Supramolecular Diversity of Oxabicyclo[2.2.2]octenes Formed between Substituted 2H-Pyran-2-ones and Vinyl-Moiety-Containing Dienophiles



Abstract
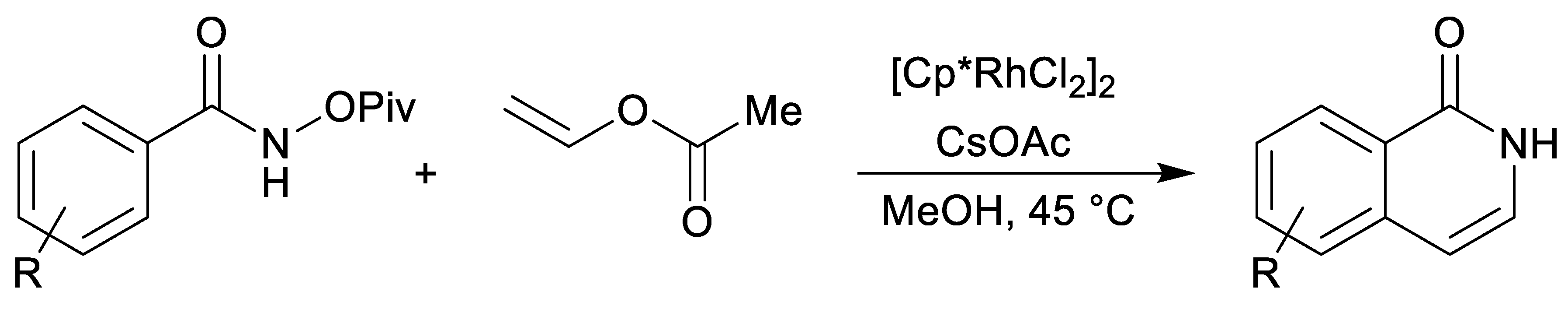
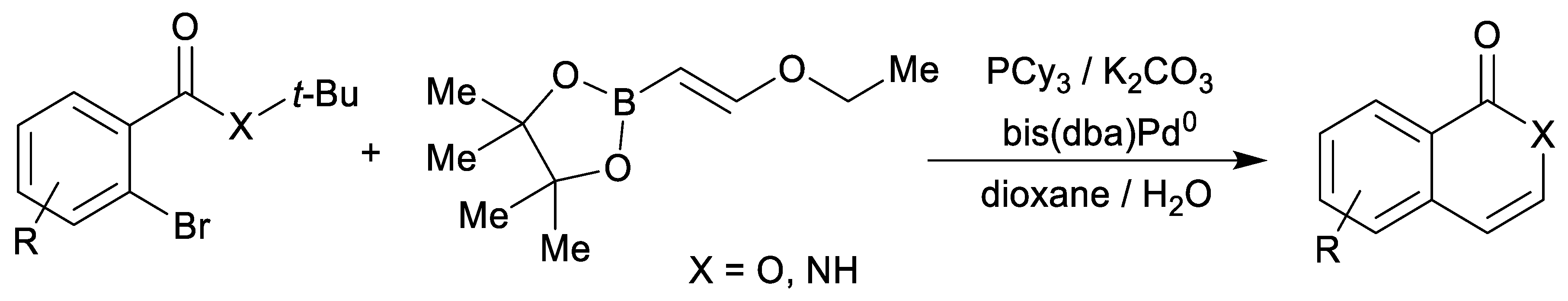
1. Introduction
2. Materials and Methods
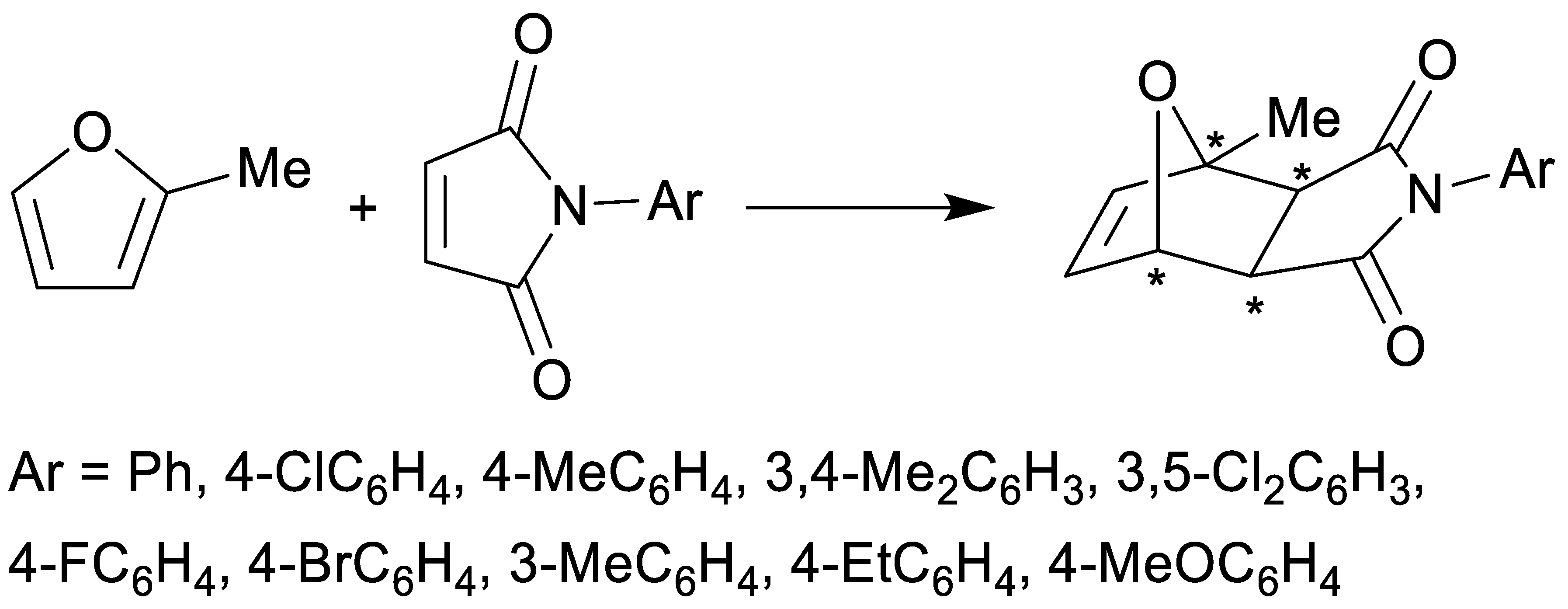
2.1. Synthesis
2.2. X-ray Single Crystal Analysis
3. Results
3.1. Chemistry
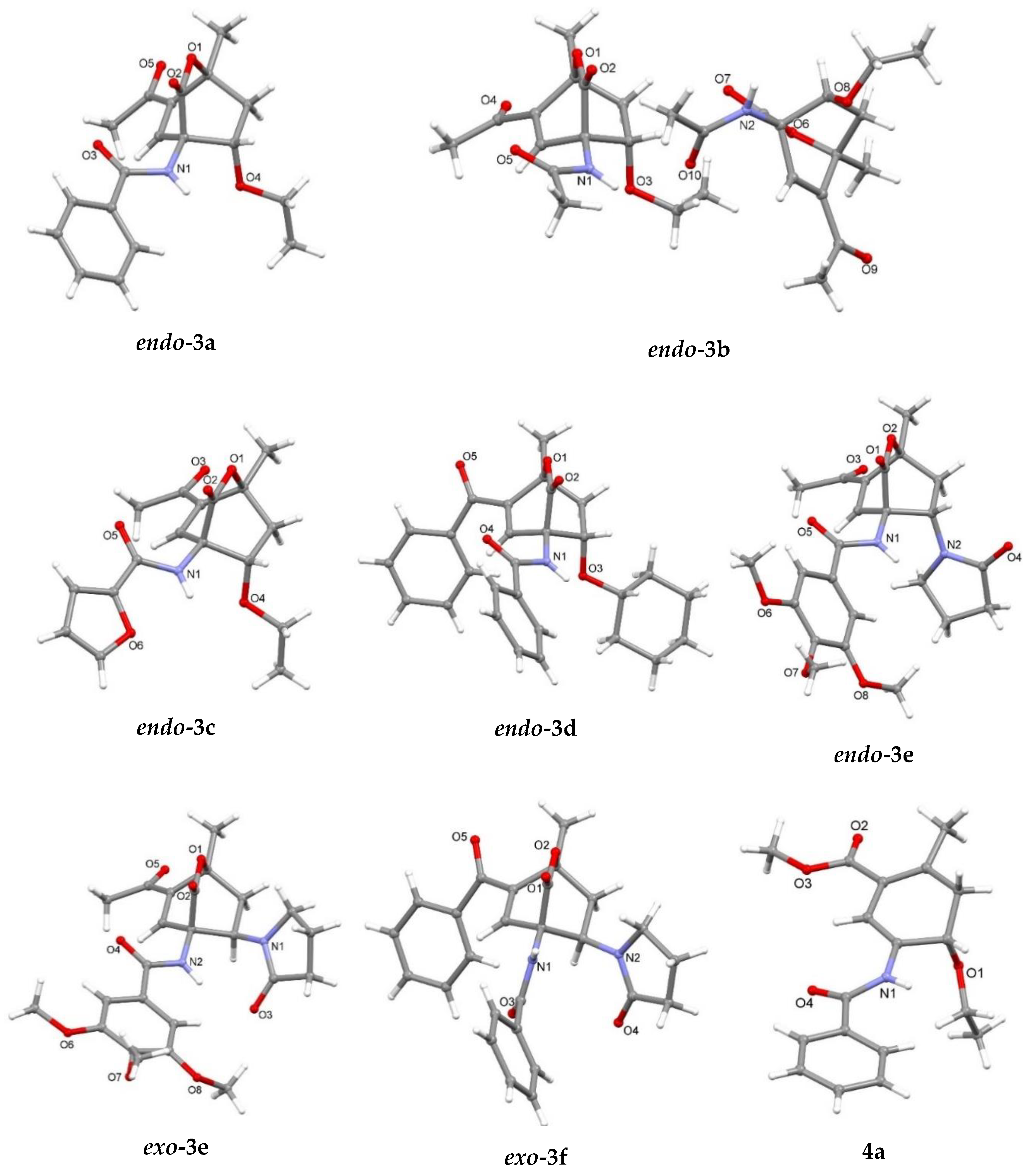
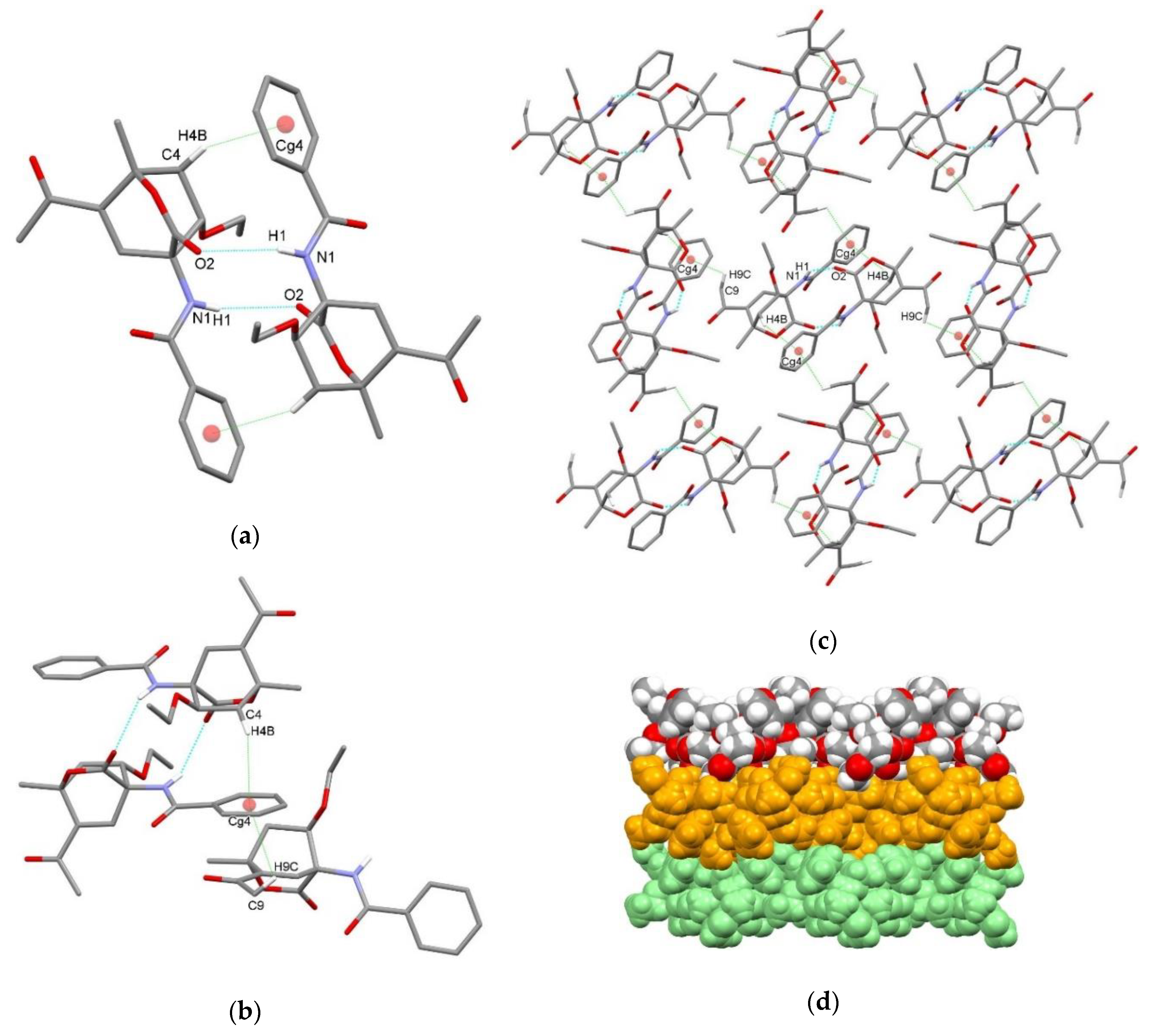
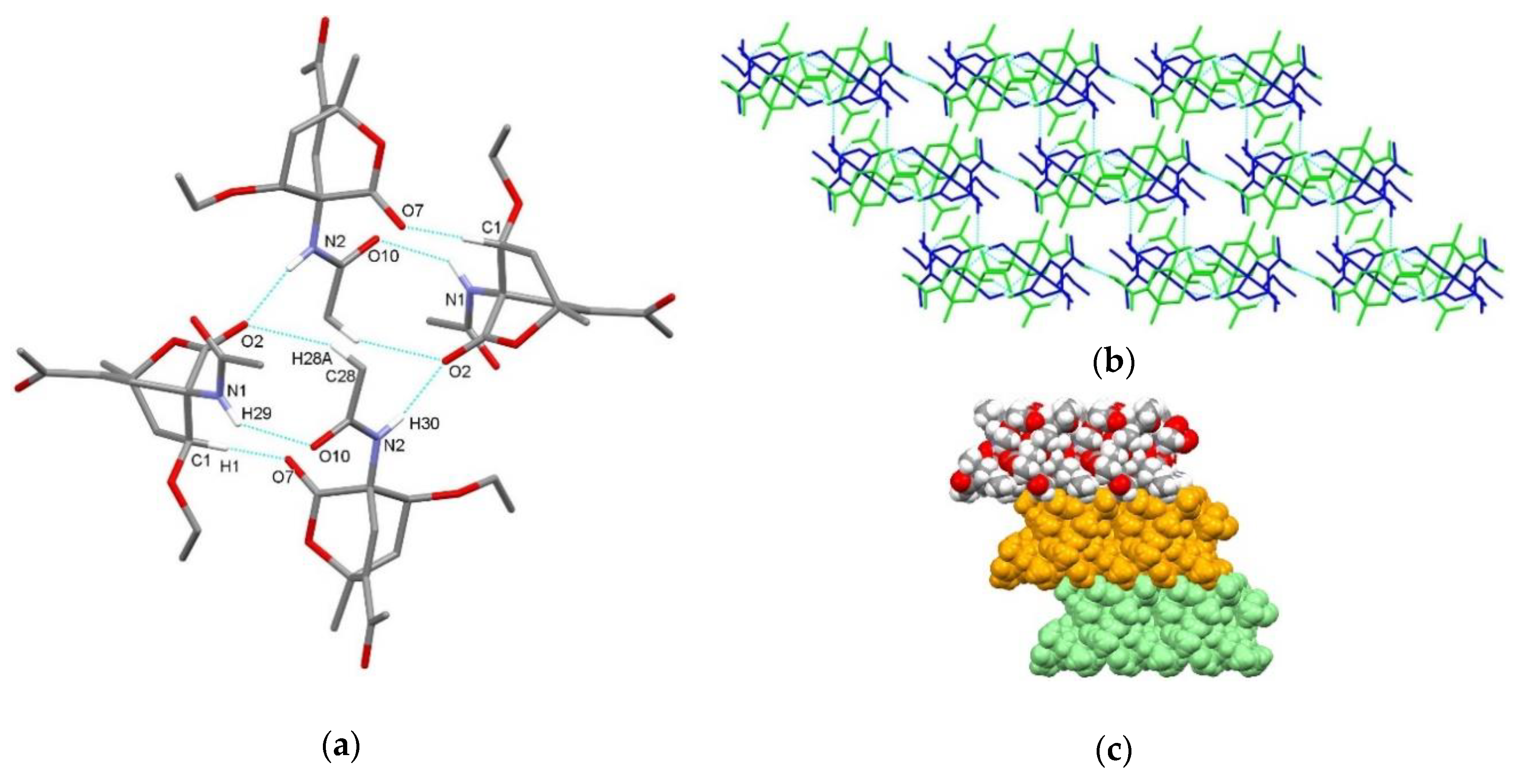
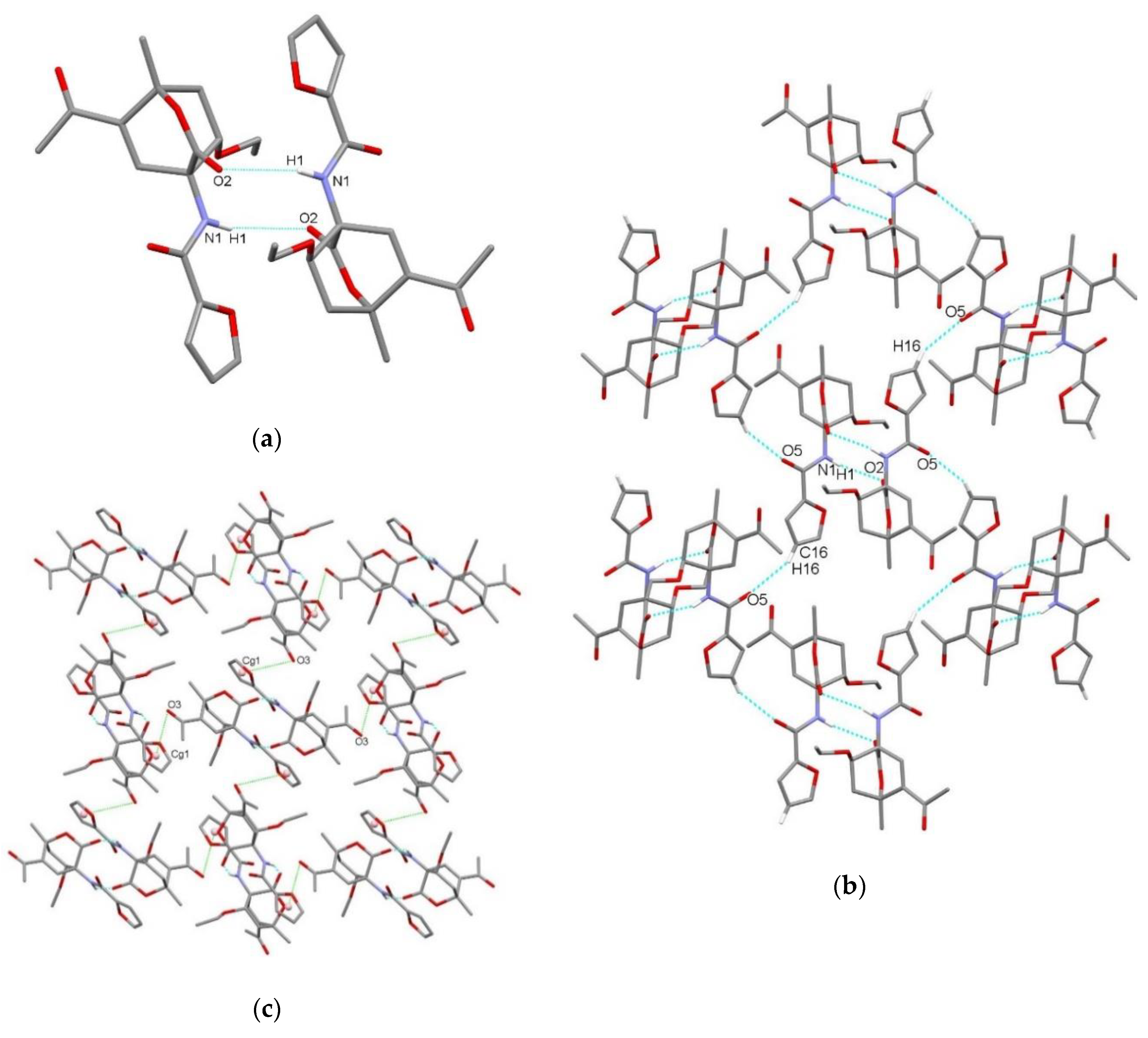
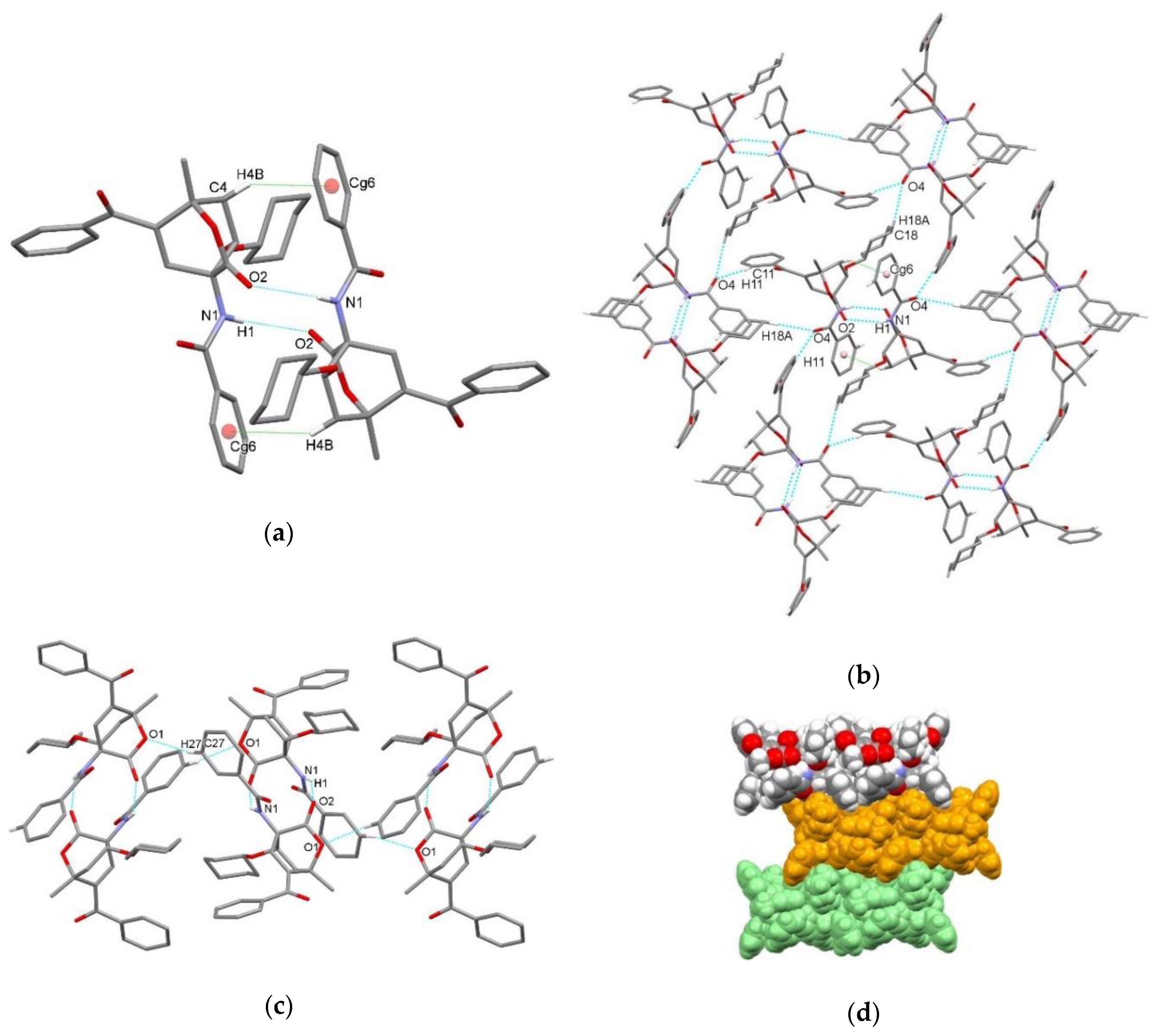
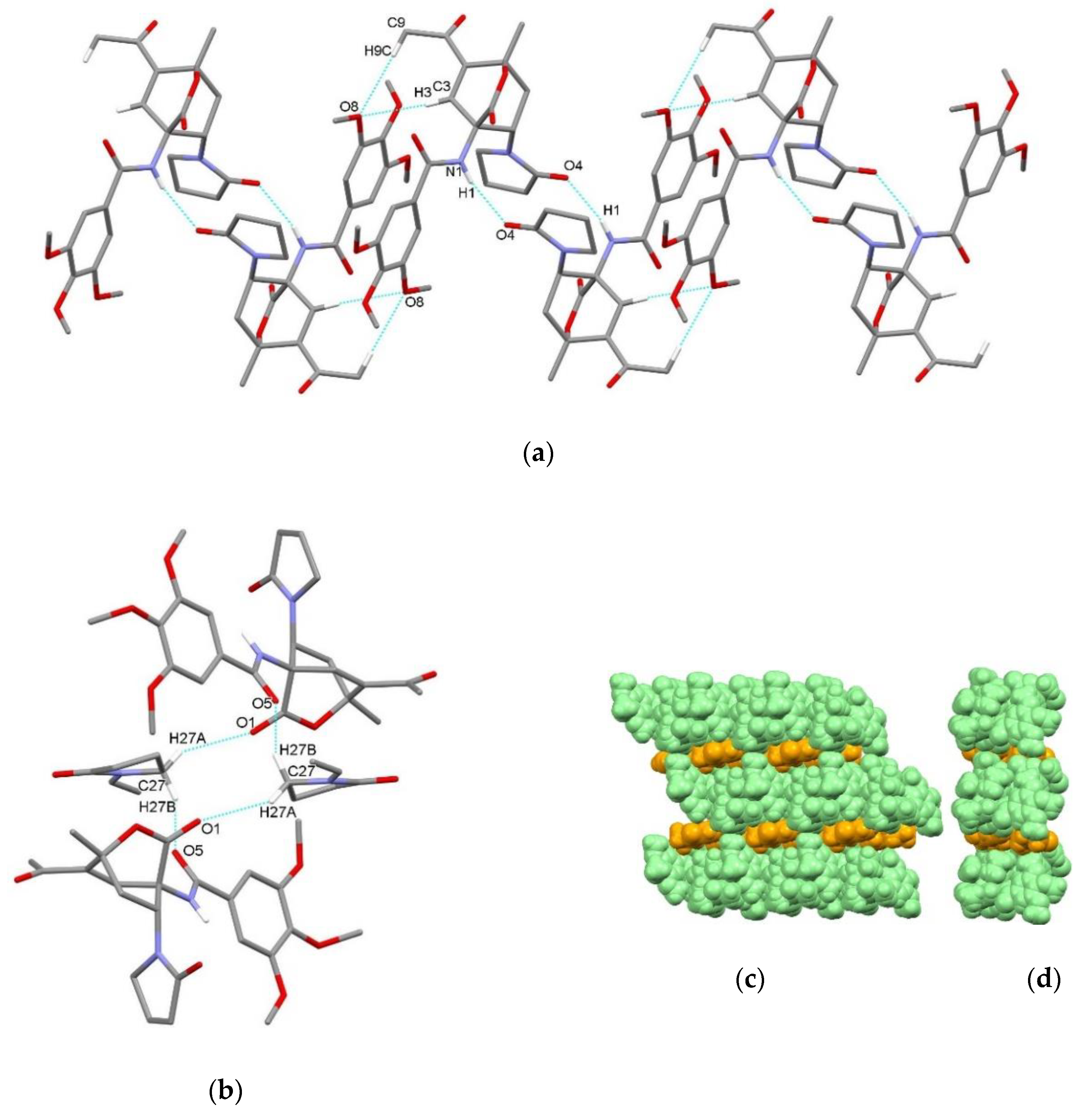
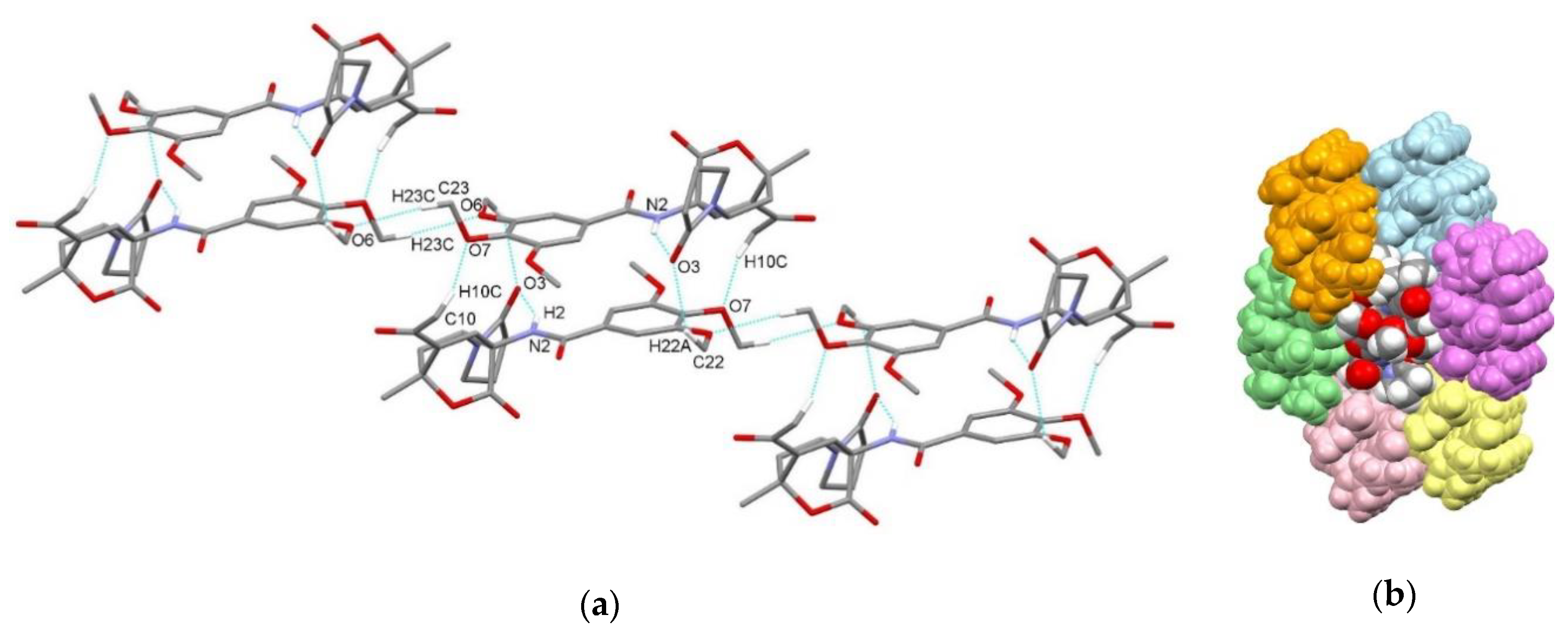
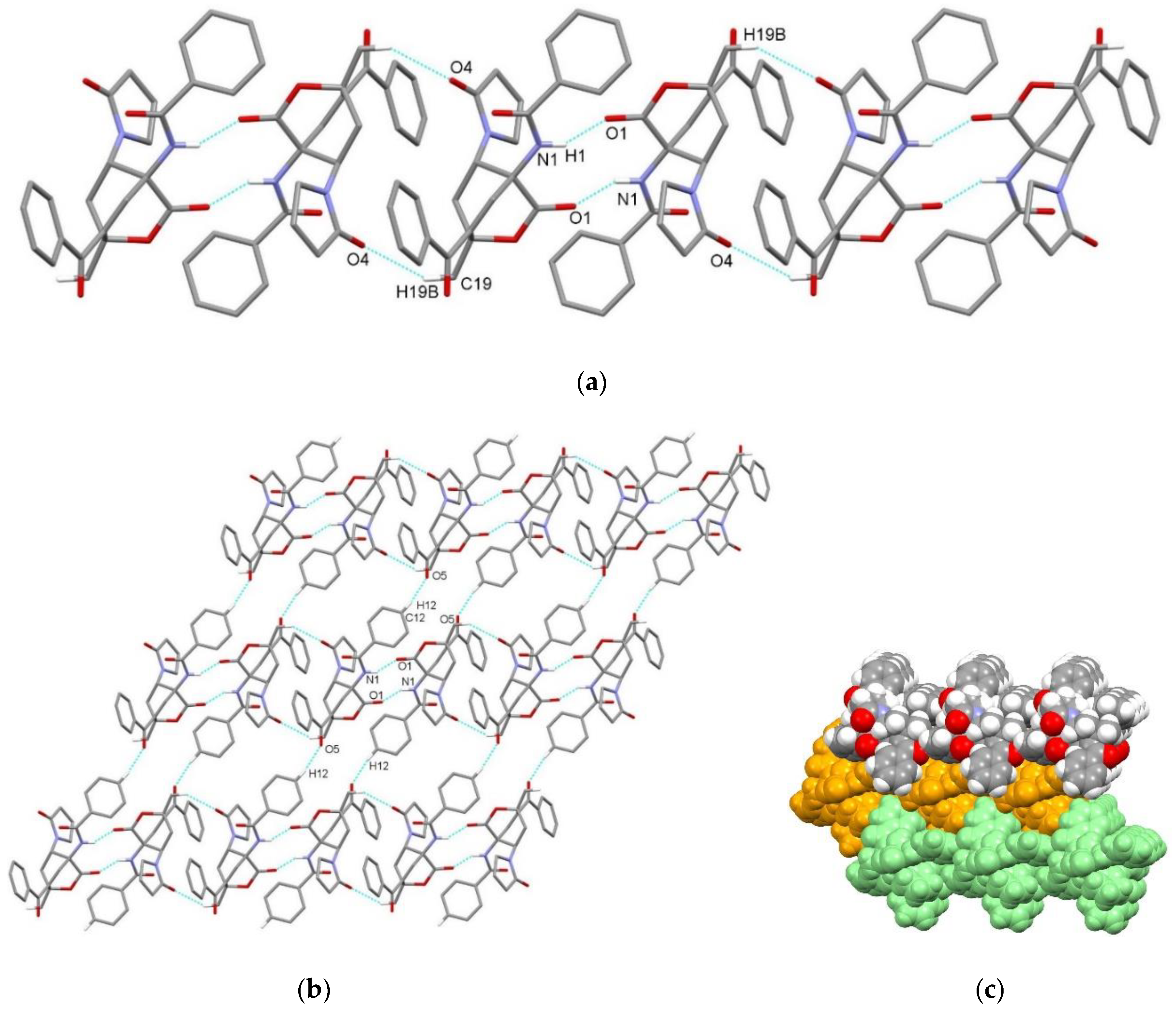
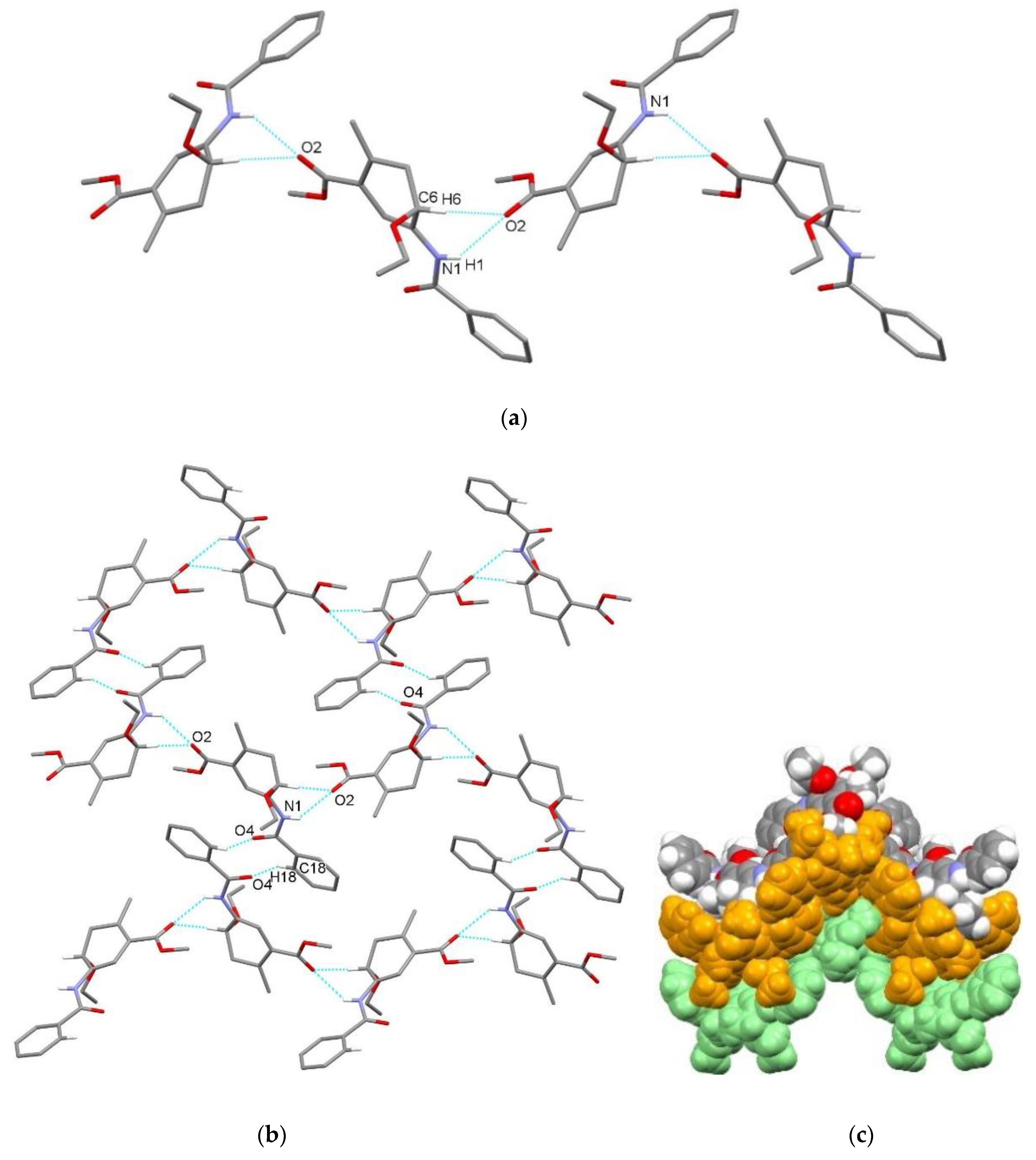
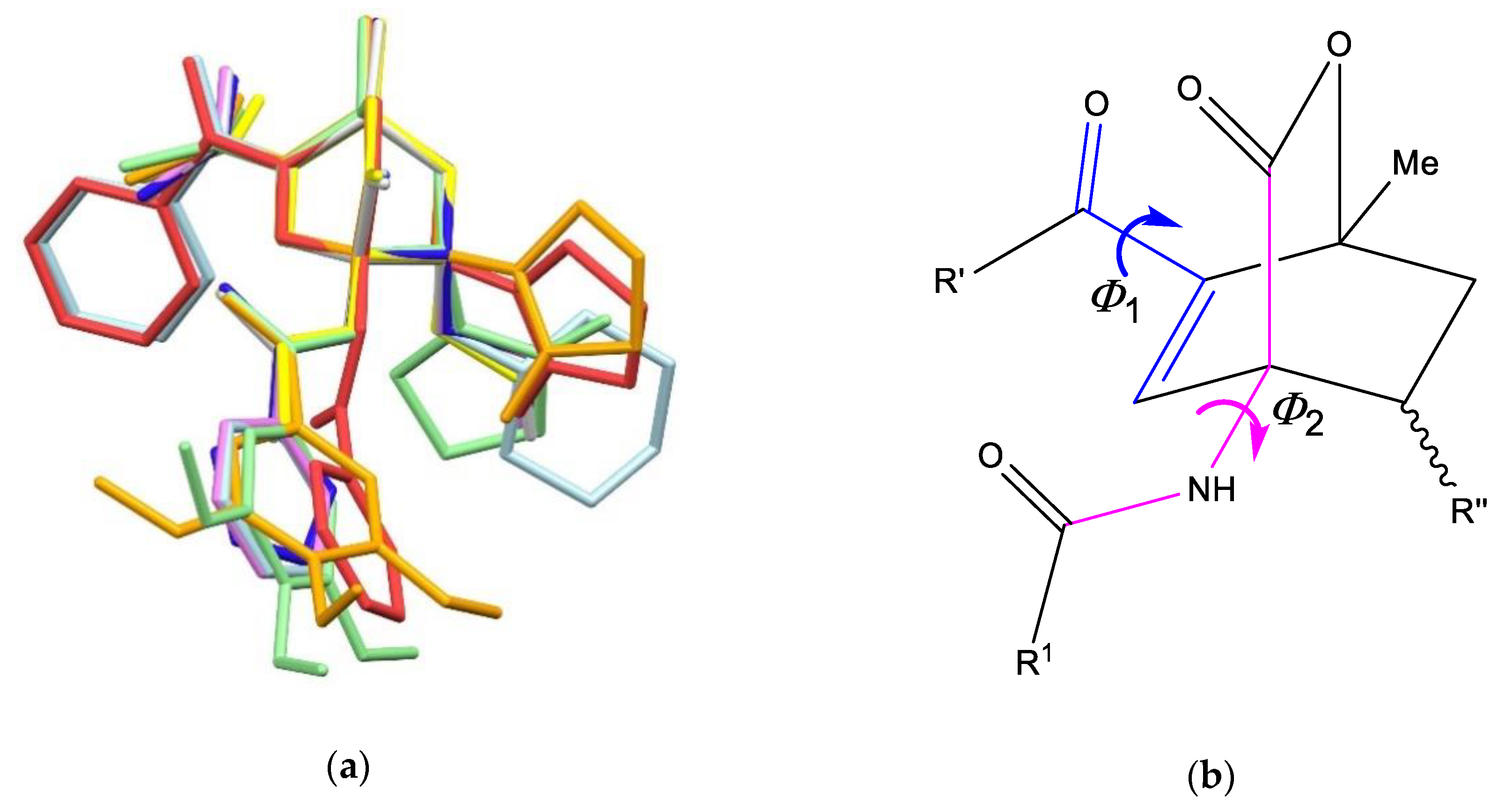
3.2. X-Ray Single Crystal Determination
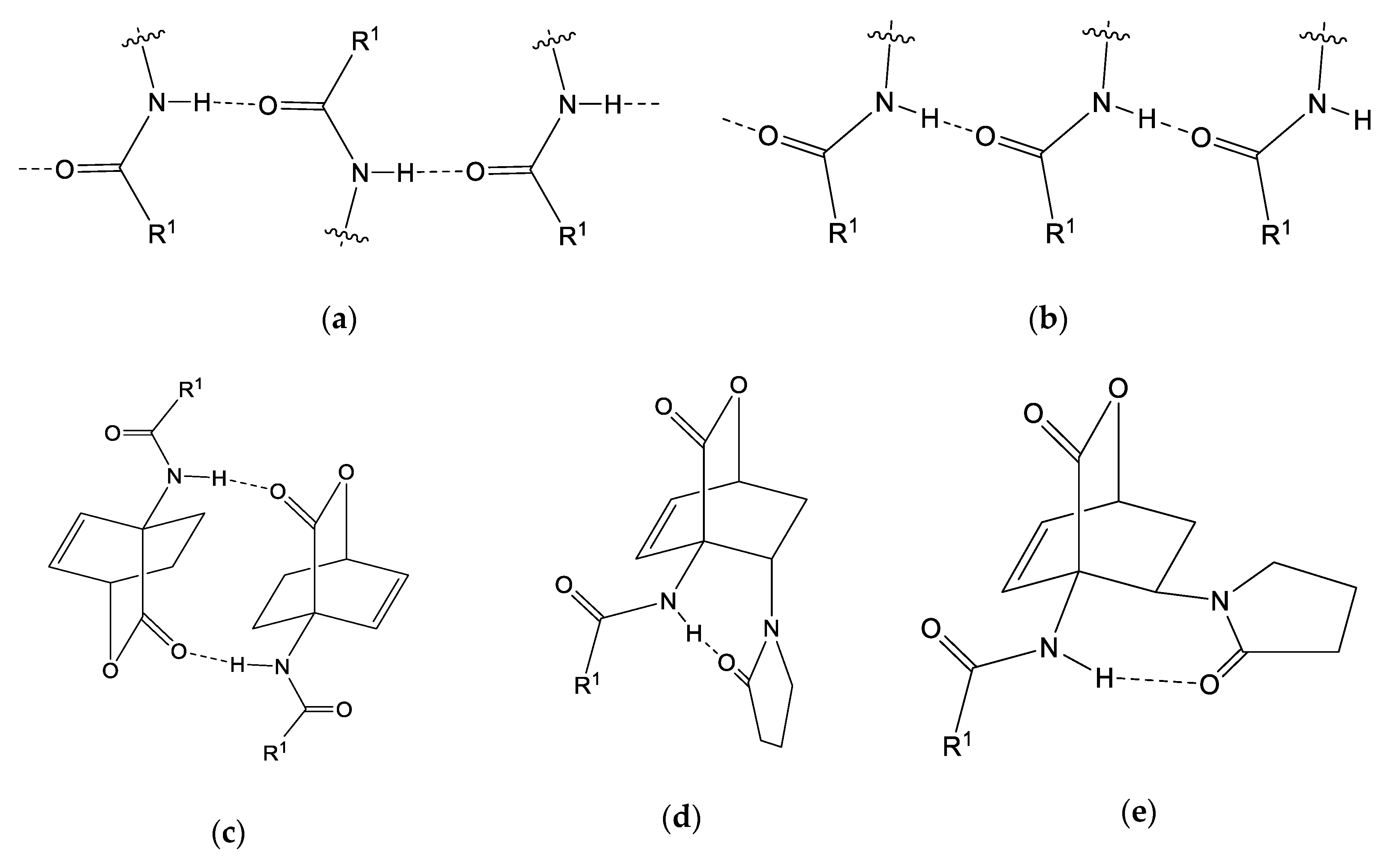
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dyan, O.T.; Borodin, G.I.; Zaikin, P.A. The Diels–Alder reaction for the synthesis of polycyclic aromatic compounds. Eur. J. Org. Chem. 2019, 7271–7306. [Google Scholar] [CrossRef]
- Mancini, P.M.E.; Ormachea, C.M.; Kneeteman, M.N. Polar Diels–Alder reactions under microwave irradiation employing different heterocyclic compounds as electrophiles. Mini-Rev. Org. Chem. 2019, 16, 527–543. [Google Scholar] [CrossRef]
- Harada, S.; Nishida, A. Catalytic and enantioselective Diels–Alder reactions of siloxydienes. Asian J. Org. Chem. 2019, 8, 732–745. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Asymmetric organocatalytic reactions of α,β-unsaturated cyclic ketones. Symmetry 2011, 3, 84–125. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Koprowski, M.; Drzazga, Z.; Parcheta, R.; Łastawiecka, E.; Demchuk, O.M.; Justyniak, I. Efficient oxidative resolution of 1-phenylphosphol-2-ene and Diels–Alder synthesis of enantiopure bicyclic and tricyclic P-stereogenic C-P heterocycles. Symmetry 2020, 12, 346. [Google Scholar] [CrossRef]
