
Tendencies in ABO3 Perovskite and SrF2, BaF2 and CaF2 Bulk and Surface F-Center Ab Initio Computations at High Symmetry Cubic Structure




Abstract
1. Introduction
2. Details of Ab Initio Computations of the F-Centers in ABO3 Perovskites, as Well as in SrF2, BaF2 and CaF2 Fluorites
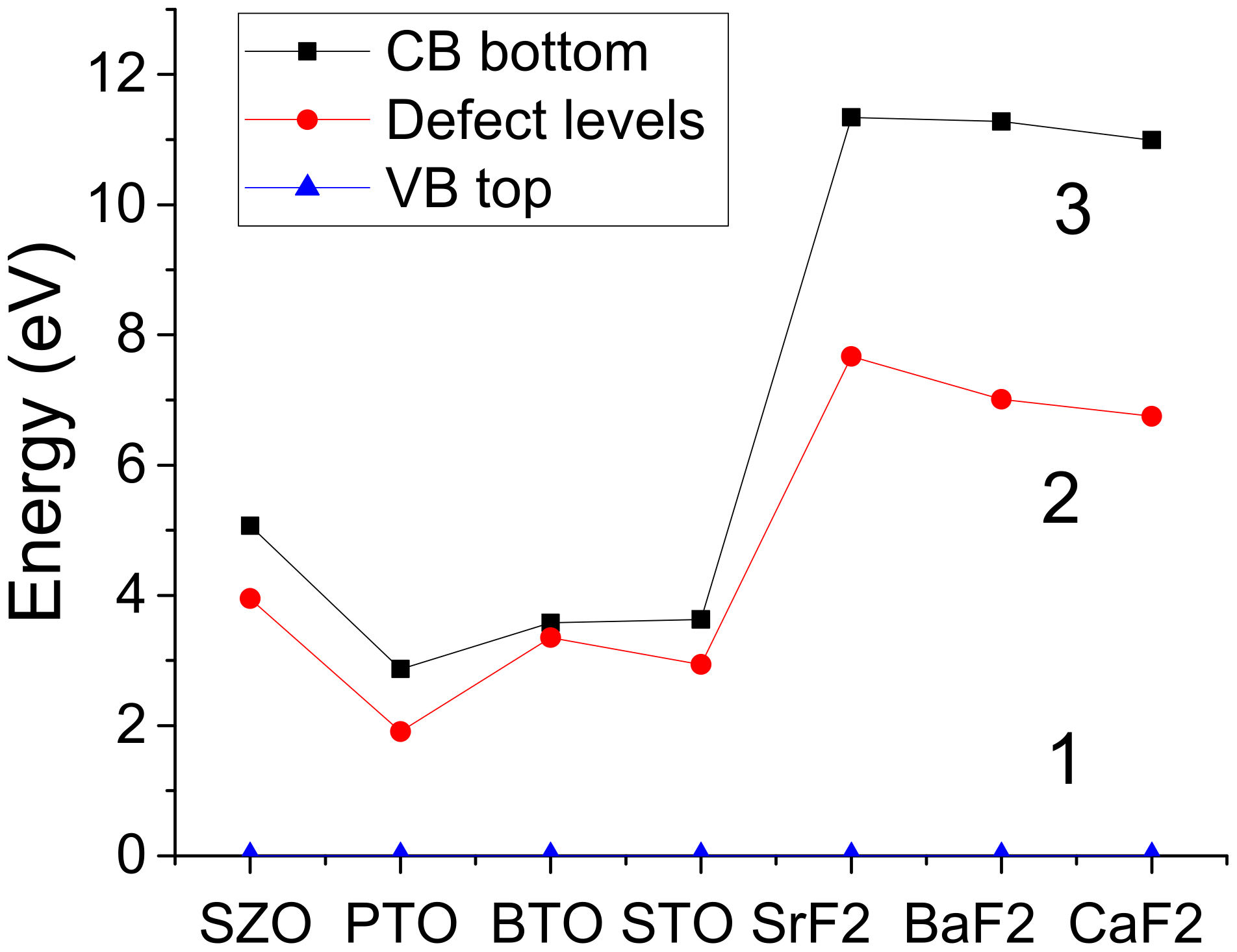
3. Ab Initio Computation Results for F-Centers in ABO3 Perovskites, as Well as in SrF2, BaF2 and CaF2
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kotomin, E.A.; Popov, A.I. Radiation-induced point defects in simple oxides. Nucl. Instrum. Methods Phys. Res. Sect. B 1998, 141, 1–15. [Google Scholar] [CrossRef]
- Popov, A.I.; Kotomin, E.A.; Maier, J. Basic properties of the F-type centers in halides, oxides and perovskites. Nucl. Instrum. Methods Phys. Res. Sect. B 2010, 268, 3084–3089. [Google Scholar] [CrossRef]
- Itoh, N. Creation of lattice defects by electronic excitation in alkali halides. Adv. Phys. 1982, 31, 491–551. [Google Scholar] [CrossRef]
- Itoh, N.; Tanimura, K. Formation of interstitial-vacancy pairs by electronic excitation in pure ionic-crystals. J. Phys. Chem. Solids 1990, 51, 717–735. [Google Scholar] [CrossRef]
- Lushchik, C.; Lushchik, A. Evolution of Anion and Cation Excitons in Alkali Halide Crystals. Phys. Solid State 2018, 60, 1487–1505. [Google Scholar] [CrossRef]
- Dauletbekova, A.; Akilbekov, A.; Elango, A. Thermo-and Photostimulated Recombinations of F-HA and α-IA Centres in KBr with Large Na Concentration. Phys. Stat. Sol. B 1982, 112, 445–452. [Google Scholar] [CrossRef]
- Kirm, M.; Lushchik, A.; Lushchik, C.; Martinson, I.; Nagirnyi, V.; Vasil’chenko, E. Creation of groups of spatially correlated defects in a KBr crystal at 8 K. J. Phys. Condens. Matter 1998, 10, 3509–3521. [Google Scholar] [CrossRef]
- Weinstein, I.A.; Kortov, V.S.; Vohmintsev, A.S. The compensation effect during luminescence of anion centers in aluminum oxide. J. Lumin. 2007, 122, 342–344. [Google Scholar] [CrossRef]
- Feldbach, E.; Toldsepp, E.; Kirm, M.; Lushchik, A.; Mizohata, K.; Raisanen, J. Radiation resistance diagnostics of wide-gap optical materials. Opt. Mater. 2016, 55, 164–167. [Google Scholar] [CrossRef]
- Ananchenko, D.V.; Nikiforov, S.V.; Konev, S.F.; Ramazanova, G.R. ESR and luminescent properties of anion-deficient α-Al2O3 single crystals after high-dose irradiation by a pulsed electron beam. Opt. Mater. 2019, 90, 118–122. [Google Scholar] [CrossRef]
- Lushchik, A.; Karner, T.; Lushchik, C.; Vasilchenko, E.; Dolgov, S.; Issahanyan, V.; Liblik, P. Dependence of long-lived defect creation on exciton density in MgO single crystals. Phys. Stat. Sol. C 2007, 4, 1084–1087. [Google Scholar]
- Myasnikova, L.; Shunkeyev, K.; Zhanturina, N.; Ubaev, Z.; Barmina, A.; Sagimbaeva, S.; Aimaganbetova, Z. Luminescence of self-trapped excitons in alkali halide crystals at low temperature uniaxial deformation. Nucl. Instrum. Methods Phys. Res. Sect. B 2020, 464, 95–99. [Google Scholar] [CrossRef]
