Abstract
Diethanolamine is a tridentate symmetric ligand that is used for organic synthesis to increase metal chelation or alter the molecular polarities. Prenylated flavonoids are well known for their anticancer properties even in colon cancer. Colorectal cancer is a major threat to society causing death through metastasis to several patients with stage IV. Here, we provided altered structures of xanthohumol and 8-prenylanaringenin of the symmetric ligand diethanolamine, based on theoretical studies that are showing better binding affinities to several colon cancer-related proteins. Using molecular docking and dynamics, alongside density function theory and ADMET studies we are representing these two new derivatives of prenylated flavonoids having promising results against this disease.
1. Introduction
The high mortality rate (90%) amongst patients with stage IV colorectal cancer indicates a necessity in new chemotherapeutic agents with increased selectivity. In such ways, the scientific community may be in a position to decrease the risk of metastasis which sometimes is associated with high resistances in monotherapy [1]. It seems that nowadays drugs are not yet in a position to withstand the resistant mechanisms of tumor cells, which are linked to the tumor microenvironment such as the accumulation of phospholipids [2].
Prenylated flavonoids are secondary metabolites occurring in hops. This group of chemicals is showing a cytotoxic potential even in multi resistant cancer cells [3]. More specifically, 8-prenylnaringenin inhibits breast cancer cells and recently has been evaluated for its antiproliferative and apoptotic activities against human colon cancer cells [4] showing remarkable results. Additionally, symmetric ligands in organic synthesis could provide enhanced chelating properties if they have donor atoms and change the polarity of the bioactive molecule. This function is very important since the binding affinity of the ligand molecules on related proteins it is affected drastically by the polarity index. Therefore, an increasing or decreasing binding affinity could be decisive for the selection of the molecule as drug inhibitor. The anticancer action of the flavonoid family is established since several studies are showing that flavonoids can regulate cell death-related cellular signaling via ROS in human colon cancer cells [5,6], oral cancer cells [7], inhibiting enzymes responsible for this disease such as reductases and dehydrogenases [8] and mediated SIRT1 signaling activation in hepatic disorders [9]. Recently natural flavonoids and chalcones were even tested against several COVID-19 related proteins with promising results [10,11].
Molecular docking is a computational strategy that resulted in predicting binding site complementarity between a drug and its therapeutic target and has been massively used to assist drug repositioning for several diseases including cancer [12,13,14]. Additionally, the DFT computational approach under the B3LYP/6 311++G (d, p) level of theory by using a one-dimensional potential energy surface is another tool that helps the researchers to predict the reactivity of the design molecules [15,16]. On the other hand, molecular dynamics are well used for predicting protein–ligand binding sites, including binding pockets and the binding residues in each pocket [17,18].
Herein, we performed two major computation experiments, molecular docking and molecular dynamics on several colon cancer-related proteins [19], to evaluate the anticancer activity of 6-prenylnaringenin, 8-prenylnaringenin, xanthohumol, and isoxanthohumol, compared to established anticancer agents such as Rolipram, Oxaliplatin, and Xeloda (Figure 1). Based on the best two candidates of that group molecules, we altered their structure to increase their binding affinities with the related proteins. The small molecules were also evaluated using density functional theory studies. Finally, the two new molecules were evaluated theoretically for their potential drug use, with the help of ADMET studies.
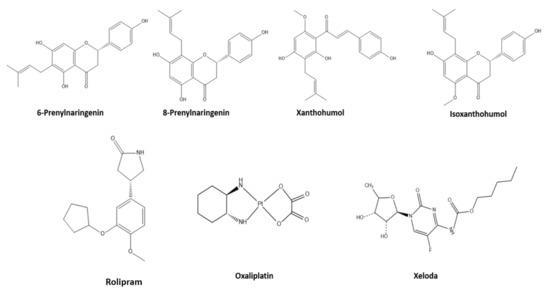
Figure 1.
Representation of the studied molecules.