- Rádai, Z.; Bagi, P.; Czugler, M.; Karaghiosoff, K.; Keglevich, G. Optical resolution of dimethyl α-hydroxy-arylmethylphosphonates via diastereomer complex formation using calcium hydrogen O,O′-dibenzoyl-(2R,3R)-tartrate; X-ray analysis of the complexes and products. Symmetry 2020, 12, 758. [Google Scholar] [CrossRef]
- Soai, K.; Kawasaki, T.; Matsumoto, A. Role of asymmetric autocatalysis in the elucidation of origins of homochirality of organic compounds. Symmetry 2019, 11, 694. [Google Scholar] [CrossRef]
- Hoffmann, R.; Woodward, R.B. The conservation of orbital symmetry. Acc. Chem. Res. 1968, 1, 17–22. [Google Scholar] [CrossRef]
- Woodward, R.B.; Hoffmann, R. Selection rules for concerted cycloaddition reactions. J. Am. Chem. Soc. 1965, 87, 2046–2048. [Google Scholar] [CrossRef]
- Sauer, J.; Sustmann, R. Mechanistic aspects of Diels–Alder reactions: A critical survey. Angew. Chem. Int. Ed. 1980, 19, 779–807. [Google Scholar] [CrossRef]
- Uemura, N.; Toyoda, S.; Shimizu, W.; Yoshida, Y.; Mino, T.; Sakamoto, M. Absolute asymmetric synthesis involving chiral symmetry breaking in Diels–Alder reaction. Symmetry 2020, 12, 910. [Google Scholar] [CrossRef]
- Kitagawa, D.; Bardeen, C.J.; Kobatake, S. Symmetry breaking and photomechanical behavior of photochromic organic crystals. Symmetry 2020, 12, 1478. [Google Scholar] [CrossRef]
- Ram, V.J.; Srivastava, P. Pyran-2-ones as a versatile synthon for the synthesis of diverse arenes and heteroarenes. Curr. Org. Chem. 2001, 5, 571–599. [Google Scholar] [CrossRef]
- Afarinkia, K.; Vinader, V.; Nelson, T.D.; Posner, G.H. Diels–Alder cycloadditions of 2-pyrones and 2-pyridones. Tetrahedron 1992, 48, 9111–9171. [Google Scholar] [CrossRef]
- Štefane, B.; Perdih, A.; Pevec, A.; Šolmajer, T.; Kočevar, M. The participation of 2H-pyran-2-ones in [4 + 2] cycloadditions: An experimental and computational study. Eur. J. Org. Chem. 2010, 5870–5883. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. Ethyl vinyl ether as a synthetic equivalent of acetylene in a DABCO-catalyzed microwave-assisted Diels–Alder–elimination reaction sequence starting from 2H-pyran-2-ones. Synlett 2008, 2613–2616. [Google Scholar] [CrossRef]