- Schweizer, S.; Secu, M.; Spaeth, J.M.; Hobs, L.W.; Edgar, A.; Williams, G.V.M. New developments in X-ray storage phosphors. Radiat. Meas. 2004, 38, 633–638. [Google Scholar] [CrossRef]
- Elango, A.; Sagimbaeva, S.; Sarmukhanov, E.; Savikhina, T.; Shunkeev, K. Effect of uniaxial stree on luminescence of X- and VUV-irradiated NaCl and NaBr crystals. Radiat. Meas. 2001, 33, 823–827. [Google Scholar] [CrossRef]
- Kortov, V.; Lushchik, A.; Nagirnyi, V.; Ananchenko, D.; Romet, I. Spectrally resolved thermally stimulated luminescence of irradiated anion-defective alumina single crystals. J. Lumin. 2017, 186, 189–193. [Google Scholar] [CrossRef]
- Koyama, T.; Suemoto, T. Dynamics of nuclear wave packets at the F center in alkali halides. Rep. Prog. Phys. 2011, 74, 076502. [Google Scholar] [CrossRef]
- Skuratov, V.A.; O’Connell, J.; Kirilkin, N.S.; Neethling, J. On the threshold of damage formation in aluminium oxide via electronic excitations. Nucl. Instrum. Methods Phys. Res. Sect. B 2014, 326, 223–227. [Google Scholar] [CrossRef]
- Van Vuuren, J.A.; Saifulin, M.M.; Skuratov, V.A.; O’Connell, J.H.; Aralbayeva, G.; Dauletbekova, A.; Zdorovets, M. The influence of stopping power and temperature on latent track formation in YAP and YAG. Nucl. Instrum. Methods Phys. Res. Sect. B 2019, 460, 67–73. [Google Scholar] [CrossRef]
- Mota, F.; Ortiz, C.J.; Vila, R.; Casal, N.; Garcia, A.; Ibarra, A. Calculation of damage function of Al2O3 in irradiation facilities for fusion reactor applications. J. Nucl. Mater. 2013, 442, S699–S704. [Google Scholar] [CrossRef]
- Dukenbayev, K.; Kozlovskiy, A.; Kenzhina, I.; Berguzinov, A.; Zdorovets, M. Study of the effect of irradiation with Fe7+ ions on the structural properties of thin TiO2 foils. Mater. Res. Express 2019, 6, 046309. [Google Scholar] [CrossRef]
- Zdorovets, M.; Dukenbayev, K.; Kozlovskiy, A.; Kenzhina, I. Defect formation in AlN after irradiation with He2+ ions. Ceram. Int. 2019, 45, 8130–8137. [Google Scholar] [CrossRef]
- Janesco, B.G.; Jones, S.I. Quantifying the delocalization of surface and bulk F-centers. Surf. Sci. 2017, 659, 9–15. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Christensen, N.E.; Kotomin, E.A.; Postnikov, A.V.; Borstel, G. First-principles and semiempirical calculations for F center in KNbO3. Phys. Rev. B 1997, 56, 8599–8604. [Google Scholar] [CrossRef]
- Carballo-Cordova, D.A.; Ochoa-Lara, M.T.; Olive-Mendez, S.F.; Espinosa-Magana, F. First-principles calculations and Bader analysis of oxygen-deficient induced magnetism in cubic BaTiO3-x and SrTiO3-x. Philos. Mag. 2019, 99, 181–187. [Google Scholar] [CrossRef]
- Ojha, S.K.; Gogoi, S.K.; Mandal, P.; Kaushik, S.D.; Freeland, J.W.; Jain, M.; Middey, S. Oxygen vacancy induced electronic structure modification of KTaO3. Phys. Rev. B 2021, 103, 085102. [Google Scholar] [CrossRef]
