
Covalently Bonded Fullerene Nano-Aggregates (C60)n: Digitalizing Their Energy–Topology–Symmetry




Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
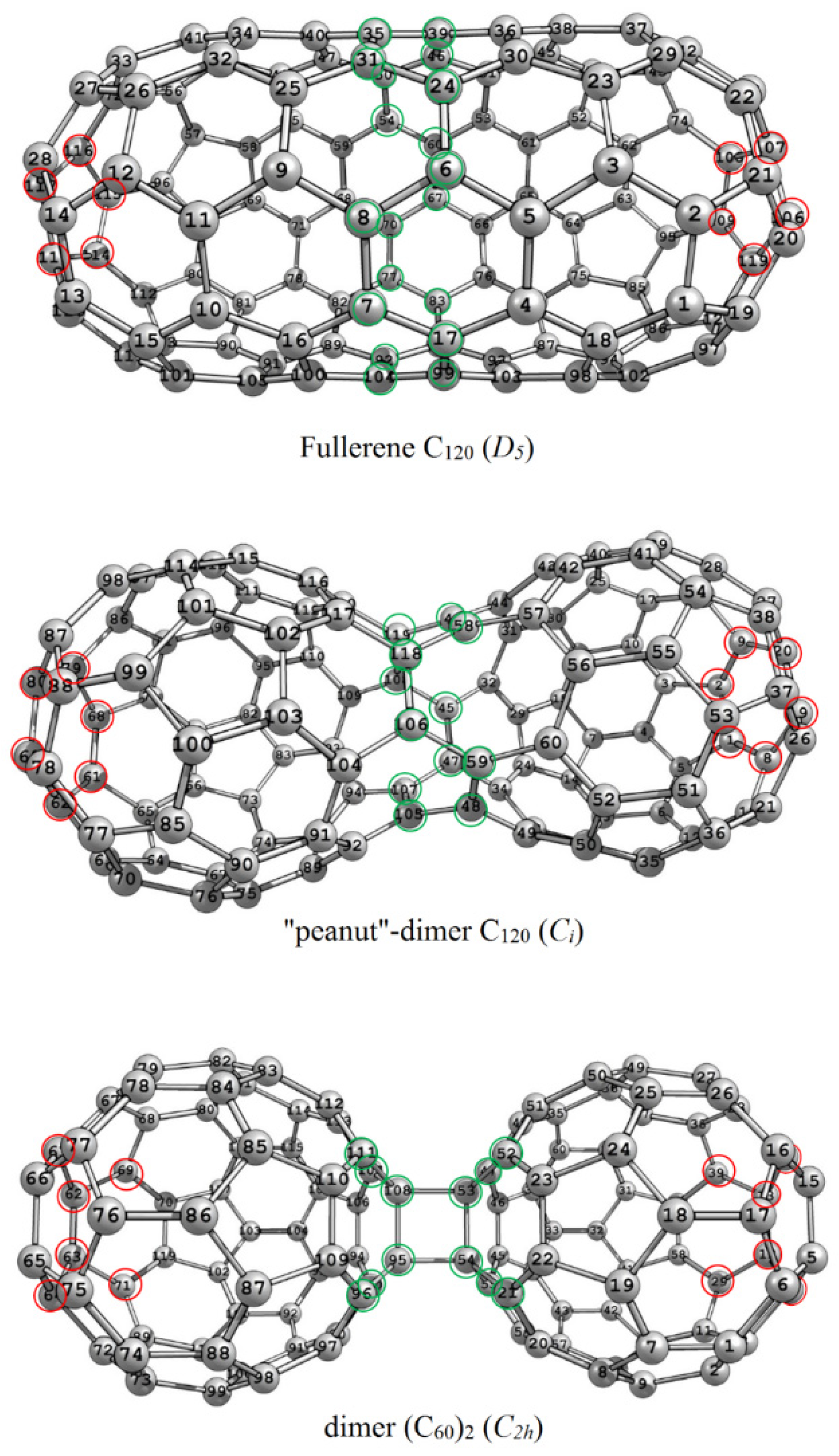
3.1. The C120 Isomers: A Fullerene and Two Dimers
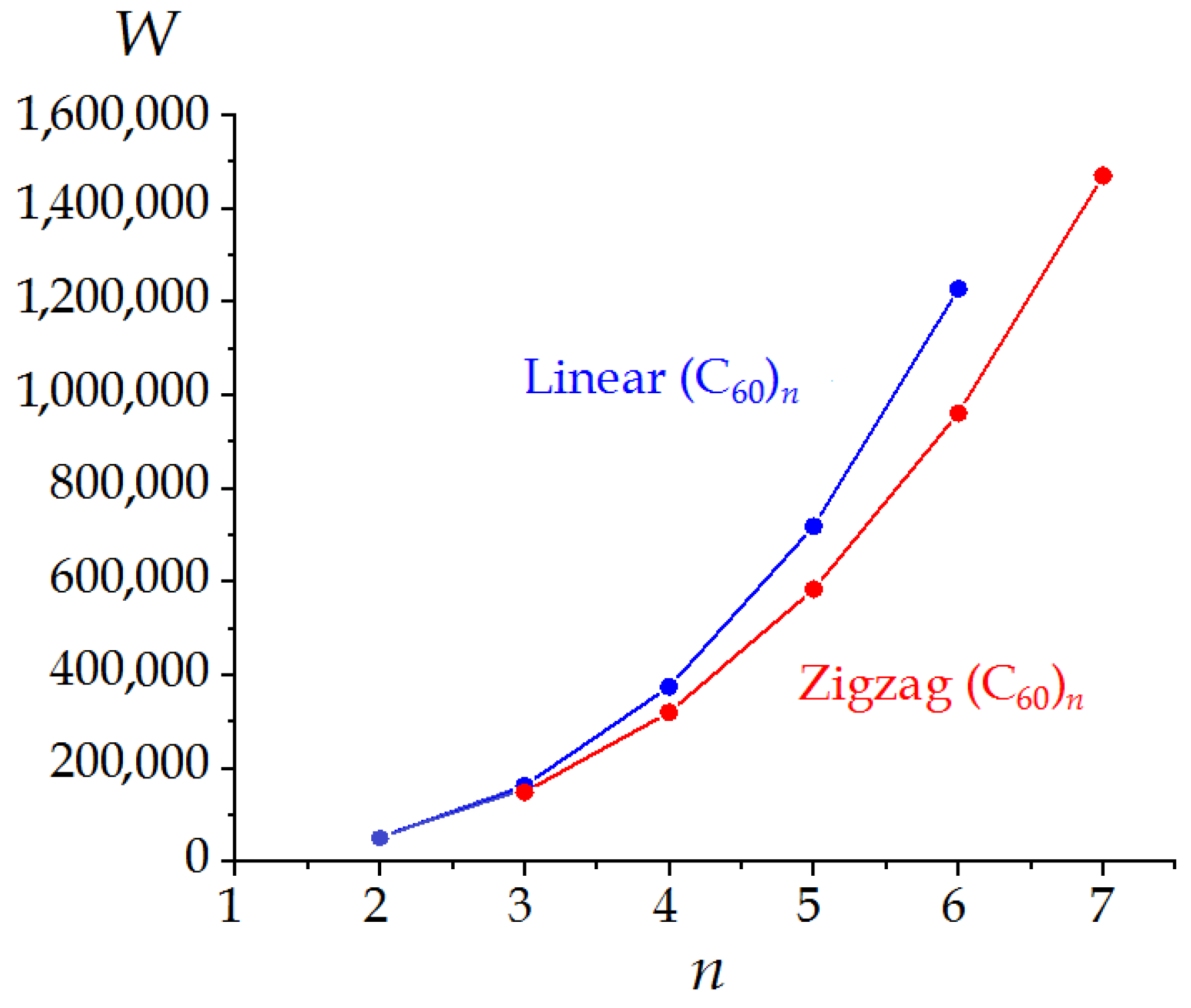
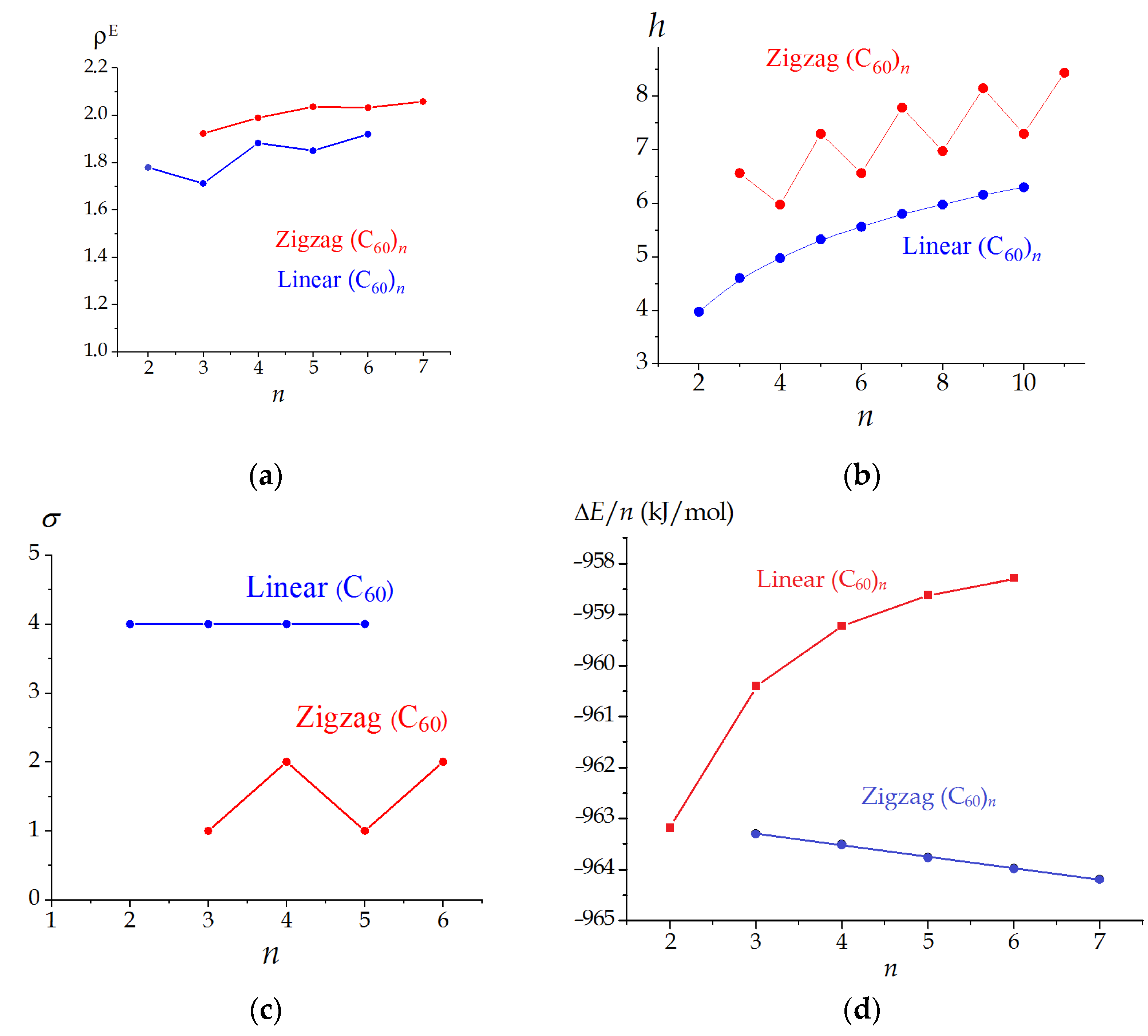
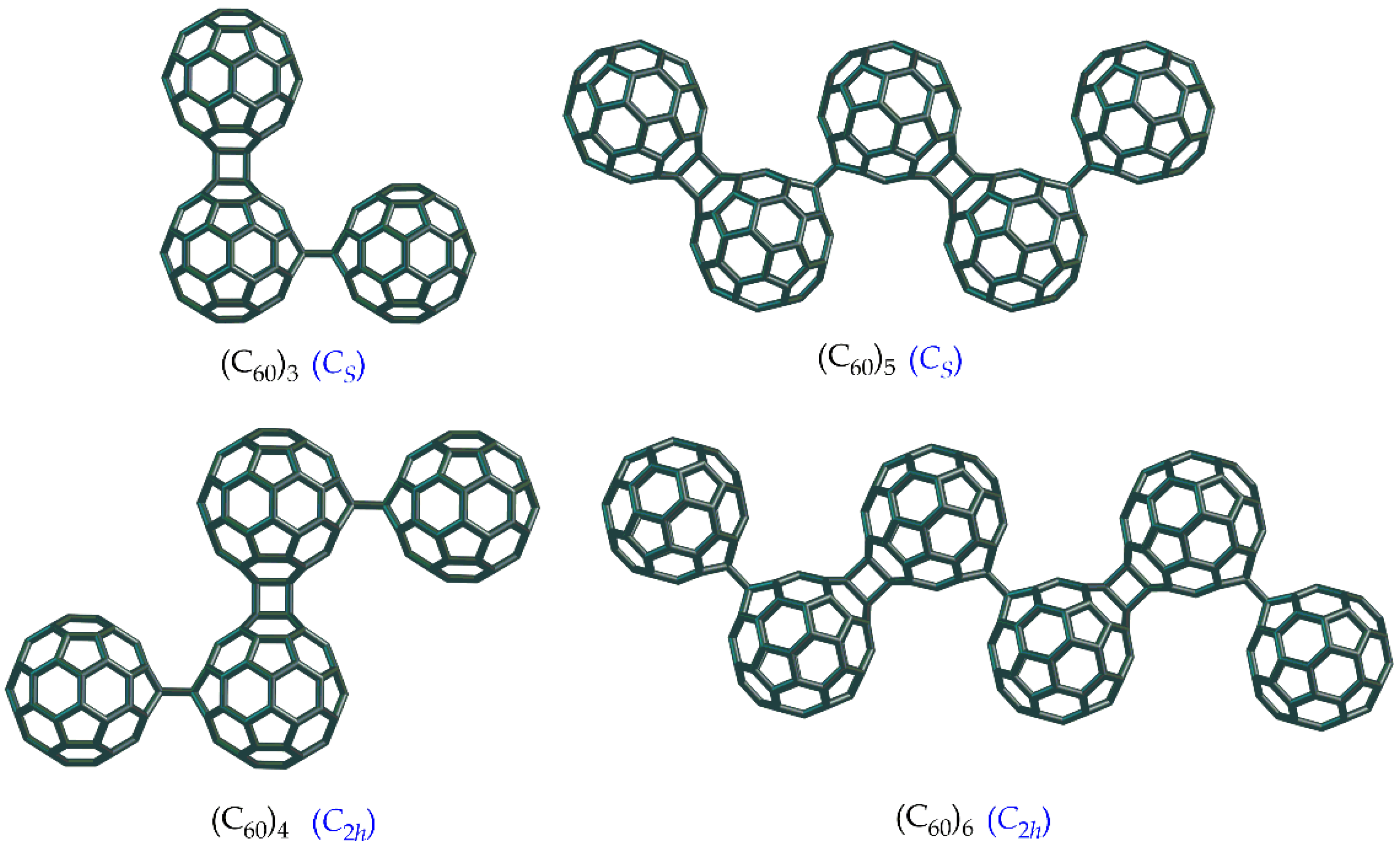
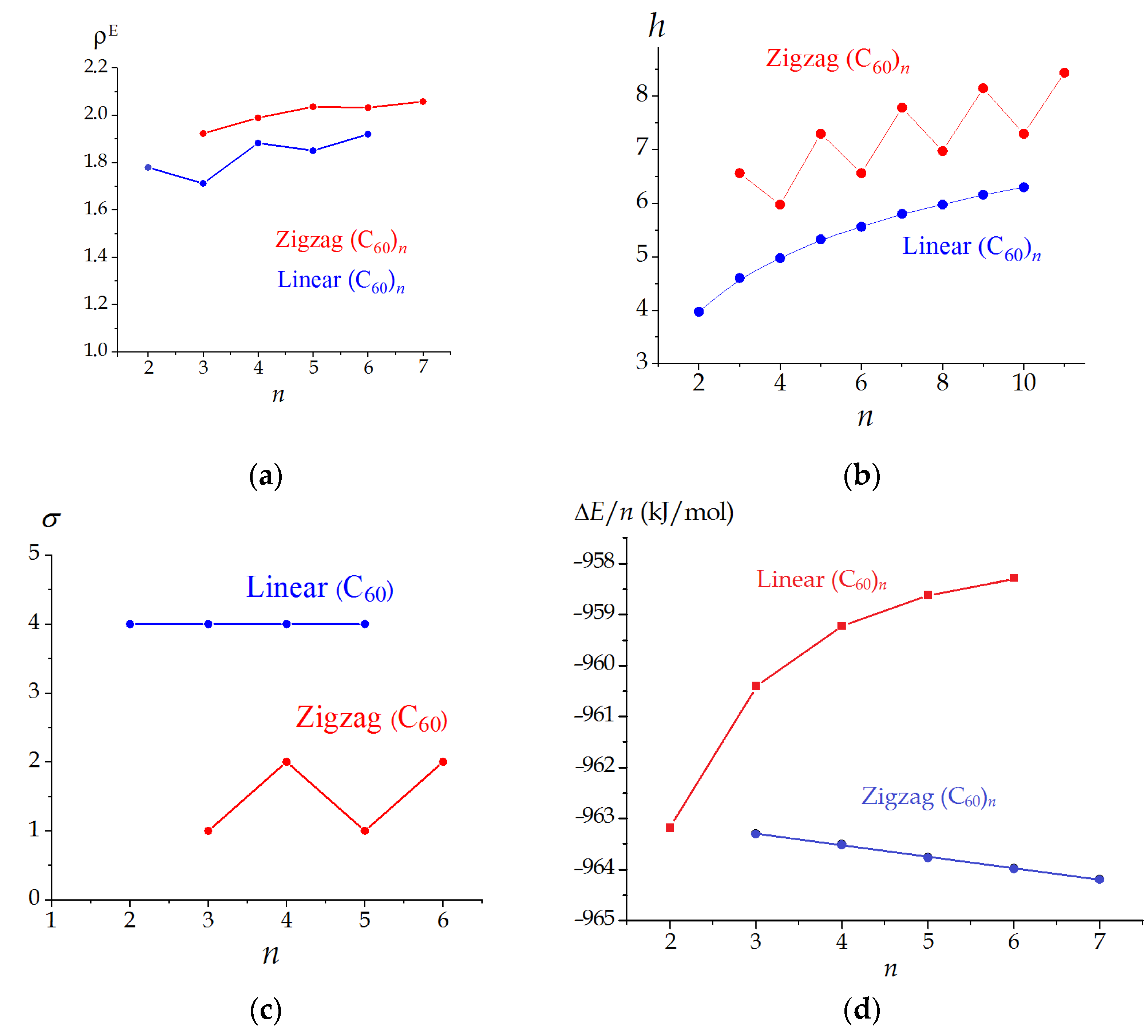
3.2. Linear and Zigzag [2+2]-Linked Oligomers (C60)n
4. Conclusions and Prospective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhechkov, L.; Heine, T.; Seifert, G. D5h C50 fullerene: A building block for oligomers and solids? J. Phys. Chem. A 2004, 108, 11733–11739. [Google Scholar] [CrossRef]
- Ariga, K.; Shrestha, L.K. Fullerene nanoarchitectonics with shape-shifting. Materials 2020, 13, 2280. [Google Scholar] [CrossRef] [PubMed]
- Segura, J.L.; Martin, N. [60] Fullerene dimers. Chem. Soc. Rev. 2000, 29, 13–25. [Google Scholar] [CrossRef]
- Komatsu, K.; Wang, G.-W.; Murata, Y.; Tanaka, T.; Fujiwara, K. Mechanochemical synthesis and characterization of the fullerene dimer C120. J. Org. Chem. 1998, 63, 9358–9366. [Google Scholar] [CrossRef]
- Komatsu, K.; Fujiwara, K.; Tanaka, T.; Murata, Y. The fullerene dimer C120 and related carbon allotropes. Carbon 2000, 38, 1529–1534. [Google Scholar] [CrossRef]