- Juranovič, A.; Kranjc, K.; Perdih, F.; Polanc, S.; Kočevar, M. Comparison of the reaction pathways and intermediate products of a microwave-assisted and high-pressure-promoted cycloaddition of vinyl-moiety-containing dienophiles on 2H-pyran-2-ones. Tetrahedron 2011, 67, 3490–3500. [Google Scholar] [CrossRef]
- Cole, C.J.F.; Fuentes, L.; Snyder, S.A. Asymmetric pyrone Diels–Alder reactions enabled by dienamine catalysis. Chem. Sci. 2020, 11, 2175–2180. [Google Scholar] [CrossRef]
- Geist, E.; Berneaud-Kötz, H.; Baikstis, T.; Dräger, G.; Kirschning, A. Toward chromanes by de novo construction of the benzene ring. Org. Lett. 2019, 21, 8930–8933. [Google Scholar] [CrossRef]
- Grant, P.S.; Brimble, M.A.; Furkert, D.P. Synthesis of the bicyclic lactone core of leonuketal, enabled by a telescoped Diels–Alder reaction sequence. Chem. Asian J. 2019, 14, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Kondratov, I.S.; Tolmachova, N.A.; Dolovanyuk, V.G.; Gerus, I.I.; Daniliuc, C.-G.; Haufe, G. Synthesis of trifluoromethyl-containing polysubstituted aromatic compounds by Diels–Alder reaction of ethyl 3-benzamido-2-oxo-6-(trifluoromethyl)-2H-pyran-5-carboxylate. Eur. J. Org. Chem. 2015, 2482–2491. [Google Scholar] [CrossRef]
- Kondratov, I.S.; Tolmachova, N.A.; Dolovanyuk, V.G.; Gerus, I.I.; Bergander, K.; Daniliuc, C.-G.; Haufe, G. Diels–Alder reaction of ethyl 3-benzamido-2-oxo-6-(trifluoromethyl)-2H-pyran-5-carboxylate with alkoxyalkenes as an effective approach to trifluoromethyl-containing 3-aminobenzoic acid derivatives. Eur. J. Org. Chem. 2014, 2443–2450. [Google Scholar] [CrossRef]
- Burch, P.; Binaghi, M.; Scherer, M.; Wentzel, C.; Bossert, D.; Eberhardt, L.; Neuburger, M.; Scheiffele, P.; Gademann, K. Total synthesis of gelsemiol. Chem. Eur. J. 2013, 19, 2589–2591. [Google Scholar] [CrossRef] [PubMed]
- Bartelson, K.J.; Singh, R.P.; Foxman, B.M.; Deng, L. Catalytic asymmetric [4 + 2] additions with aliphatic nitroalkenes. Chem. Sci. 2011, 2, 1940–1944. [Google Scholar] [CrossRef]