- Osinkin, D.A.; Khodimchuk, A.V.; Porotnikova, N.M.; Bogdanovich, N.M.; Fetisov, A.V.; Ananyev, M.V. Rate-Determining Steps of Oxygen Surface Exchange Kinetics on Sr2Fe1.5Mo0.5O6−δ. Energies 2020, 13, 250. [Google Scholar] [CrossRef]
- Farlenkov, A.S.; Ananyev, M.V.; Eremin, V.A.; Porotnikova, N.M.; Kurumchin, E.K.; Melehov, B.T. Oxygen isotope exchange in doped calcium and barium zirconates. Solid State Ion. 2016, 290, 108–115. [Google Scholar] [CrossRef]
- Mueller, D.N.; Machala, M.L.; Bluhm, H.; Chuech, W.C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 2015, 6, 6097. [Google Scholar] [CrossRef]
- Ji, Q.; Bi, L.; Zhang, J.; Cao, H.; Zhao, X.S. The role of oxygen vacancies of ABO3 perovskite oxydes in the oxygen reduction reaction. Energy Environ. Sci. 2020, 13, 1408–1428. [Google Scholar] [CrossRef]
- Su, H.Y.; Sun, K. DFT study of the stability of oxygen vacancy in cubic ABO3 perovskites. J. Mater. Sci. 2015, 50, 1701–1709. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio calculations of SrTiO3, BaTiO3, PbTiO3, CaTiO3, SrZrO3, PbZrO3 and BaZrO3 (001), (011) and (111) surfaces as well as F centers, polarons, KTN solid solutions and Nb impurities therein. Int. J. Mod. Phys. B 2014, 28, 1430009. [Google Scholar] [CrossRef]
- Crespillo, M.L.; Graham, J.T.; Agullo-Lopez, F.; Zhang, Y.W.; Weber, W.J. Real-Time Identification of Oxygen Vacancy Centers in LiNbO3 and SrTiO3 during Irradiation with High Energy Particles. Crystals 2021, 11, 315. [Google Scholar] [CrossRef]
- Jiao, X.P.; Liu, T.Y.; Lu, Y.Z.; Li, Q.Y.; Guo, R.; Wang, X.L.; Xu, X. Optical Properties of the Oxygen Vacancy in KNbO3 Crystal. J. Electron. Mater. 2020, 49, 2137–2143. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I.; Purans, J.; Jia, R. First principles hybrid Hartree-Fock-DFT calculations of bulk and (001) surface F centers in oxide perovskites and alkali-earth fluorides. Low Temp. Phys. 2020, 46, 1206–1212. [Google Scholar] [CrossRef]
- Gu, X.K.; Samira, S.; Nikolla, E. Oxygen Sponges for Electrocatalysis: Oxygen Reduction/Evolution on Nonstoichiometric, Mixed Metal Oxides. Chem. Mater. 2018, 30, 2860–2872. [Google Scholar] [CrossRef]
- Shi, H.; Eglitis, R.I.; Borstel, G. Ab initio calculations of the CaF2 electronic structure and F centers. Phys. Rev. B 2005, 72, 045109. [Google Scholar] [CrossRef]
- Wen, C.; Lanza, M. Calcium fluoride as high-k dielectric for 2D electronics. Appl. Phys. Reviews 2021, 8, 021307. [Google Scholar]
- Laanaiya, M.; Bouibes, A.; Zaoui, A. Ground state properties of fluorine from DFT-hybrid functional. Vacuum 2021, 187, 110118. [Google Scholar] [CrossRef]
- Weng, J.P.; Gao, S.P. Layer-dependent band gaps and dielectric constants of ultrathin fluorite crystals. J. Phys. Chem. Solids 2021, 148, 109738. [Google Scholar] [CrossRef]