2. Materials and Methods
2.1. Molecular Dynamics
The protein–ligand binding site prediction was undertaken by using the COACH server (https://zhanglab.ccmb.med.umich.edu/COACH/(accessed on 8 January 2021)). The prediction occurred using two comparative methods, TM-SITE and S-SITE, which recognize ligand-binding templates from the BioLiP protein function database by binding-specific substructure and sequence profile comparisons [20]. The molecular dynamic simulation was done to evaluate the molecular docking study. The testing dataset consists of 500 nonredundant proteins that harbor 814 ligands (410 natural ligands, 238 drug-like ligands, and 164 metal ions). Besides, a set of 400 nonredundant proteins with at least 5 residues bound to known ligands were collected from BioLiP as the training set to train the method. The match between each pair of query and template structures was evaluated by a composite scoring function which counts for both global and local, structural and sequence similarities [21,22]. I-TASSER was the force field using QUARK as a computer algorithm for ab initio protein structure prediction. QUARK models are built from small fragments, (1–20 residues), by replica-exchange Monte Carlo simulation where the energy is defined as RMSD of the domain models to the full chain. The simulation length stands for approximately 4 h for each protein set structure, using temperature range Tmin = 1 and Tmax 60 kcal mol−1 R−1, pressure 1 atm, and 1fs time step [23].
After the prediction of the binding pocket, we performed energy minimization of the ligands as an implicit. The molecular dynamics took place using Amber force field at 298 K, 1 atm, 2.0 fs time per step, and duration 100,000.0 ps to the proteins and the complexes to calculate the RMSD value and re-evaluate the docking procedure. Evidence can be found in Figure S1.
2.2. Molecular Docking
Molecular docking studies were administered by using iGEMDOCK 2.1 software [24]. The 4P75, 4UYA and 6MFQ proteins coded crystal structures were selected from the Protein Data Bank (www.rcsb.org (accessed on: 10 January 2021)). Ligand molecules were collected by Drug Bank (www.drugbank.ca (accessed on: 8 January 2021). The novel ligands were drawn with the help of ChemDraw Ultra 12.0, Chem3D Pro 12.0, and Avogadro software [25]. The scoring function consisted of an easy empirical scoring function and a pharmacophore-based scoring function to scale back the number of false positives. The energy function is often dissected into the following terms:
where Ebind is the empirical binding energy used during the molecular docking; Epharma is the energy of binding-site pharmacophores; Eligpre is a penalty value if the ligand unsatisfied with the ligand preferences. Epharma and Eligpre were used to improve the number of true positives by discriminating active compounds from hundreds of thousands of nonactive compounds. The empirical binding energy (Ebind) is given as:
where Einter and Eintra are the intermolecular and intramolecular energy, respectively, and Epenal may be a large penalty value if the ligand is out of the range of the search box. In this paper, Epenal is set to 10,000. For screening: the population size was = 200, generations = 70, number of solutions = 3. Fitness is the total energy of a predicted pose within the binding site [26]. The docking procedure was validated using two methods: (i) the protein inhibitors from the studied proteins were removed and re-docked into the active site using AutoDock 4.2.6 and (ii) molecular docking performed using both auto dock and iGEMDOCK. Furthermore, the binding pockets are predicted both by the COACH server and iGEMDOCK as well [27]. Screenshots of the ligand–amino acid residue interactions were created by CHIMERA software [28]. As was mentioned above, molecular dynamics simulations were also used to re-evaluate the docking predictions.
Etot = Ebind + Epharma + Eligpre
Ebind = Einter + Eintra + Epenal
2.3. Density Function Theory Studies
Geometric optimization calculations were performed following the DFT method using ORCA software [29,30]. Frequency calculations were performed to obtain thermodynamic properties and to verify that each optimization achieved an energy minimum. The quantum chemical descriptors extracted directly from the ORCA output file were total energy, Huckel atomic charges, electronic density, dipole moment, Mayer population analysis, the energy of the highest occupied molecular orbital (HOMO), and the energy of the lowest unoccupied molecular orbital (LUMO) [31,32].
2.4. Structural Modification
The best two candidates according to their binding affinities to the related proteins, Xanthohumol and 8-Prenylnaringenin were altered chemically in structure, adding at first a prenylated group unit to the second ring of their moieties. After that addition, we realized that the binding affinities declined, showing that the increase of the lipophilicity of the second ring of the chalcones altered negatively the chemical relativity with the protein. After that, we decided to introduce diethanolamine structure moiety to the second ring, increasing successfully their binding affinities, even more, compared to the original molecules. The structure modification was performed using the Chem3D Pro software and structures were optimized with ORCA software.