- Afarinkia, K.; Abdullahi, M.H.; Scowen, I.J. A new, general method for the synthesis of carbasugar-sugar pseudodisaccharides. Org. Lett. 2009, 11, 5182–5184. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Wang, Y.-Q.; Liu, Y.; Foxman, B.M.; Deng, L. Asymmetric Diels–Alder reactions of 2-pyrones with a bifunctional organic catalyst. J. Am. Chem. Soc. 2007, 129, 6364–6365. [Google Scholar] [CrossRef]
- Shimo, T.; Yasutake, M.; Shinmyozu, T.; Somekawa, K. Crystal structure of methyl endo-8-cyano-exo-8-methyl-3-oxo-2-oxabicyclo[2.2.2]oct-5-ene-6-carboxylate. Anal. Sci. 2003, 19, 471–472. [Google Scholar] [CrossRef]
- Zhang, H.; Appels, D.C.; Hockless, D.C.R.; Mander, L.N. A new approach to the total synthesis of the unusual diterpenoid tropone, harringtonolide. Tetrahedron Lett. 1998, 39, 6577–6580. [Google Scholar] [CrossRef]
- Hren, J.; Polanc, S.; Kočevar, M. The synthesis and transformations of fused bicyclo[2.2.2]octenes. Arkivoc 2008, i, 209–231. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. Green chemistry starting from 2H-pyran-2-one derivatives. Curr. Green Chem. 2014, 1, 202–215. [Google Scholar] [CrossRef]
- Kranjc, K.; Kočevar, M. Regio- and stereoselective syntheses and cycloadditions of substituted 2H-pyran-2-ones and their fused derivatives. Arkivoc 2013, i, 333–363. [Google Scholar] [CrossRef]
- Kranjc, K.; Polanc, S.; Kočevar, M. Diels–Alder reactions of fused pyran-2-ones with maleimides: Efficient syntheses of benz[e]isoindoles and related systems. Org. Lett. 2003, 5, 2833–2836. [Google Scholar] [CrossRef]
- Kranjc, K.; Perdih, F.; Kočevar, M. Effect of ring size on the exo/endo selectivity of a thermal double cycloaddition of fused pyran-2-ones. J. Org. Chem. 2009, 74, 6303–6306. [Google Scholar] [CrossRef] [PubMed]
- Krivec, M.; Gazvoda, M.; Kranjc, K.; Polanc, S.; Kočevar, M. A way to avoid using precious metals: The application of high-surface activated carbon for the synthesis of isoindoles via the Diels–Alder reaction of 2H-pyran-2-ones. J. Org. Chem. 2012, 77, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Gladysz, J.A.; Lee, S.J.; Tomasello, J.A.V.; Yu, Y.S. High-pressure cycloadditions of pyrones: Synthesis of highly functionalized six-membered rings by inhibition of carbon dioxide loss. J. Org. Chem. 1977, 42, 4171–4172. [Google Scholar] [CrossRef]