- Liebig, A.; Hapala, P.; Weymouth, A.J.; Giessibl, F.J. Quantifying the evolution of atomic interaction of a complex surface with a functionalized atomic force microscopy tip. Sci. Rep. 2020, 10, 14104. [Google Scholar] [CrossRef]
- Ryabochkina, P.A.; Krushchalina, S.A.; Yurlov, I.A.; Egorysheva, A.V.; Atanova, A.V.; Veselova, V.O.; Kyashkin, V.M. Blackbody emission from CaF2 and ZrO2 nanosized electric particles doped with Er3+ ions. RSC Adv. 2020, 10, 26288–26297. [Google Scholar] [CrossRef]
- Shi, H.; Eglitis, R.I.; Borstel, G. Ab initio calculations of the BaF2 bulk and surface F centres. J. Phys. Condens. Matter 2006, 18, 8367–8381. [Google Scholar] [CrossRef]
- Cappellini, G.; Bosin, A.; Serra, G.; Furthmüller, J.; Bechstedt, F.; Botti, S. Electronic and Optical Properties of Small Metal Fluoride Clusters. ACS Omega 2020, 5, 13268–13277. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, N.; Szpikowska-Sroka, B.; Goryczka, T.; Pisarski, W.A. Spectroscopic Properties of Eu3+ Ions in Sol-Gel Materials Containing Calcium Fluoride Nanocrystals. Phys. Stat. Sol. B 2020, 257, 1900478. [Google Scholar] [CrossRef]
- Matusalem, F.; Marques, M.; Teles, L.K.; Filippetti, A.; Cappellini, G. Electronic properties of fluorides by efficient approximated quasiparticle DFT-1/2 and PSIC methods. J. Phys. Condens. Matter 2018, 30, 365501. [Google Scholar] [CrossRef] [PubMed]
- Vassilyeva, A.F.; Eglitis, R.I.; Kotomin, E.A.; Dauletbekova, A.K. Ab initio calculations of MgF2 (001) and (011) surface structure. Physica B 2010, 405, 2125–2127. [Google Scholar] [CrossRef]
- Shi, H.; Chang, L.; Jia, R.; Eglitis, R.I. Ab initio calculations of the charge transfer and aggregation of F centers in CaF2. J. Phys. Chem. C 2012, 116, 4832–4839. [Google Scholar] [CrossRef]
- Häfner, M.; Bredow, T. Mobility of F Centers in Alkali Halides. J. Phys. Chem. C 2021, 125, 9085–9095. [Google Scholar] [CrossRef]
- Mir, A.; Zaoui, A.; Bensaid, D. The displacement effect of a fluorine atom in CaF2 on the band structure. Appl. Surf. Sci. 2018, 439, 1180–1185. [Google Scholar] [CrossRef]
- Ibraheem, A.M.; Khalafalla, M.A.H.; Eisa, M.H. First principle calculation of accurate native defect levels in CaF2. Eur. Phys. J. B 2017, 90, 42. [Google Scholar] [CrossRef]
- Shi, H.; Jia, R.; Eglitis, R.I. First-principles calculations of surface H centers in BaF2. Phys. Rev. B 2010, 81, 195101. [Google Scholar] [CrossRef]
- Andrade, A.B.; Ferreira, N.S.; Valerio, M.E.G. Particle size effects on structural and optical properties of BaF2 nanoparticles. RSC Adv. 2017, 7, 26839–26848. [Google Scholar] [CrossRef]
- Watanabe, M.; Azuma, J.; Asaka, S.; Tsujibayashi, T.; Arimoto, O.; Nakanishi, S.; Itoh, H.; Kamada, M. Photostimulated detection of defect formation in BaF2 under irradiation of synchrotron radiation. Phys. Stat. Sol. B 2013, 250, 396–401. [Google Scholar] [CrossRef]
