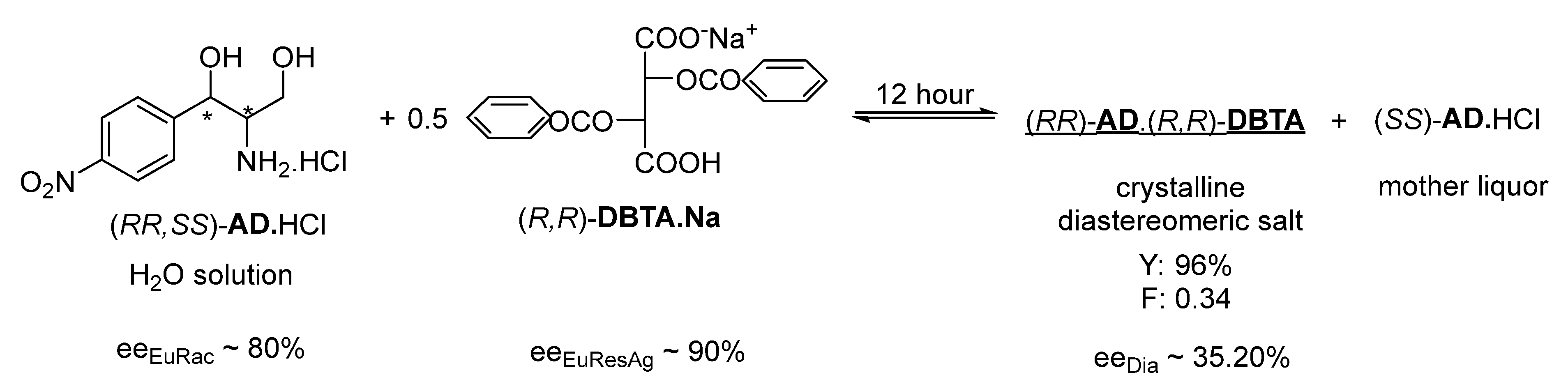
The resolution of racemic
AD.HCl was also investigated by adding to the aqueous solution an aqueous mixture of the acidic (
R,
R)-
DBTA.Na salt (that has a close molecular structure to the (
R)-
acid.Ca
1/2 salt) in a 0.5 molar equivalent; however, the purity of the salt enantiomers precipitated from the resulting solution in 1 h was barely measurable (ee
Dia of ~0.02) and was low even after 12 h of crystallization, although the eutectic composition of both the racemic
AD and the resolving agent was above 80% (
Figure 2).
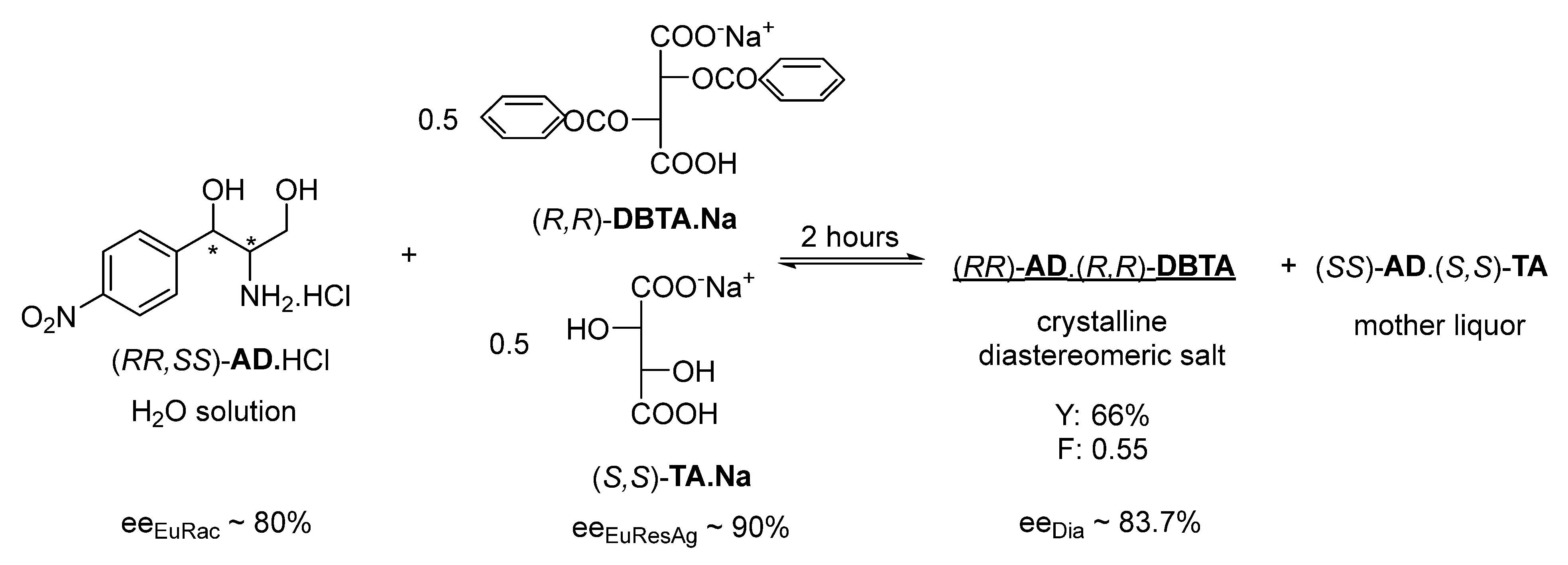
If we apply 0.5 mol of (
S,S)-
TA.Na with the same resolving agent, immediate crystallization begins and after 1 h crystallization, (
R,R)-