- Yoshida, M.; Ōsawa, E. Formalized drawing of fullerene Nets. 1. Algorithm and exhaustive generation of isomeric structures. Bull. Chem. Soc. Jpn. 1995, 68, 2073–2081. [Google Scholar] [CrossRef]
- Ueno, H.; Ōsawa, S.; Ōsawa, E.; Takeuchi, K. Stone-Wales rearrangement pathways from the hinge-opened [2+2] C60 dimer to Ipr C120 fullerenes. Vibrational analysis of intermediates. Fuller. Sci. Technol. 1998, 6, 319–338. [Google Scholar] [CrossRef]
- Takashima, A.; Nishii, T.; Onoe, J. Formation process and electron-beam incident energy dependence of one-dimensional uneven peanut-shaped C60 polymer studied using in situ high-resolution infrared spectroscopy and density-functional calculations. J. Phys. D Appl. Phys. 2012, 45, 485302. [Google Scholar] [CrossRef]
- Wang, G.; Li, Y.; Huang, Y. Structures and electronic properties of peanut-shaped dimers and carbon nanotubes. J. Phys. Chem. 2005, 109, 10957–10961. [Google Scholar] [CrossRef]
- Zhu, S.-E.; Li, F.; Wang, G.-W. Mechanochemistry of fullerenes and related materials. Chem. Soc. Rev. 2013, 42, 7535–7570. [Google Scholar] [CrossRef]
- Kunitake, M.; Uemura, S.; Ito, O.; Fujiwara, K.; Murata, Y.; Komatsu, K. Structural analysis of C60 trimers by direct observation with scanning tunneling microscopy. Angew. Chem. 2002, 114, 1011–1014. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Masumoto, K.; Tanaka, T.; Komatsu, K. Formation of dimer, trimer, and tetramer of C60 and C70 by γ-ray, charged-particle irradiation, and their HPLC separation. Chem. Phys. Lett. 1999, 300, 661–666. [Google Scholar] [CrossRef]