- Shi, H.; Jia, R.; Eglitis, R.I. First-principles simulations on the aggregation of F centers in BaF2: R centers. Solid State Ion. 2011, 187, 1–7. [Google Scholar] [CrossRef]
- Hoya, J.; Laborde, J.I.; Richard, D.; Renteria, M. Ab initio study of F-centers in alkali halides. Comput. Mater. Sci. 2017, 139, 1–7. [Google Scholar]
- Shi, H.; Chang, L.; Jia, R.; Eglitis, R.I. Ab initio calculations of hydroxyl impurities in CaF2. J. Phys. Chem. C 2012, 116, 6392–6400. [Google Scholar] [CrossRef]
- Cen, J.; Liang, F.; Chen, D.L.; Zhang, L.L.; Yang, N.; Zhu, W.D. Adsorption of water molecule on calcium fluoride and magnesium fluoride surfaces: A combined Theoretical and experimental study. J. Phys. Chem. C 2020, 124, 7853–7859. [Google Scholar] [CrossRef]
- Arends, J. Color centers in additively colored CaF2 and BaF2. Phys. Stat. Sol. B 1964, 7, 805–815. [Google Scholar] [CrossRef]
- Nepomnyashchikh, A.I.; Radzhabov, E.A.; Egranov, A.V.; Ivashechkin, V.F.; Istomin, A.S. Defect formation and VUV luminescence in BaF2. Radiat. Eff. Defects Solids 2002, 157, 715–719. [Google Scholar] [CrossRef]
- Cavenett, B.C.; Hayes, W.; Hunter, I.C.; Stoneham, A.M. Magneto optical properties of F centres in alkaline earth fluorides. Proc. R. Soc. Lond. Ser. A 1969, 309, 53–68. [Google Scholar]
- Assylbayev, R.; Lushchik, A.; Lushchik, C.; Kudryavtseva, I.; Shablonin, E.; Vasilchenko, E.; Akilbekov, A.; Zdorovets, M. Structural defects caused by swift ions in fluorite single crystals. Opt. Mater. 2018, 75, 196–203. [Google Scholar] [CrossRef]
- Batool, A.; Izerrouken, M.; Aisida, S.O.; Hussain, J.; Ahmad, I.; Afzal, M.Q. Effect of Ca colloids on in-situ ionoluminescence of CaF2 sigle crystals. Nucl. Instrum. Methods Phys. Res. Sect. B 2020, 476, 40–43. [Google Scholar] [CrossRef]
- Saunders, V.R.; Dovesi, R.; Roetti, C.; Causa, N.; Harrison, N.M.; Orlando, R.; Zicovich-Wilson, C.M. Crystal-2009 User Manual; University of Torino: Torino, Italy, 2009. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.E. Surface effects in ferroelectrics: Periodic slab computations for BaTiO3. Ferroelectrics 1997, 194, 323–342. [Google Scholar] [CrossRef]
- Cohen, R.E. Periodic slab LAPW computations for ferroelectric BaTiO3. J. Phys. Chem. Solids 1996, 57, 1393–1396. [Google Scholar] [CrossRef]
- Shang, Y. Lower bounds for Gaussian Estrada index of graphs. Symmetry 2018, 10, 325. [Google Scholar] [CrossRef]
- Alhevaz, A.; Baghipur, M.; Shang, Y. On generalized distance Gaussian Estrada index of graps. Symmetry 2019, 11, 1276. [Google Scholar] [CrossRef]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Monkhorst, H.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio calculations of the atomic and electronic structure of BaZrO3 (111) surfaces. Solid State Ion. 2013, 230, 43–47. [Google Scholar] [CrossRef]