2.5. ADMET Studies
Xanthohumol, 8-Prenylanaringenin, and their two novel derivatives were also evaluated for their pharmacological profile using the SwissADME server (http://www.swissadme.ch/ (accessed on 21 January 2021)). The predicted result consists of physiochemical properties, lipophilicity, water-solubility, pharmacokinetics, drug-likeness, and toxicity studies. The simulation provides information about the absorption, distribution, metabolism, excretion, and toxicity of the drug [33]. Moreover, to double-check the gained values, we used Biosig Tool online software [34]. The pharmacological profile of the simplest scoring inhibitors was evaluated by Toxtree software [35,36], using Cramer rules and Cytochrome P450 metabolism prediction, taking information on pKa values, logP values, solubility, refractivity, and estimated toxicity of the molecules.
3. Results and Discussion
3.1. Biological Description
Colorectal cancer is the third commonest explanation for cancer-related death in both men and ladies within the occident. Genetic alterations within the development of colorectal cancer initiate with mutations in the adenomatous polyposis (APC) gene and chromosomal instability [37]. Additionally, mutations within the PMS2 gene are reported in about 6% of families with Lynch syndrome that have an identified gene mutation. Lynch syndrome increases the risk of many sorts of cancer, particularly colorectal cancer [38,39]. People with Lynch syndrome even have an increased risk of cancers of the endometrium, ovaries, stomach, small intestine, liver, gall bladder ducts, upper urinary tract, and brain. Additionally, aminoacylation of transfer RNAs establishes the principles of the genetic code [40]. The reactions are catalyzed by a group of 20 enzymes, referred to as aminoacyl tRNA synthetases (AARSs). These connections include heritable mutations within the genes for tRNA synthetases that are causally linked to the disease. Furthermore, MLK4 may be a member of the family of kinases that regulate the JNK, p38, and ERK kinase signaling pathways [41]. MLK4 mutations are identified in various human cancers, including frequently in colorectal cancer, where their function and pathobiological importance are uncertain [42]. That is why the inhibition of protein synthesis on the above biomolecules is believed that probably would have an apoptosis activity on cancer cells, providing an anticancer mechanism based on cell proteins and not alterations on RNA structure [43,44,45].
3.2. Molecular Dynamics
The prediction of the binding sites of the related carcinoma proteins was completed after mixing of multiple prediction results of algorithms from TM-SITE, S-SITE, COFACTOR, FINDSITE, and ConCavity. The probability of a residue to be a binding residue was calculated from individual methods, which are used because of the feature vectors for the residue. The top-scoring predictions from each of the programs were combined employing a linear SVM. The detailed prediction can be found in the Supplementary material Table S1. On the other hand, in Figure 2, we can see the predicted binding sites of the studied protein structures calculated with molecular dynamics simulations as predicted by the COACH server.
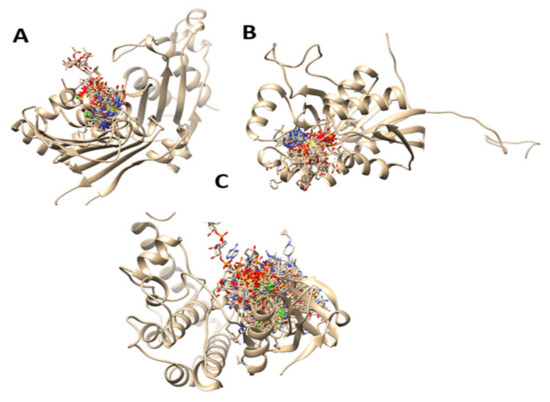
Figure 2.
Predicted binding sites of the studied protein structures calculated with molecular dynamics simulations. ((A): 6MFQ, (B): 4P75, (C): 4UYA)).