- Sun, D.; Reed, C.A. Crystal engineering a linear polymer of C60 fullerene via supramolecular pre-organization. Chem. Commun. 2000, 2391–2392. [Google Scholar] [CrossRef] [Green Version]
- Chadli, H.; Rahmani, A.; Sbai, K.; Hermet, P.; Rols, S.; Sauvajol, J.-L. Calculation of Raman-active modes in linear and zigzag phases of fullerene peapods. Phys. Rev. B 2006, 74, 205412. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, C.; Ōsawa, E.; Choi, J.W.; Sur, J.C.; Lee, K.H. Snapshots of the fragmentation for C70@single-walled carbon nanotube: Tight-binding molecular dynamics simulations. Int. J. Mol. Sci. 2021, 22, 3929. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.Y.; Ōsawa, E.; Lee, K.H. A conversion dynamics of (C60)2 dimer fullerenes to a fused dimer cage in carbon nanopeapods: Tight-binding molecular dynamics simulation. Bull. Korean Chem. Soc. 2019, 40, 1054–1055. [Google Scholar] [CrossRef]
- Glukhova, O.E.; Kolesnikova, A.S.; Kirillova, I.V. Investigation of the effect of bending on the polymerization of fullerenes inside carbon nanotubes. Fuller. Nanotub. Carbon Nanostruct. 2012, 20, 391–394. [Google Scholar] [CrossRef]