- Klärner, F.-G.; Breitkopf, V. The effect of pressure on retro Diels–Alder reactions. Eur. J. Org. Chem. 1999, 2757–2762. [Google Scholar] [CrossRef]
- Uroos, M.; Pitt, P.; Harwood, L.M.; Lewis, W.; Blake, A.J.; Hayes, C.J. Total synthesis of (–)-aritasone via the ultra-high pressure hetero-Diels–Alder dimerisation of (–)-pinocarvone. Org. Biomol. Chem. 2017, 15, 8523–8528. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.K.; Prasad, D.L.V.K.; Yadav, A.; Yadav, K. On the solvent- and temperature-driven stereoselectivity of the Diels–Alder cycloaddition reactions of furan with maleic anhydride and maleimide. J. Phys. Org. Chem. 2020, e4131. [Google Scholar] [CrossRef]
- Tsypysheva, I.P.; Borisevich, S.S.; Lobov, A.N.; Kovalskaya, A.V.; Shamukaev, V.V.; Safiullin, R.L.; Khursan, S.L. Inversion of diastereoselectivity under high pressure conditions: Diels–Alder reactions of 12-N-substituted derivatives of (–)-cytisine with N-phenylmaleimide. Tetrahedron Asymmetry 2015, 26, 732–737. [Google Scholar] [CrossRef]
- Webb, J.N.; Marsden, P.S.; Raw, A.S. Rhodium(III)-catalyzed C–H activation/annulation with vinyl esters as an acetylene equivalent. Org. Lett. 2014, 16, 4718. [Google Scholar] [CrossRef]
- Toure, M.; Jaime-Figueroa, S.; Burslem, G.M.; Crews, C.M. Expeditious synthesis of isoquinolones and isocoumarins with a vinyl borane as an acetylene equivalent. Eur. J. Org. Chem. 2016, 4171–4175. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Pillars of crystal engineering: Crystal energies and symmetry operators. CrystEngComm 2018, 20, 2511–2518. [Google Scholar] [CrossRef]