AD can be separated with 83.7% (ee
Dia) enantiomeric purity, a 66% yield, and good efficiency (F: 0.55) (
Figure 3).
Our new recognition that a quasi-racemic resolving agent mixture (see Dutch resolution [
14,
15,
16] and resolving agent mixtures with related molecular structure [
15,
16]) may be suitable for providing the terms of kinetic control, and thus the effect of the eutectic composition of the resolving agent or the racemic compound can rapidly prevail.
2.3. Liquid1 + Solid1 to Liquid2 + Solid2
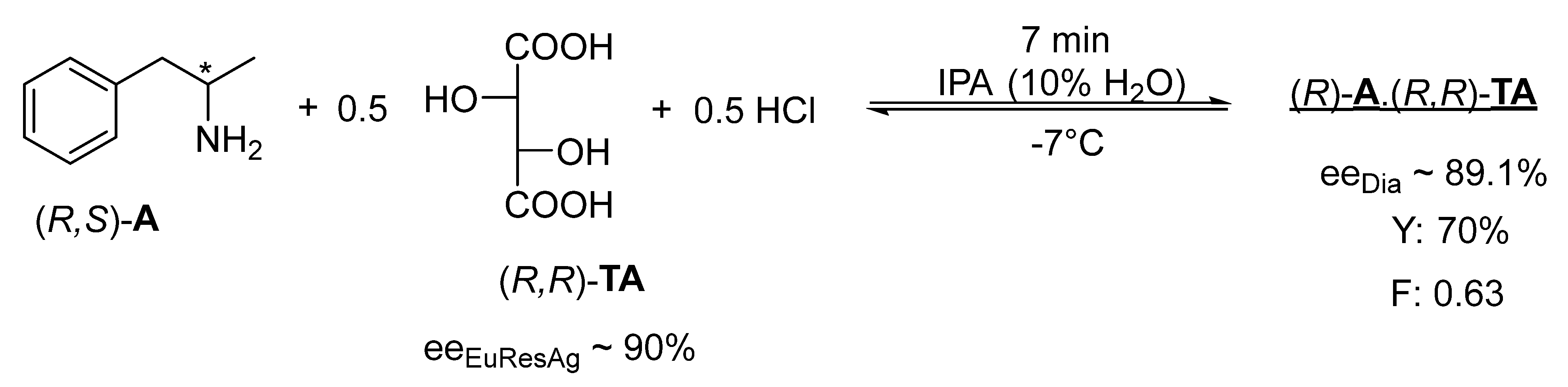
It is also a new finding that rapid kinetic control can occur if the solvent (IPA) contains a small amount of water (10%) and 0.5 mol of hydrochloric acid, as in the case of the racemic base (
Figure 4).