- Ōsawa, S.; Sakai, M.; Ōsawa, E. Nature of cyclobutane bonds in the neutral [2+2] dimer of C60. J. Phys. Chem. A 1997, 101, 1378–1383. [Google Scholar] [CrossRef]
- Ōsawa, S.; Ōsawa, E.; Hirose, Y. Doubly bonded C60 dimers and congeners: Computational studies of structures, bond energies and transformations. Fuller. Sci. Technol. 1995, 3, 565–585. [Google Scholar] [CrossRef]
- Bihlmeier, A.; Samson, C.C.M.; Klopper, W. DFT study of fullerene dimers. ChemPhysChem 2005, 6, 2625–2632. [Google Scholar] [CrossRef]
- Sabirov, D.S. Polarizability of C60 fullerene dimer and oligomers: The unexpected enhancement and its use for rational design of fullerene-based nanostructures with adjustable properties. RSC Adv. 2013, 3, 19430–19439. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Terentyev, A.O.; Bulgakov, R.G. Polarizability of fullerene [2 + 2]-dimers: A DFT study. Phys. Chem. Chem. Phys. 2014, 16, 14594–14600. [Google Scholar] [CrossRef] [PubMed]
- Pankratyev, E.Y.; Tukhbatullina, A.A.; Sabirov, D.S. Dipole polarizability, structure, and stability of [2+2]-linked fullerene nanostructures (C60)n (n ≤ 7). Phys. E 2017, 86, 237–242. [Google Scholar] [CrossRef]
- Ma, F.; Li, Z.-R.; Zhou, Z.-J.; Wu, D.; Li, Y.; Wang, Y.-F.; Li, Z.-S. Modulated nonlinear optical responses and charge transfer transition in endohedral fullerene dimers Na@C60C60@F with n-fold covalent bond (n = 1, 2, 5, and 6) and long range ion bond. J. Phys. Chem. C 2010, 114, 11242–11247. [Google Scholar] [CrossRef]