- Lengauer, H.; Makuc, D.; Šterk, D.; Perdih, F.; Pichler, A.; Trdan Lušin, T.; Plavec, J.; Časar, Z. Co-crystals, salts or mixtures of both? The case of tenofovir alafenamide fumarates. Pharmaceutics 2020, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, D.S.; Il’in, M.V.; Suslonov, V.V.; Novikov, A.S. Symmetrical noncovalent interactions Br⋯Br observed in crystal structure of exotic primary peroxide. Symmetry 2020, 12, 637. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Kappe, C.O. My twenty years in microwave chemistry: From kitchen ovens to microwaves that aren’t microwaves. Chem. Rec. 2019, 19, 15–39. [Google Scholar] [CrossRef]
- Sarotti, A.M.; Pisano, P.L.; Pellegrinet, S.C. A facile microwave-assisted Diels–Alder reaction of vinylboronates. Org. Biomol. Chem. 2010, 8, 5069–5073. [Google Scholar] [CrossRef]
- Tajti, Á.; Szatmári, E.; Perdih, F.; Keglevich, G.; Bálint, E. Microwave-assisted Kabachnik–Fields reaction with amino alcohols as the amine component. Molecules 2019, 24, 1640. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Afarinkia, K.; Abdullahi, M.H.; Scrowen, I.J. A synthesis of carbasugar–sugar pseudodisaccharides via a cycloaddition–cycloreversion reaction of 2H-pyran-2-ones. Org. Lett. 2010, 12, 5564–5566. [Google Scholar] [CrossRef]
- Drljaca, A.; Hubbard, C.D.; van Eldik, R.; Asano, T.; Basilevsky, M.V.; le Noble, W.J. Activation and reaction volumes in solution. 3. Chem. Rev. 1998, 98, 2167–2289. [Google Scholar] [CrossRef] [PubMed]
- Van Eldik, R.; Asano, T.; le Noble, W.J. Activation and reaction volumes in solution. 2. Chem. Rev. 1989, 89, 549–688. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
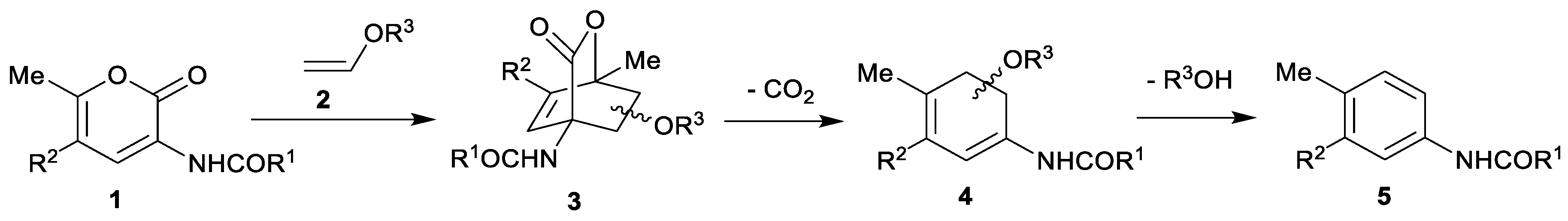{kind=link}
{kind=link}
{kind=link}
| endo-3a | endo-3b | endo-3c | endo-3d | exo-3e | endo-3e∙6a | exo-3f | 4a | |
|---|---|---|---|---|---|---|---|---|
| CCDC number | 2034987 | 2034988 | 2034989 | 2034990 | 2034991 | 2034992 | 2034993 | 2034994 |
| Formula | C19H21NO5 | C14H19NO5 | C17H19NO6 | C28H29NO5 | C24H28N2O8 | C30H37N3O9 | C26H24N2O5 | C18H21NO4 |
| Mr (g mol−1) | 343.37 | 281.3 | 333.33 | 459.52 | 472.48 | 583.63 | 444.47 | 315.36 |
| Crystal size (mm) | 0.50 × 0.38 × 0.25 | 0.25 × 0.25 × 0.25 | 0.70 × 0.50 × 0.50 | 0.45 × 0.25 × 0.20 | 0.35 × 0.10 × 0.10 | 0.50 × 0.13 × 0.13 | 0.60 × 0.30 × 0.18 | 0.50 × 0.20 × 0.20 |