The racemic 1-methyl-2-phenylethylamine ((R,S)-A) began to crystallize slowly in the IPA solvent when an equivalent (R,R) TA was added, where the efficiency and enantiomeric purity of the resolution is low.
If the IPA solution of the racemic 1-methyl-2-phenylethylamine (
A) also contains 0.5 mol of concentrated aqueous hydrochloric acid, and 0.5 mol of (
R,
R)-
TA is also added, rapid crystallization begins and occurs within 7 min (
Figure 4). After cooling (−7 °C), (
R)-
A with high enantiomeric purity (ee
Dia ~ 89.1%) can be isolated from the filtered crystalline diastereomeric salt, while in 15 min at room temperature (25 °C), the purity of (
R)-
A decreases (ee
Dia ~ 83.5%), which, if the diastereomer is not filtered off, decreases further in 12 h (ee
Dia ~ 44.0%) [
17].
2.4. The Use of Amphoteric Resolving Agents (Liquid1 + Solid1 to Liquid2 + Solid2)
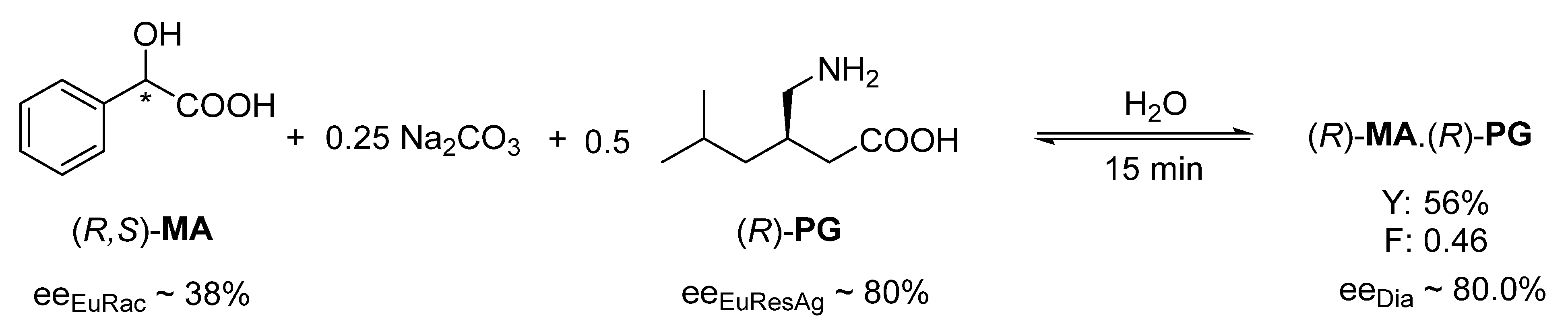
The occurrence of rapid kinetic control was also observed when an amphoteric resolving agent was used. Thus, resolution of racemic mandelic acid (
MA) in aqueous solution by the addition of 0.25 mol of Na
2CO
3 and 0.5 mol of (
R)-pregabalin (
PG) gave the highest enantiomeric purity (ee
Dia ~ 80.0%) with a crystallization time of 15 min (
Figure 5), but this value deteriorates sharply (after 168 h the ee
Dia is 62%) [
18] with a longer crystallization time.