- Tukhbatullina, A.A.; Shepelevich, I.S.; Sabirov, D.S. Exaltation of polarizability as a common property of fullerene dimers with diverse intercage bridges. Fuller. Nanotub. Carbon Nanostruct. 2018, 26, 661–666. [Google Scholar] [CrossRef]
- Kaur, S.; Sharma, A.; Sharma, H.; Dhiman, S.; Mudahar, I. Substitutional doping of symmetrical small fullerene dimers. Int. J. Quantum Chem. 2019, 119, e26019. [Google Scholar] [CrossRef]
- Swart, M.; van Duijnen, P.T. Rapid determination of polarizability exaltation in fullerene-based nanostructures. J. Mater. Chem. C 2015, 3, 23–25. [Google Scholar] [CrossRef]
- Silva, R.A.L.; de Brito, S.F.; Machado, D.F.S.; Carvalho-Silva, V.H.; de Oliveira, H.C.B.; Ribeiro, L. The influence of the configuration of the (C70)2 dimer on its rovibrational spectroscopic properties: A theoretical survey. J. Mol. Model. 2018, 24, 235. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Wirz, L.N.; Avery, J. The topology of fullerenes. WIRES Comput. Mol. Sci. 2015, 5, 96–145. [Google Scholar] [CrossRef]
- Sure, R.; Hansen, A.; Schwerdtfeger, P.; Grimme, S. Comprehensive theoretical study of all 1812 C60 isomers. Phys. Chem. Chem. Phys. 2017, 19, 14296–14305. [Google Scholar] [CrossRef]
- Ori, O.; Cataldo, F.; Graovac, A. Topological ranking of C28 fullerenes reactivity. Fuller. Nanotub. Carbon Nanostruct. 2009, 17, 308–323. [Google Scholar] [CrossRef]