| Crystal color | colorless | colorless | colorless | colorless | colorless | colorless | colorless | colorless |
| Crystal system | orthorhombic | triclinic | orthorhombic | monoclinic | monoclinic | triclinic | triclinic | orthorhombic |
| Space group | Pcab | P–1 | Pcab | C2/c | P21/n | P–1 | P–1 | Pcab |
| a (Å) | 13.6258(2) | 11.1004(9) | 13.2171(2) | 25.6982(6) | 10.1996(4) | 11.2314(3) | 9.2483(3) | 12.1827(3) |
| b (Å) | 15.0585(3) | 11.3611(8) | 14.2221(3) | 13.3577(2) | 19.6440(11) | 11.6094(3) | 11.4819(3) | 14.8081(4) |
| c (Å) | 17.0887(3) | 13.2166(9) | 17.1809(4) | 18.3592(4) | 12.5325(6) | 12.1689(3) | 12.1508(3) | 18.6214(4) |
| α (°) | 90 | 65.350(3) | 90 | 90 | 90 | 85.027(2) | 112.763(2) | 90 |
| β (°) | 90 | 89.871(4) | 90 | 127.2900(10) | 113.734(3) | 73.439(2) | 93.341(2) | 90 |
| γ (°) | 90 | 81.713(3) | 90 | 90 | 90 | 78.714(2) | 106.199(2) | 90 |
| V (Å3) | 3506.33(11) | 1495.97(19) | 3229.58(11) | 5013.85(18) | 2298.65(19) | 1490.66(7) | 1122.23(5) | 3359.35(14) |
| Z | 8 | 4 | 8 | 8 | 4 | 2 | 2 | 8 |
| T (K) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) |
| Dc (g cm−3) | 1.301 | 1.249 | 1.371 | 1.218 | 1.365 | 1.300 | 1.315 | 1. 247 |
| F(000) | 1456 | 600 | 1408 | 1952 | 1000 | 620 | 468 | 1344 |
| Reflections collected | 7555 | 6695 | 6922 | 10443 | 8146 | 12041 | 8995 | 7151 |
| Rint | 0.0132 | 0.0459 | 0.0143 | 0.0192 | 0.0551 | 0.0232 | 0.0271 | 0.0285 |
| Data/restraints/parameters | 3990/0/230 | 3976/38/402 | 3663/0/221 | 5708/0/309 | 4170/0/313 | 6796/0/385 | 5103/0/300 | 3783/0/212 |
| R1, wR2 [I > 2σ(I)] a | 0.0442, 0.1092 | 0.0576, 0.1418 | 0.0471, 0.1293 | 0.0551, 0.1413 | 0.0573, 0.1252 | 0.0482, 0.1219 | 0.0535, 0.1356 | 0.0586, 0.1431 |
| R1, wR2 (all data) b | 0.0595, 0.1206 | 0.0926, 0.1650 | 0.0618, 0.1429 | 0.0874, 0.1683 | 0.1184, 0.1572 | 0.0715, 0.1385 | 0.0768, 0.1547 | 0.0940, 0.1709 |
| GOF, S c | 1.050 | 1.040 | 1.044 | 1.023 | 1.038 | 1.017 | 1.025 | 1.046 |