6MFQ, 4P75 and 4UYA are 2-chain structure proteins and biomarkers for colon cancer. Defects in these proteins are the cause of hereditary non-polyposis colorectal cancer type 4 (HNPCC4). HNPCC is an autosomal, dominantly inherited disease associated with a marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic cancers of the gastrointestinal [46]. After the prediction of the binding pocket areas of the structures, we proceeded with molecular docking studies using ligand prenylated molecules, with in vitro proven anti colon cancer activity, common anticancer agents that are in use for that disease, and two novel derivatives of the best candidates in docking studies.
3.3. Molecular Docking
Using the predicted binding pockets of the studied structures, we performed molecular docking studies with the ligands 6-Prenylnaringenin, 8-Prenylnaringenin, Isoxanthohumol, Xanthohumol, 8-Prenylnaringenin derivative, Xanthohumol derivative, Rolipram, Oxylplatin, and Xeloda. Rolipram, Oxaliplatin, and Xeloda are common anticancer drugs that are in use for colon cancer treatment. 8-Prenylnaringenin and Xanthohumol exhibited relatively high binding affinities with the selected proteins, so we decided to alter their structures chemically, by substituting the second ring of the prenylated chalcone molecules with diethanolamine. As we can see in Table 1, the binding affinities of the molecules increased after the substitution. In general, all the prenylated molecules when compared to the commercial anticancer drugs, showed similar, or even better in some cases, binding affinities. The two substituted novel chalcone molecules exhibited even higher binding affinities with the selected proteins. This is due to the alteration of energies attributed to hydrogen bonds and van der Waals interactions. Hydrophobic interactions are contacts between nonpolar parts of the molecule. In protein–ligand complexes, nonpolar parts are buried upon binding. This causes a displacement of water molecules and an increase in entropy. Therefore, the hydrophobic interactions are entropy-driven and have been shown to play a significant role in ligand binding. In Figure 3 we can observe depicted structures of the binding protein–drug complexes. On the other hand, hydrogen bonds also contribute to the binding affinity energies of the ligands into the proteins. A hydrogen bond occurs between two electronegative atoms. The hydrogen is normally covalently attached to one atom (donor) but interacts electrostatically with the other (acceptor). This interaction is happening because of the dipole between the electronegative atoms and the proton. Alterations in ligands, such as substitutions, are also altering the polarity thus, it may fit better to the binding sites of the proteins.

Table 1.
Binding affinities and energy distribution of the studied protein–ligand complexes.
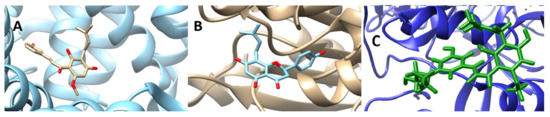
Figure 3.
Screenshots of the binding protein–drug complexes. ((A): 6MFQ-8Prenylnaringenin, (B): 4P75-Xanthohumol derivative, (C): 6MFQ-8Prenylnaringenin derivative)).
3.4. Density Function Theory Studies
Using density functional theory studies, we were able to determine the optimized 3D structures of the two novel derivatives. Selected bond lengths and angles alongside atomic charges, can be found in Supplementary Tables S1–S3, respectively. The optimized structures and van der Waals sphere structures of the two novel molecules can be found in Figure 4. The quality B3LYP method was used throughout for the geometries, with the fraction of exact exchange equal to 20%. For the final energies, B3LYP was modified by using also 10% and 15%. For the optimized structures, single-point calculations were done with a large basis set, with cc-pvtz (-f) [47].
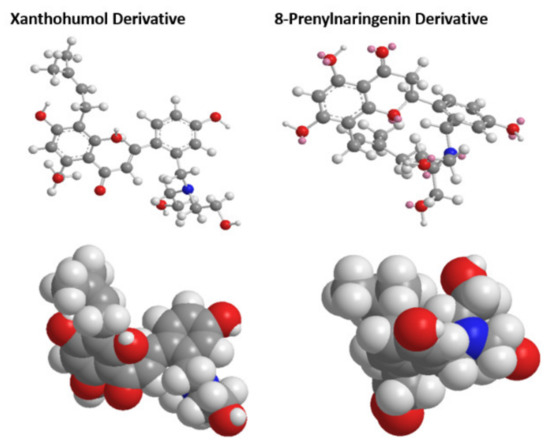
Figure 4.