- Vukicevic, D.; Cataldo, F.; Ori, O.; Graovac, A. Topological efficiency of C66 fullerene. Chem. Phys. Lett. 2011, 501, 442–445. [Google Scholar] [CrossRef]
- Ori, O.; D’Mello, M. A topological study of the structure of the C76 fullerene. Chem. Phys. Lett. 1992, 197, 49–54. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Ori, O.; Laszlo, I. Isomers of the C84 fullerene: A theoretical consideration within energetic, structural, and topological approaches. Fuller. Nanotub. Carbon Nanostruct. 2018, 26, 100–110. [Google Scholar] [CrossRef]
- Dobrynin, A.A.; Ori, O.; Putz, M.V.; Vesnin, A.Y. Generalized topological efficiency—Case study with C84 fullerene. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 545–550. [Google Scholar] [CrossRef]
- Bultheel, A.; Ori, O. Topological modeling of 1-Pentagon carbon nanocones—Topological efficiency and magic sizes. Fuller. Nanotub. Carbon Nanostruct. 2018, 26, 291–302. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Ori, O. Skeletal rearrangements of the C240 fullerene: Efficient topological descriptors for monitoring Stone–Wales transformations. Mathematics 2020, 8, 968. [Google Scholar] [CrossRef]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Chemcraft. Available online: http://www.chemcraftprog.com (accessed on 7 October 2021).