2.5. Achiral Crystalline Reagent, Tandem Resolution
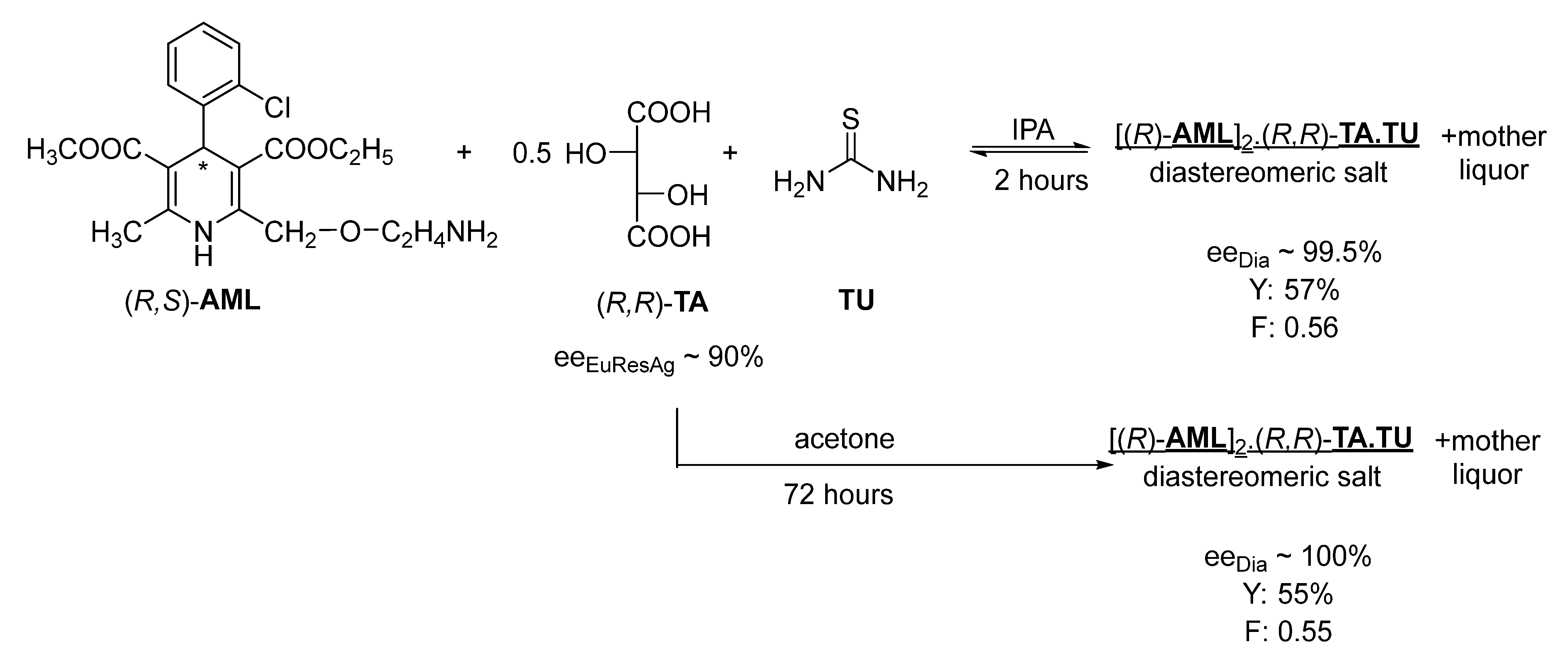
It has been observed that during the fractional crystallization of diastereomers in the presence of co-crystals, high-purity diastereomeric salt precipitates from one of the solvents under kinetic control, while, from another solvent, high purity diastereomers can only be separated under the influence of thermodynamic control, which is not suitable for developing a continuous resolution.
During the resolution of racemic amlodipine (
AML) with tartaric acid (
TA) in the presence of thiourea (
TU) in the IPA solvent (
Figure 6), the neutral
AML salt of
TA precipitates with thiourea. An almost enantiomerically pure diastereomer is then precipitated with a crystallization time of 2 h, although the crystallization time can be reduced; however, it may still be suitable for developing a continuous process.