- Mesquita, W.D.; Oliveira, M.C.; Assis, M.; Ribeiro, R.A.P.; Eduardo, A.C.; Teordoro, M.D.; Marques, G.E.; Júnior, M.G.; Longo, E.; Gurgel, M.F.C. Unraveling the relationship between bulk structure and exposed surfaces and its effect on the electronic structure and photoluminescent properties of Ba0.5Sr0.5TiO3: A joint experimental and theoretical approach. Mater. Res. Bull. 2021, 143, 111442. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Kleperis, J.; Purans, J.; Popov, A.I.; Jia, R. Ab initio calculations of CaZrO3 (011) surfaces: Systematic trends in polar (011) surface calculations of ABO3 perovskites. J. Mater. Sci. 2020, 55, 203–217. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Gabrusenoks, J.; Popov, A.I.; Jia, R. Comparative ab initio calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces. Crystals 2020, 10, 745. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Piskunov, S. First principles calculations of SrZrO3 bulk and ZrO2-terminated (001) surface F centers. Comput. Condens. Matter 2016, 7, 1–6. [Google Scholar] [CrossRef]
- Zhukovskii, Y.F.; Kotomin, E.A.; Piskunov, S.; Ellis, D.E. A comparative ab initio study of bulk and surface oxygen vacancies in PbTiO3, PbZrO3 and SrTiO3 perovskites. Solid State Commun. 2009, 149, 1359–1362. [Google Scholar] [CrossRef]
- Sokolov, M.; Eglitis, R.I.; Piskunov, S.; Zhukovskii, Y.F. Ab initio hybrid DFT calculations of BaTiO3 bulk and BaO-terminated (001) surface F-centers. Int. J. Mod. Phys. B 2017, 31, 1750251. [Google Scholar] [CrossRef]
- Carrasco, J.; Illas, F.; Lopez, N.; Kotomin, E.A.; Zhukovskii, Y.F.; Evarestov, R.A.; Mastrikov, Y.A.; Piskunov, S.; Maier, J. First-principles calculations of the atomic and electronic structure of F centers in the bulk and on the the (001) surface of SrTiO3. Phys. Rev. B 2006, 73, 064106. [Google Scholar] [CrossRef]
- Jia, R.; Shi, H.; Borstel, G. Ab initio calculations for SrF2 with F and M centers. Comput. Mater. Sci. 2008, 43, 980–988. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, J.S.; Noh, T.W.; Byun, D.Y.; Yoo, K.S.; Yamaura, K.; Takayama-Muromachi, E. Systematic trends in the electronic structure parameters of the 4d transition-metal oxides SrMO3 (M = Zr, Ru and Rh). Phys. Rev. B 2003, 67, 113101. [Google Scholar] [CrossRef]
- Yoshino, M.; Yukawa, H.; Morinaga, M. Modification of local electronic structures due to phase transition in perovskite-type oxides, SrBO3 (B = Zr, Ru, Hf). Mater. Trans. 2004, 45, 2056–2061. [Google Scholar] [CrossRef][Green Version]
- Kennedy, B.J.; Howard, C.J.; Chakoumakos, B.C. High-temperature phase transitions in SrZrO3. Phys. Rev. B 1999, 59, 4023–4027. [Google Scholar] [CrossRef]
- Robertson, J. Band offsets of wide-band-gap oxides and implications for future electronic devices. J. Vacuum Sci. Technol. 2000, 18, 1785–1791. [Google Scholar] [CrossRef]
- Nelmes, R.J.; Kuhs, W.F. The crystal structure of tetragonal PbTiO3 at room temperature and at 700 K. Solid State Commun. 1985, 54, 721–723. [Google Scholar] [CrossRef]