Optimized structures and van der Waals sphere structures of the two novel chalcone molecules.
The distribution and energy of HOMO is a crucial parameter to elucidate the antioxidant potential of phenolic antioxidants. The electron-donating capacity of the molecule can be predicted by looking at the energy values of HOMO. Therefore, in Figure 5, we can see the HOMO and LUMO orbitals of the derivatives indicating ΔGAP for Xanthohumol derivative equal to 11.265 eV and ΔGAP for 8-Prenylnaringenin derivative equal to 11.403 eV. The results are in good agreement with the docking studies revealing that the 8-Prenylnaringenin derivative has higher activity than the Xanthohumol derivative.
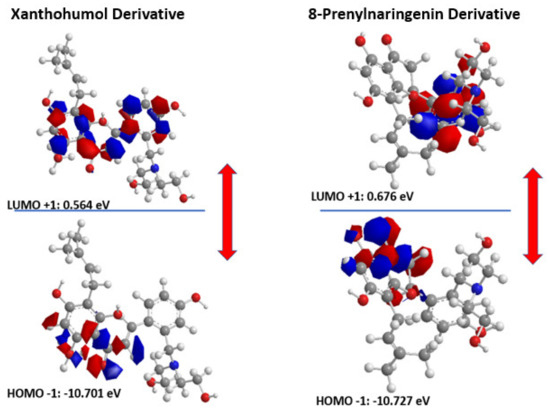
Figure 5.
HOMO-LUMO orbitals of the two novel chalcone molecules.
3.5. Structural Modification
At first, we substituted the two molecules with an additional prenylated group leading to an increase in lipophilicity of the molecules. This resulted in a decrease in the binding affinities of the molecules on the selected proteins. After this step we substituted the second ring of the derivatives with a diethanolamine, a strategic synthesis reported previously in the literature [48]. By doing this we increased the hydrophilicity of the molecules and their binding affinities to the studied proteins. This will probably increase their biological activity as well, a fact that should be studied further in vitro in another work. The substitution of the two molecules can be found in Figure 6. This structural modification had an impact on the amino acid residue that the molecules interacted with as well. Detailed information of the binding amino acid residues with the molecules Xanthohumol, Xanthohumol derivative, 8-Prenylnaringenin, and 8-Prenylnaringenin, can be found in Table 2.
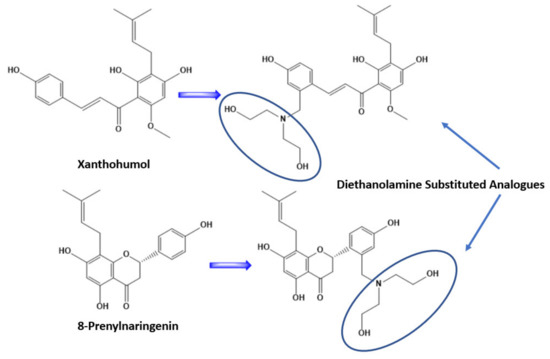
Figure 6.
Structural modification of Xanthohumol and 8-Prenylnaringenin, after substitution with diethanolamine on the second ring.
Table 2.
Amino acid residue of the binding complexes with the two novel chalcone molecules.
3.6. ADMET Studies
Our final approach was to gauge theoretically the pharmacological properties of the two novel molecules. The anticipated pharmacological properties of those two candidate molecules are depicted in Table 3. The pharmacological properties of the drugs, including the absorption, distribution, metabolism, excretion, and toxicity, are also presented. It is worthy to say that these two derivatives have very similar pharmacological values and better than the in vitro evaluated Xanthohumol and 8-Prenylnaringenin, a fact that has to do probably with their increase in hydrophilicity. We believe though that further studies should be done for these molecules to be fully evaluated as drug candidates. Regarding their toxicity their increase in polarity decreases their toxicity values as well, increasing more their potential use.
Table 3.
Pharmacokinetic characteristics of the design molecules compared with their parent ones.