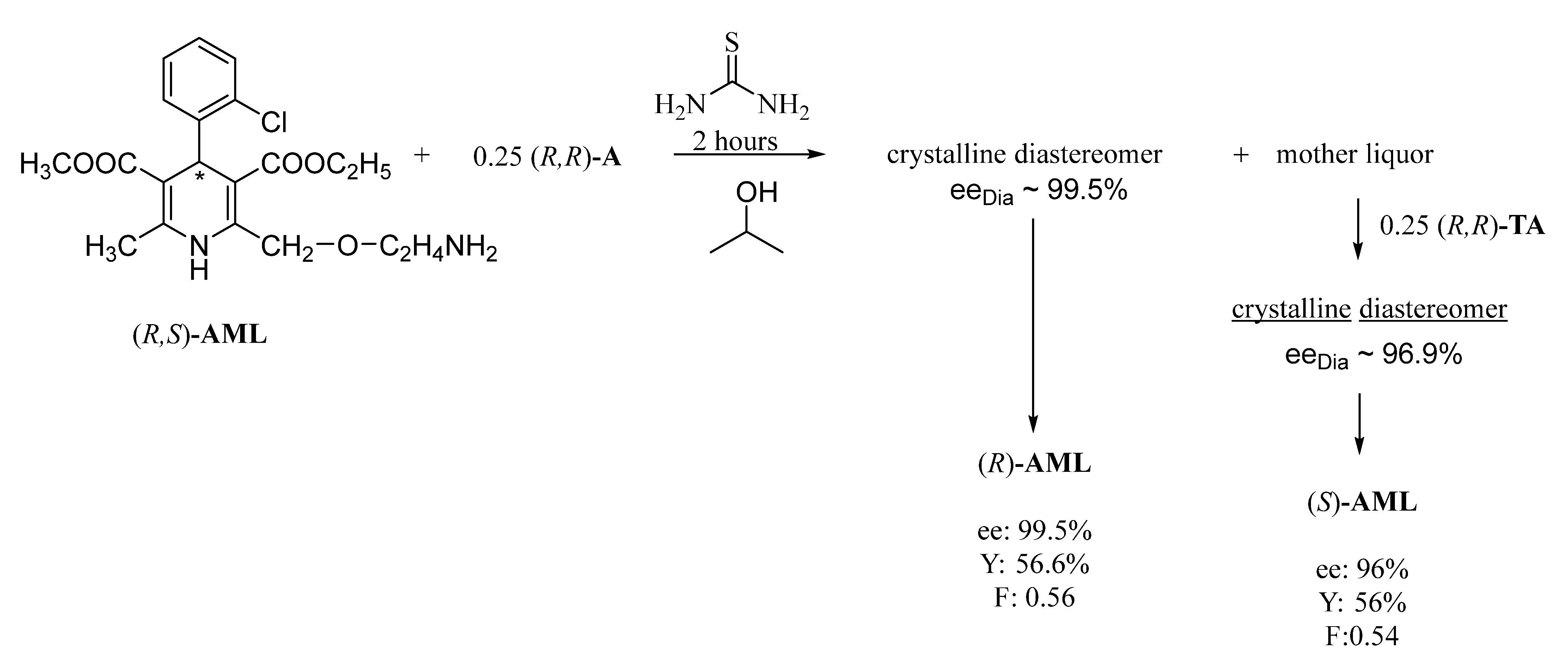
With the further addition of (
R,
R)-tartaric acid to the mother liquor, the diastereomeric salt containing the other enantiomer also crystallizes from the solvent within a short time (
Figure 7).
The continuous separation of each amlodipine enantiomer, in this case, allows the rapid reuse of the unseparated racemic fraction [
19,
20].
3. Experimental Procedure
In the method used to determine the enantiomeric excess values (ee), the obtained enantiomeric purity (specific rotation) was compared with the literature values of the corresponding pure enantiomer. Optical rotations were determined on a PerkinElmer 241 polarimeter. Chemicals were the products of Sigma-Aldrich, and the organic solvents used were freshly distilled.
3.1. Resolution of Racemic Aminodiol
DL-threo-1-(4′-nitrophenyl)-2-aminopropane-1,3-diol hydrochloride monohydrate was added to the upper part of the first cell of a three-liter, five-cell foam column divided into five sections with vertical partitions. Purified compressed air was passed as a blowing gas, thereby also ensuring agitation. A vibrating powder feeder at a rate of 580.0 g/h was added the racemic aminodiol and another vibrating powder feeder delivered the resolving agent dibenzoyl-D-tartaric acid. Furthermore, N-dimethyl-halfamide calcium salt was added at a rate of 493.0 g/h simultaneously. The cells were maintained at 65 °C, 64 °C, 62 °C, 38 °C, and 25 °C respectively. The average residence time of the reaction mixture was 45 min and the slurry was continuously filtered. The dibenzoyl-d-tartaric acid salt of D-(−)-threoaminodidol with N,N-dimethyl halfamide precipitates in the form of a dense white crystalline precipitate, while L-(+)-threoaminodiol remains in solution. The precipitated diastereomeric salt was washed with water (3 × 50 mL) and then decomposed with a mixture of water (100 mL) and cc. HCl (15 mL) at 60 °C. The mixture was cooled to 25 °C, where the insoluble dibenzoyl-D-tartaric acid and N,N-dimethyl halfamide was filtered off and washed with water (3 × 30 mL). Concentrated aqueous ammonium hydroxide (20–25 mL, pH 9.2) was added to the combined mother liquors at 60 °C and cooled to 20 °C. The majority of D-(−)-threo-1-(4′-nitrophenyl-(-2-aminopropane-1,3-diol) precipitates were filtered off and washed with 3 × 30 mL water. As such, 221.0 g (Y: 96.0%) of D-(−)-threo-1-(4′-nitrophenyl)-2-aminopropane-1,3-diol was obtained per hour; m.p.: 164–166 °C; = −29.3°.