- Mabud, S.A.; Glazer, A.M. Lattice parameters and birefringence in PbTiO3 single crystals. J. Appl. Cryst. 1979, 12, 49–53. [Google Scholar] [CrossRef]
- Wemple, S.H. Polarization fluctuations and the optical-absorption edge in BaTiO3. Phys. Rev. B 1970, 2, 2679–2689. [Google Scholar] [CrossRef]
- Meyer, B.; Padilla, J.; Vanderbilt, D. Theory of PbTiO3, BaTiO3 and SrTiO3 surfaces. Faraday Discuss. 1999, 114, 395–405. [Google Scholar] [CrossRef]
- Edwards, J.W.; Speiser, R.; Johnston, H.L. Structure of barium titanate at elevated temperatures. J. Amer. Chem. Soc. 1951, 73, 2934–2935. [Google Scholar] [CrossRef]
- van Benthem, K.; Elsasser, C.; French, R.H. Bulk electronic structure of SrTiO3: Experiment and theory. J. Appl. Phys. 2001, 90, 6156–6164. [Google Scholar] [CrossRef]
- Sato, M.; Soejima, Y.; Ohama, N.; Okazaki, A.; Scheel, H.J.; Müller, K.A. The lattice constant vs temperature relation around the 105 K transition of a flux-grown SrTiO3 crystal. Phase Trans. 1985, 5, 207–218. [Google Scholar] [CrossRef]
- Rubloff, G.W. Far-Ultraviolet Reflectence Spectra and the Electronic Structure of Ionic Crystals. Phys. Rev. B 1972, 5, 662–684. [Google Scholar] [CrossRef]
- Hayes, W. Crystals with the Fluorite Structure; Clarendon Press: Oxford, UK, 1974. [Google Scholar]
- Leger, J.M.; Haines, J.; Atouf, A.; Schulte, O. High-pressure x-ray and neutron-diffraction studies of BaF2: An example of a coordination number of 11 in AX2 compounds. Phys. Rev. B 1995, 52, 13247. [Google Scholar] [CrossRef] [PubMed]
- Nicolav, M. Shaped single crystals of CaF2. J. Cryst. Growth 2000, 218, 62–66. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Structure at Room Temperature | Band Gap (eV), Room Temperature | Transition T to Cubic Phase (K) | Experimental Lattice Constant at Cubic Phase (Å) |
|---|---|---|---|---|
| SrZrO3 | Orthorhombic | 5.6 [80] | 1433 [81] | 4.154 [82] |
| PbTiO3 | Tetragonal | 3.4 [83] | 763 [84] | 3.970 [85] |
| BaTiO3 | Tetragonal to orthorhombic | 3.38 (// c); 3.27 (⊥ c) [86] | 403 [87] | 4.0037 [88] |
| SrTiO3 | Cubic | 3.75 [89] | 110 [87] | 3.89845 [90] |
| SrF2 | Cubic | 11.25 [91] | - | 5.799 [92] |
| BaF2 | Cubic | 11.00 [91] | - | 6.20 [93] |
| CaF2 | Cubic | 12.1 [91] | - | 5.46 [94] |
| Computed Characteristics | SrZrO3 | PbTiO3 | BaTiO3 | SrTiO3 |
|---|---|---|---|---|
| Bulk lattice constant (Å) | 4.163 | 3.936 | 4.008 | 3.904 |
| Bulk oxygen vacancy in SrZrO3, PbTiO3, BaTiO3 and SrTiO3 perovskites | ||||
| B atom shift (% of a0) | +3.68 | +1.63 | +1.06 | +7.76 |
| O atom shift (% of a0) | −2.63 | −0.88 | −0.71 | −7.79 |
| A atom shift (% of a0) | +0.46 | −2.58 | −0.08 | +3.94 |
| Oxygen vacancy on SrZrO3, PbTiO3, BaTiO3 and SrTiO3 (001) surface | ||||
| B atom shift (% of a0) | +9.17 | +9.98 | +0.1 | +14.0 |
| O atom shift (% of a0) | −4.16 | −5.58 | −1.4 | −8.0 |
| A atom shift (% of a0) | +7.68 | - | +1.0 | - |