- Stück, D.; Baker, T.A.; Zimmerman, P.; Kurlancheek, W.; Head-Gordon, M. On the nature of electron correlation in C60. J. Chem. Phys. 2011, 135, 194306. [Google Scholar] [CrossRef]
- Chai, J.-D. Density functional theory with fractional orbital occupations. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef]
- Chai, J.-D. Thermally-assisted-occupation density functional theory with generalized-gradient approximations. J. Chem. Phys. 2014, 140, 18A521. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D. Role of exact exchange in thermally-assisted-occupation density functional theory: A proposal of new hybrid schemes. J. Chem. Phys. 2017, 146, 044102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheka, E.F.; Zaets, V.A. The radical nature of fullerene and its chemical activity. Russ. J. Phys. Chem. A 2005, 79, 2009–2015. [Google Scholar]
- Tuktarov, A.R.; Akhmetov, A.R.; Sabirov, D.S.; Khalilov, L.M.; Ibragimov, A.G.; Dzhemilev, U.M. Catalytic [2+1] cycloaddtion of diazo compounds to [60] fullerene. Russ. Chem. Bull. 2009, 58, 1724–1730. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Terentyev, A.O.; Bulgakov, R.G. Counting the isomers and estimation of anisotropy of polarizability of the selected C60 and C70 bisadducts promising for organic solar cells. J. Phys. Chem. A 2015, 119, 10697–10705. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.V. Stereochemistry of simple molecules inside nanotubes and fullerenes: Unusual behavior of usual systems. Molecules 2020, 25, 2437. [Google Scholar] [CrossRef]
- Diniakhmetova, D.R.; Friesen, A.K.; Kolesov, S.V. Quantum chemical modeling of the addition reactions of 1-n-phenylpropyl radicals to C60 fullerene. Int. J. Quantum Chem. 2016, 116, 489–496. [Google Scholar] [CrossRef]
- Shestakov, A.F. Role of fullerene–nitrogen complexes of alkali metals in C60-catalyzed nitrogen fixation. Russ. J. Phys. Chem. A 2020, 94, 919–924. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Tulyabaev, A.R.; Khalilov, L.M. How reliable are GIAO calculations of 1H and 13C NMR chemical shifts? A statistical analysis and empirical corrections at DFT (PBE/3z) level? J. Comput. Chem. 2011, 32, 1993–1997. [Google Scholar] [CrossRef]
- Iranmanesh, A.; Ashrafi, A.R.; Graovac, A.; Cataldo, F.; Ori, O. Wiener index role in topological modeling of hexagonal systems–from fullerenes to graphene. In Distance in Molecular Graphs—Applications; Gutman, I., Furtula, B., Eds.; University Kragujevac: Kragujevac, Serbia, 2012; pp. 135–155. [Google Scholar]
- Ōsawa, E. Formation mechanism of C60 under nonequilibrium and irreversible conditions—An annotation. Fuller. Nanotub. Carbon Nanostruct. 2012, 20, 299–309. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Bulgakov, R.G.; Khursan, S.L. Indices of the fullerene reactivity. ARKIVOC 2011, 2011, 200–224. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, D.S.; Khursan, S.L.; Bulgakov, R.G. 1,3-Dipolar addition reactions to fullerenes: The role of the local curvature of carbon surface. Russ. Chem. Bull. 2008, 57, 2520–2525. [Google Scholar] [CrossRef]
- Graovac, A.; Ashrafi, A.R.; Ori, O. Topological efficiency approach to fullerene stability—Case study with C50. Adv. Math. Chem. Appl. 2015, 2, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Chen, Z. Curved pi-conjugation, aromaticity, and the related chemistry of small fullerenes (<C60) and single-walled carbon nanotubes. Chem. Rev. 2005, 105, 3643–3696. [Google Scholar] [CrossRef] [PubMed]