3.2. Resolution of Aminodiol with the Sodium Salt of (R,R)-DBTA
NaOH (0.35 g, 8.75 mmol) and (R,R)-DBTA (3.13 g, 8.75 mmol) were dissolved in 10 cm3 of distilled water and added to 30 cm3 of a stock solution of racemic AD.HCl (containing 4.35 g, i.e., 17.5 mmol). The mixture was stirred for 12 h.
The precipitated diastereomeric salt was then filtered off, washed twice with a small amount of water, and then concentrated NH4OH was added with a mass equal to two times the wet weight of the diastereomeric salt. After 2 h of crystallization, the mixture was filtered and dried.
From the mother liquor, the other AD enantiomer was precipitated with concentrated NH4OH with a mass equal to three times the weight of the wet diastereomeric salt. After 20 h of crystallization, the enantiomer was filtered and dried. The obtained (R,R)-AD was 2.088 g (Y: 96%), F: 0.34.
3.3. Resolution of Racemic Aminodiol with a Quasi-Racemic Resolving Agent Mixture of (R,R)-DBTA and (S,S)-TA Salts
Next, 0.35 g (8.75 mmol) of NaOH and 1.31 g (8.75 mmol) of (S,S)-TA were combined and dissolved in 5 cm3 of distilled water, to which 30 cm3 of the stock solution of racemic AD.HCl (containing: 4.35 g, i.e., 17.5 mmol) was added. NaOH (0.35 g, 8.75 mmol) and (R,R)-DBTA (3.13 g, 8.75 mmol) were dissolved in distilled water (10 cm3) and the two solutions were combined. The mixture was stirred and, after 2 h, the precipitated diastereomeric salt was filtered, washed twice with a small amount of water, and then concentrated NH4OH was added with a mass equal to four times the wet weight. After 2 h of crystallization, the enantiomer was filtered and dried. The obtained (R,R)-AD was 1.44 g (Y: 66%, F: 0.55).
From the mother liquor, the other AD enantiomer was precipitated with concentrated NH4OH with a mass equal to six times the weight of the wet diastereomeric salt. After 20 h of crystallization, the enantiomer was filtered and dried.
3.4. Resolution of (−) (1-Methyl-2-phenyl)-Ethylamine
We dissolved 5 g of the racemic compound (2-phenyl-1methyl)-ethylamine in a mixture of 12 cm3 of IPA and 1.53 cm3 of aqueous hydrochloric acid (37%), which was heated to boiling. Then, 2.81 g (−)-tartaric acid was added to the hot mixture. The obtained diastereomeric salt was filtrated quickly and washed with 2 × 4 cm3 of IPA. The obtained salt was suspended with 10 cm3 of hexane, filtrated, and then dried on air. The weight of the obtained salt was 4.62 g (87.5%), = −25.3 (c = 1, water), and ee: 83.5%. The diastereomeric salt obtained was suspended in a solution of 11 cm3 of IPA and 0.3 cm3 of diluted hydrochloride acid (37%) while stirring and heating to boil twice (the diastereomeric salt remained in suspension). The mixture was cooled to 30 °C in 10 min. The obtained salt was suspended with 10 cm3 of hexane, filtrated, and dried on air. The weight of the obtained salt was 3.7 g (70.0%), = −28.8 (c = 1, water), and ee: 95%.