| Computed Characteristics | SrZrO3 | PbTiO3 | BaTiO3 | SrTiO3 |
|---|---|---|---|---|
| Bulk oxygen vacancy in SrZrO3, PbTiO3, BaTiO3 and SrTiO3 perovskites | ||||
| O atom net charge (e) | −2.0 | −2.0 | −2.0 | −2.0 |
| O atom charge (e) in ABO3 | −1.351 | −1.232 | −1.388 | −1.407 |
| F-center charge (e) | −1.25 | −0.85 | −1.103 | −1.10 |
| F-cent. format. energy (eV) | 7.55 | 7.82 | 10.30 | 7.10 |
| Band gap with F-cent. (eV) | 5.07 | 2.87 | 3.58 | 3.63 |
| F-center under CB (eV) | 1.12 | 0.96 | 0.23 | 0.69 |
| Oxygen vacancy on SrZrO3, PbTiO3, BaTiO3 and SrTiO3 (001) surface | ||||
| F-center charge (e) | −1.10 | - | −1.052 | - |
| F-cent. format. energy (eV) | 7.52 | 5.99 | 10.20 | 6.22 |
| F-center under CB (eV) | 0.93 | 0.71 | 0.07 | 0.25 |
| Computed Bulk Fluorine Vacancy Properties | SrF2 | BaF2 | CaF2 |
|---|---|---|---|
| B3PW computed lattice constant a0 (Å) | 5.845 | 6.26 | 5.50 |
| F atom net charge (e) | −1.0 | −1.0 | −1.0 |
| F atom charge (e) in SrF2, BaF2 and CaF2 | −0.954 | −0.923 | −0.902 |
| Charge inside the fluorine vacancy (e) | −0.848 | −0.801 | −0.752 |
| A atom shift (% of a0) | −0.02 | +0.03 | +0.15 |
| F atom shift (% of a0) | −0.27 | −0.23 | +0.28 |
| Fluorine vacancy formation energy (eV) | 10.33 | 7.82 | 7.87 |
| B3PW calc. band gap, perfect crystal (eV) | 11.31 | 11.30 | 10.96 |
| B3PW calc. band gap with F-center (eV) | 11.34 | 11.28 | 10.99 |
| Fluorine vacancy ind. level under CB (eV) | 3.67 | 4.27 | 4.24 |
| Computed (111) surface fluorine vacancy | |||
| Ba atom shift (% of a0) | - | −0.13 | - |
| F atom shift (% of a0) | - | −0.37 | - |
| Charge inside the fluorine vacancy (e) | - | −0.790 | - |
| Fluorine vacancy formation energy (eV) | - | 7.48 | - |
| Fluorine vacancy ind. level under CB (eV) | - | 4.11 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eglitis, R.I.; Purans, J.; Popov, A.I.; Jia, R. Tendencies in ABO3 Perovskite and SrF2, BaF2 and CaF2 Bulk and Surface F-Center Ab Initio Computations at High Symmetry Cubic Structure. Symmetry 2021, 13, 1920. https://doi.org/10.3390/sym13101920
Eglitis RI, Purans J, Popov AI, Jia R. Tendencies in ABO3 Perovskite and SrF2, BaF2 and CaF2 Bulk and Surface F-Center Ab Initio Computations at High Symmetry Cubic Structure. Symmetry. 2021; 13(10):1920. https://doi.org/10.3390/sym13101920
Chicago/Turabian StyleEglitis, Roberts I., Juris Purans, Anatoli I. Popov, and Ran Jia. 2021. "Tendencies in ABO3 Perovskite and SrF2, BaF2 and CaF2 Bulk and Surface F-Center Ab Initio Computations at High Symmetry Cubic Structure" Symmetry 13, no. 10: 1920. https://doi.org/10.3390/sym13101920
APA StyleEglitis, R. I., Purans, J., Popov, A. I., & Jia, R. (2021). Tendencies in ABO3 Perovskite and SrF2, BaF2 and CaF2 Bulk and Surface F-Center Ab Initio Computations at High Symmetry Cubic Structure. Symmetry, 13(10), 1920. https://doi.org/10.3390/sym13101920