4. Conclusions
In this work, we investigated the binding pockets of several colon cancer-related proteins using molecular dynamics and we evaluated theoretically the binding affinities of a family of prenylated molecules with the help of molecular docking studies. The prenylated molecules were used in the literature against colon cancer previously in vitro with promising results. Therefore, Xanthohumol and 8-Prenylnaringenin were computationally revealed as promising candidates evenly compared with traditionally anticancer agents. The molecules were then studied with density functional theory and finally, we managed to increase the binding affinity of the molecules on cancer proteins by substitution with diethanolamine molecule, attached on the second phenolic ring. We do believe that these two novel derivatives will show promising results when tested in vitro and should be used in further studies against colon cancer.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/sym13060948/s1, Figure S1: Root-mean-square deviation (RMSD) values (y-axis) along with the time frame (x-axis) for atoms of the protein–8Prenylnaringenin derivative complex and protein–Xanthohumol derivative complex; Table S1: Bond lengths of the Xanthohumol and 8-Prenylnaringenin derivatives; Table S2: Bond angles of the Xanthohumol and 8-Prenylnaringenin derivatives; Table S3: Atomic charges of the Xanthohumol and 8-Prenylnaringenin derivatives.
Author Contributions
Conceptualization, M.C.V.; methodology, M.C.V.; software, C.C.P.; validation, Y.S., resources, C.C.P.; writing—original draft preparation, K.S.P.; writing—review and editing, M.C.V.; visualization, K.S.P.; supervision, M.C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scagliarini, A.; Mathey, A.; Aires, V.; Delmas, D. Xanthohumol, a Prenylated Flavonoid from Hops, Induces DNA Damages in Colorectal Cancer Cells and Sensitizes SW480 Cells to the SN38 Chemotherapeutic Agent. Cells 2020, 9, 932. [Google Scholar] [CrossRef] [PubMed]
- Anna, A.; Marusiak, N.L.; Stephenson, H.B.; Eleanor, W.T.; Yaoyong, L.; Karen, B.; Susan, M.; Phil, C.; Lorena, A.; Puto, J.A.; et al. Recur-rent MLK4 Loss-of-Function Mutations Suppress JNK Signaling to Promote Colon Tumorigenesis. Cancer Res. 2016, 76, 724–735. [Google Scholar]
- Urmann, C.; Riepl, H. Semi-Synthetic Approach Leading to 8-Prenylnaringenin and 6-Prenylnaringenin: Optimization of the Microwave-Assisted Demethylation of Xanthohumol Using Design of Experiments. Molecules 2020, 25, 4007. [Google Scholar] [CrossRef]
- Koosha, S.; Mohamed, Z.; Sinniah, A.; Ibrahim, Z.A.; Seyedan, A.; Alshawsh, M.A. Antiproliferative and apoptotic activities of 8-prenylnaringenin against human colon cancer cells. Life Sci. 2019, 232, 116633. [Google Scholar] [CrossRef]
- Dükel, M.; Tavsan, Z.; Kayali, H.A. Flavonoids regulate cell death-related cellular signaling via ROS in human colon cancer cells. Process. Biochem. 2021, 101, 11–25. [Google Scholar] [CrossRef]
- Seo, H.W.; No, H.; Cheon, H.J.; Kim, J.-K. Sappanchalcone, a flavonoid isolated from Caesalpinia sappan L., induces caspase-dependent and AIF-dependent apoptosis in human colon cancer cells. Chem. Interact. 2020, 327, 109185. [Google Scholar] [CrossRef]
- Kode, J.; Kovvuri, J.; Nagaraju, B.; Jadhav, S.; Barkume, M.; Sen, S.; Kasinathan, N.K.; Chaudhari, P.; Mohanty, B.S.; Gour, J.; et al. Synthesis, biological evaluation, and molecular docking analysis of phenstatin based indole linked chalcones as anticancer agents and tubulin polymerization inhibitors. Bioorganic Chem. 2020, 105, 104447. [Google Scholar] [CrossRef]