3.5. Resolution of Mandelic Acid
1.52 g (10 mmol) of racemic mandelic acid (MA), 0.27 g (2.5 mmol) of Na
2CO
3, and 0.80 g (5.0 mmol) of (
R)-(−)-3-(aminomethyl)-5-methylhexanoic acid ((
R)-PREG) was dissolved in 6.1 mL of hot water. The crystalline diastereomeric salt appeared by gradually cooling down the solution to 26 °C and was then separated from the mother liquor by filtration after 15 min. To decompose the diastereomer, a mixture of 1.9 mL of 25% aqueous ammonia and 2.0 mL of water was added to the crystals. Next, 0.70 g (4.4 mmol) of crystalline (
R)-PREG was filtered off after 3 h of crystallization, after which 1.5 mL of 37% HCl was added to the mother liquor to afford 0.43 g (56%) of (
R)-MA with an ee
D of 80% that was separated from the mother liquor after 2 h of crystallization where
= −121.6 (c = 1, water) and ee: 80% [
21].
3.6. Resolution of Racemic Amlodipine with (R,R)-Tartaric Acid in a Solution of Acetone and Thiourea in a Tandem Resolution
The mixture of 2.00 g (4.9 mmol) racemic amlodipine, 0.18 g (1.2 mmol) (R,R)-tartaric acid, and 1.12 g (15 mmol) thiourea was dissolved in 10 cm3 of acetone. After first warming and stirring, a solution was formed while cooling to room temperature and the diastereomeric salt was crystallized. The mixture was stirred for 1 day, where then the crystalline precipitate was filtered, washed with 3 × 1 cm3 of acetone, and then dried to afford 1.04 g of diastereomeric salt. Then, 10 cm3 of a 10% NaOH solution and 40 cm3 of dichloromethane was added to the mixture of the salt and stirred for 1 h. The organic phase was separated, washed with 1 × 5 cm3 of water and then concentrated. To the organic phase, 1.5–2 mmol of fumaric acid was added in 10 cm3 of acetone, where the racemic amlodipine-fumarate salt was then filtered and the enantiomeric mixture remaining in the mother liquor was crystallized from 40 cm3 of hexane. The obtained (R)-amlodipine was obtained as follows: 0.63 g (Y: 63%), ee: 100%, F: 0.63.
To the mother liquor of the diastereomeric salt, 0.18 g (1.2 mmol) of (R,R)-tartaric acid was added, and the procedure described above was accomplished. The obtained (S)-amlodipine was obtained as follows: 0.6 g (Y: 60%) ee: 100%, F: 0.6.
The mixture of the salt, 10 cm3 of a 10% NaOH solution and 40 cm3 of dichloromethane were stirred for 1 h. The organic phase was separated, washed with 1 × 5 cm3 of water and then concentrated. Following this, it was stirred with 5 cm3 of hexane for 1 h, then filtered to give (R)-amlodipine as follows: 0.55 g (56.6%), ee: 99.5%, F: 0.56.
3.7. Regeneration of the Racemic Amlodipine and Separation of the (S) Enantiomer
The enantiomeric amlodipine mixtures were purified by crystallization from 14× acetone with fumaric acid equivalent to half the racemic fraction. Here, the neutral fumaric acid salt of the racemic portion crystallized. From the residue obtained by evaporating the mother liquor, the amlodipine liberated with aqueous alkali was extracted with DCM and evaporated. The enantiomeric purity (ee) of the resulting amlodipine and its production relative to the starting mixture depends on the purity (ee0) of the starting mixture.
4. Conclusions
A resolving agent with a high eutectic composition (eeEuResAg) is favorable for the rapid crystallization of a crystalline precipitating diastereomeric salt and allows kinetic resolution. It may be advantageous to add a crystalline resolving agent directly to the solution of the racemic compound.
In addition to the above, the use of a quasi-racemic resolving agent or an amphoteric resolving agent can support kinetic resolution. Thus, in given cases, continuous fractional diastereomeric crystallization requires a resolving agent (or a salt thereof, i.e., Ca, Na, etc.)) or another achiral additive (thiourea) that induces rapid crystallization, thus providing high diastereomeric purity. A further advantage can be the sequential use of the same resolving agent that crystallizes with both enantiomers when using kinetic control (tandem resolution).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