- Bonchev, D.; Mekenyan, O. A topological approach to the calculation of the π-electron energy and energy gap of infinite conjugated polymers. Z. Nat. A 1980, 35, 739–747. [Google Scholar] [CrossRef]
- Cataldo, F.; Ori, O.; Iglesias-Groth, S. Topological lattice descriptors of graphene sheets with fullerene-like nanostructures. Mol. Simul. 2010, 36, 341–353. [Google Scholar] [CrossRef]
- Sabirov, D.S.; Terentyev, A.O.; Sokolov, V.I. Activation energies and information entropies of helium penetration through fullerene walls. Insights into the formation of endofullerenes nX@C60/70 (n = 1 and 2) from the information entropy approach. RSC Adv. 2016, 6, 72230–72237. [Google Scholar] [CrossRef] [Green Version]
- Ghobani, M.; Dehmer, M.; Emmert-Streib, F. Properties of entropy-based topological measures of fullerenes. Mathematics 2020, 8, 740. [Google Scholar] [CrossRef]
- Sabirov, D.S. Information entropy changes in chemical reactions. Comput. Theor. Chem. 2018, 1123, 167–179. [Google Scholar] [CrossRef]
- Sabirov, D.S. Information entropy change in [2+2]-oligomerization of the C60 fullerene. Int. J. Chem. Model. 2017, 9, 203–213. [Google Scholar]
- Feng, B.; Zhuang, X. Carbon-enriched meso-entropy materials: From theory to cases. Acta Chim. Sin. 2020, 78, 833–847. [Google Scholar] [CrossRef]
- Diana, N.; Yamada, Y.; Gohda, S.; Ono, H.; Kubo, S.; Sato, S. Carbon materials with high pentagon density. J. Mater. Sci. 2021, 56, 2912–2943. [Google Scholar] [CrossRef]
- Champion, Y.; Thurieau, N. The sample size effect in metallic glass deformation. Sci. Rep. 2020, 10, 10801. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isomer | ΔE (kJ/mol) | W | M | ρ | ρE | ||
|---|---|---|---|---|---|---|---|
| С120 (D5) | −1170.1 | 48,820 | 15 | 366 | 460 | 1.1116 | 1.2568 |
| С120 (Ci) | −149.2 | 49,362 | 16 | 333 | 497 | 1.2353 | 1.4925 |
| (C60)2 (С2h) | +11.4 | 51,912 | 18 | 308 | 548 | 1.4045 | 1.7793 |
| n | Symmetry Point Group | σ | W | ρ | ρE | h (Bits) |
|---|---|---|---|---|---|---|
| 2 | D2h | 4 | 51,912 | 1.4045 | 1.7792 | 3.974 |
| 3 | D2h | 4 | 163,116 | 1.2639 | 1.7113 | 4.603 |
| 4 | D2h | 4 | 374,352 | 1.3493 | 1.8824 | 4.974 |
| 5 | D2h | 4 | 718,020 | 1.3043 | 1.8501 | 5.322 |
| 6 | D2h | 4 | 1,226,520 | 1.3392 | 1.9198 | 5.559 |
| n | Symmetry Point Group | σ | W | ρ | ρE | h (Bits) |
|---|---|---|---|---|---|---|
| 3 | CS | 1 | 149,004 | 1.4150 | 1.9231 | 6.559 |
| 4 | C2h | 2 | 319,776 | 1.4451 | 1.9892 | 5.974 |
| 5 | CS | 1 | 582,948 | 1.4277 | 2.0360 | 7.295 |
| 6 | C2h | 2 | 960,120 | 1.4339 | 2.0323 | 6.559 |
| 7 | CS | 1 | 1,468,572 | 1.4173 | 2.0580 | 7.781 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabirov, D.S.; Ori, O.; Tukhbatullina, A.A.; Shepelevich, I.S. Covalently Bonded Fullerene Nano-Aggregates (C60)n: Digitalizing Their Energy–Topology–Symmetry. Symmetry 2021, 13, 1899. https://doi.org/10.3390/sym13101899
Sabirov DS, Ori O, Tukhbatullina AA, Shepelevich IS. Covalently Bonded Fullerene Nano-Aggregates (C60)n: Digitalizing Their Energy–Topology–Symmetry. Symmetry. 2021; 13(10):1899. https://doi.org/10.3390/sym13101899
Chicago/Turabian StyleSabirov, Denis Sh., Ottorino Ori, Alina A. Tukhbatullina, and Igor S. Shepelevich. 2021. "Covalently Bonded Fullerene Nano-Aggregates (C60)n: Digitalizing Their Energy–Topology–Symmetry" Symmetry 13, no. 10: 1899. https://doi.org/10.3390/sym13101899
APA StyleSabirov, D. S., Ori, O., Tukhbatullina, A. A., & Shepelevich, I. S. (2021). Covalently Bonded Fullerene Nano-Aggregates (C60)n: Digitalizing Their Energy–Topology–Symmetry. Symmetry, 13(10), 1899. https://doi.org/10.3390/sym13101899