- Seliger, J.M.; Martin, H.-J.; Maser, E.; Hintzpeter, J. Potent inhibition of human carbonyl reductase 1 (CBR1) by the prenylated chalconoid xanthohumol and its related prenylflavonoids isoxanthohumol and 8-prenylnaringenin. Chem. Interact. 2019, 305, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.M.; Hassanein, E.H.; Salem, S.H.; Hussein, O.E.; Mahmoud, A.M. Flavonoids-mediated SIRT1 signaling activation in hepatic disorders. Life Sci. 2020, 259, 118173. [Google Scholar] [CrossRef]
- Vijayakumar, B.G.; Ramesh, D.; Joji, A.; Prakashan, J.J.; Kannan, T. In silico pharmacokinetic and molecular docking studies of natural flavonoids and synthetic indole chalcones against essential proteins of SARS-CoV-2. Eur. J. Pharmacol. 2020, 886, 173448. [Google Scholar] [CrossRef]
- Vlasiou, M.C.; Pafti, K.S. Screening possible drug molecules for Covid-19. The example of vanadium (III/IV/V) complex molecules with computational chemistry and molecular docking. Comput. Toxicol. 2021, 18, 100157. [Google Scholar] [CrossRef]
- Wesley, B.; Cardoso, S.; Mendanha, A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. [Google Scholar]
- Stefaniu, A.; Pirvu, L.; Albu, B.; Pintilie, L. Molecular Docking Study on Several Benzoic Acid Derivatives against SARS-CoV-2. Molecules 2020, 25, 5828. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Chaudhary, M.K.; Srivastava, A.; Singh, K.K.; Tandon, P.; Joshi, B.D. Computational evaluation on molecular stability, reactivity, and drug potential of frovatriptan from DFT and molecular docking approach. Comput. Theor. Chem. 2020, 1191, 113031. [Google Scholar] [CrossRef]
- Seema, A.; Kulkarni; Nagarajan, S.K.; Ramesh, V.; Palaniyandi, V.; Periyar Selvam, S.; Madhavan, T. Computational evaluation of major components from plant essential oils as potent inhibitors of SARS-CoV-2 spike protein. J. Mol. Struct. 2020, 1221, 128823. [Google Scholar]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved protein-ligand binding site prediction with refined lig-and-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, 438–442. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformation 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Ren, J.; Shang, L.; Wang, Q.; Li, J. Ranking Cancer Proteins by Integrating PPI Network and Protein Expression Profiles. BioMed Res. Int. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2012, 41, D1096–D1103. [Google Scholar] [CrossRef]
- Yang, J.-M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, K.I.; Pafiti, K.S. Molecular Docking, DFT Studies and ADMET Simulations for Evaluating Already Approved FDA Drugs as Inhibitors for SARS-Cov-2 RNADependent Polymerase. Lett. Drug Des. Discov. 2020, 17, 1–14. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Green-Blatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, 1327. [Google Scholar] [CrossRef]
- Rocha, J.A.; Rego, N.C.S.; Carvalho, B.T.S.; Silva, F.I.; Sousa, J.A.; Ramos, R.M.; Passos, I.N.G.; de Moraes, J.; Leite, J.R.S.A.; Lima, F.C.A. Computational quantum chemistry, molecular docking, and ADMET predictions of imidazole alkaloids of Pilocarpus microphyllus with schistosomicidal properties. PLoS ONE 2018, 13, e0198476. [Google Scholar] [CrossRef]
- Çakmak, E.; Işın, D.Ö. A theoretical evaluation on free radical scavenging activity of 3-styrylchromone derivatives: The DFT study. J. Mol. Model. 2020, 26, 98. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Vidaurre, C.; Sander, T.H.; Schlögl, A. BioSig: The Free and Open Source Software Library for Biomedical Signal Processing. Comput. Intell. Neurosci. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Patlewicz, G.; Jeliazkova, N.; Safford, R.J.; Worth, A.P.; Aleksiev, B. An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ. Res. 2008, 19, 495–524. [Google Scholar] [CrossRef]
- Rydberg, P.; Gloriam, D.E.; Zaretzki, J.; Breneman, C.; Olsen, L. SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug Metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. [Google Scholar] [CrossRef]
- Christophe, R.; Berney, R.J.; Fisher, J.-Y.; Pamela, R.J.; Philip, J. Crowe, Protein Markers in Colorec-tal Cancer, Predictors of Liver Metastasis. Ann. Surg. 1999, 230, 179–184. [Google Scholar]
- Ercan, S.; Şenses, Y. Design and molecular docking studies of new inhibitor candidates for EBNA1 DNA binding site: A computational study. Mol. Simul. 2020, 46, 332–339. [Google Scholar] [CrossRef]
- Protocol for MM/PBSA Molecular Dynamics Simulations of Proteins, Federico Fogolari, Alessandro Brigo, Henriette Molinari. Biophys. J. 2003, 85, 159–166. [CrossRef]
- Sen Gupta, P.S.; Bhat, H.R.; Biswal, S.; Rana, M.K. Computer-aided discovery of bis-indole derivatives asmulti-target drugs against cancer and bacterial infections: DFT, docking, virtual screening, and molecular dynamics studies. J. Mol. Liquids. 2020, 320, 114375. [Google Scholar] [CrossRef]
- Shivanika, C.; Kumar, S.D.; Ragunathan, V.; Tiwari, P.; Sumitha, A.; Devi, P.B. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020, 83, 1–27. [Google Scholar] [CrossRef]
- Narayan, S.; Roy, D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol. Cancer 2003, 2, 41. [Google Scholar] [CrossRef]
- D’Arcy, B.M.; Blount, J.; Prakash, A. Biochemical and structural characterization of two variants of uncertain significance in the PMS2 gene. Hum. Mutat. 2019, 40, 458–471. [Google Scholar] [CrossRef]
- Abibi, A.; Andrew, D.; Fer-guson, P.R.; Fleming, N.G.; Laurel, I.; Hajec, J.H.; Valerie, A.; Laganas, D.C.; McKinney, S.M.; McLeod, D.; et al. The Role of a Novel Auxiliary Pocket in Bacterial Phenylalanyl-tRNA Synthetase Druggability. J. Biol. Chem. 2014, 289, 21651–21662. [Google Scholar] [CrossRef]
- Amy, E.; Schmidt, K.P.; Matthew, C.; Underwood, W.B.; Timothy Mather, S.; Bajaj, P. Thermodynamic Linkage between the S1 Site, the Na+ Site, and the Ca2+ Site in the Protease Domain of Human Activated Pro-tein C (APC). THE J. Biol. Chem. 2002, 277, 28987–28995. [Google Scholar]
- Park, S.G.; Schimmel, P.; Kim, S. Aminoacyl tRNA synthetases and their connections to disease. PNAS 2008, 105, 11043–11049. [Google Scholar] [CrossRef] [PubMed]
- Per, E.; Siegbahn, M. A quantum chemical approach for the mechanisms of redox-active metalloenzymes. RSC Adv. 2021, 11, 3495. [Google Scholar]
- Janoš, P.; Spinello, A.; Magistrato, A. All-atom simulations to studying metallodrugs/target interactions. Curr. Opin. Chem. Biol. 2021, 61, 1–8. [Google Scholar] [CrossRef]
- Zhang, W.; Bell, E.W.; Yin, M.; Zhang, Y. EDock: Blind protein–ligand docking by replica-exchange monte carlo simulation. J. Chemin. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Ueno, S.; Yamazaki, R.; Ikeda, T.; Yaegashi, T.; Matsuzaki, T. Antitumor effect of a novel phenanthroindolizidine alkaloid derivative through inhibition of protein synthesis. Anticancer. Res. 2014, 34, 3391–3398. [Google Scholar]
- Schmidt, S.; Denk, S.; Wiegering, A. Targeting Protein Synthesis in Colorectal Cancer. Cancers 2020, 12, 1298. [Google Scholar] [CrossRef]
- Hadjiadamou, I.; Vlasiou, M.; Spanou, S.; Simos, Y.; Papanastasiou, G.; Kontargiris, E.; Dhima, I.; Ragos, V.; Karkabounas, S.; Drouza, C.; et al. Synthesis of vitamin E and aliphatic lipid vanadium(IV) and (V) complexes, and their cytotoxic properties. J. Inorg. Biochem. 2020, 208, 111074. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).