| Run | Starting 2H-pyran-2-ones 1 | Dienophile | R3 | Reaction Time | Product | Yield (%) a | Solvent for Crystallization | ||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | ||||||||
| 1 | Ph | Me | 1a | 2a | Et | 336 h b | endo-3a | 73 | MeOH |
| 2 | Me | Me | 1b | 2a | Et | 408 h b | endo-3b | 66 | EtOH |
| 3 | 2-furyl | Me | 1c | 2a | Et | 384 h b | endo-3c | 70 | EtOH |
| 4 | Ph | Ph | 1d | 2b | cyclohexyl | 408 h b | endo-3d | 65 | EtOH |
| 5 | 3,4,5-(OMe)3-C6H2- | Me | 1e | 6a | 2-oxopyrrolidin-1-yl | 408 h b | exo-3e | 68 | CH2Cl2 |
| 6 | 3,4,5-(OMe)3-C6H2- | Me | 1e | 6a | 2-oxopyrrolidin-1-yl | 384 h b | endo-3e | 0.5 | CH2Cl2 |
| 7 | Ph | Ph | 1d | 6a | 2-oxopyrrolidin-1-yl | 384 h b | exo-3f | 68 | i-Pr2O |
| 8 | Ph | OMe | 1f | 2a | Et | 2 h c | 4a | 35 | EtOH |
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | <(DHA) |
|---|---|---|---|---|
| endo-3a | ||||
| N1–H1⋯O2i | 0.86 | 2.31 | 3.0684(15) | 147.2 |
| endo-3b | ||||
| N1–H29⋯O10 | 0.86 | 2.32 | 2.937(3) | 128.7 |
| N2–H30⋯O2i | 0.86 | 2.06 | 2.909(3) | 170.1 |
| C1–H1⋯O7 | 0.98 | 2.33 | 3.223(4) | 152 |
| C12–H12A⋯O9ii | 0.96 | 2.60 | 3.528(6) | 163 |
| C16–H16B⋯O7iii | 0.97 | 2.57 | 3.482(5) | 157 |
| C28–H28A⋯O2 | 0.96 | 2.59 | 3.461(5) | 151 |
| endo-3c | ||||
| N1–H1⋯O2i | 0.86 | 2.33 | 3.1063(17) | 151 |
| C16–H16⋯O5ii | 0.93 | 2.51 | 3.369(2) | 154 |
| endo-3d | ||||
| N1–H1⋯O2i | 0.86 | 2.36 | 3.121(2) | 148.3 |
| C11–H11⋯O4ii | 0.93 | 2.49 | 3.363(5) | 157 |
| C18–H18A⋯O4iii | 0.97 | 2.56 | 3.471(3) | 157 |
| C27–H27⋯O1iv | 0.93 | 2.48 | 3.350(3) | 156 |
| endo-3e∙6a | ||||
| N1–H1⋯O4i | 0.86 | 2.08 | 2.8979(16) | 158.6 |
| C3–H3⋯O8ii | 0.93 | 2.56 | 3.490(2) | 174 |
| C9–H9C⋯O8ii | 0.96 | 2.60 | 3.536(2) | 166 |
| C27–H27A⋯O1 | 0.97 | 2.51 | 3.216(3) | 130 |
| C27–H27B⋯O5iii | 0.97 | 2.60 | 3.287(3) | 128 |
| exo-3e | ||||
| N2–H2⋯O3 | 0.86 | 2.10 | 2.891(3) | 152.9 |
| C10–H10C⋯O7i | 0.96 | 2.36 | 3.242(5) | 152 |
| C22–H22A⋯O3i | 0.96 | 2.49 | 3.323(4) | 145 |
| C23–H23C⋯O6ii | 0.96 | 2.47 | 3.424(5) | 171 |
| exo-3f | ||||
| N1–H1⋯O1i | 0.86 | 2.32 | 3.0620(18) | 145.2 |
| C12–H12⋯O5ii | 0.93 | 2.32 | 3.198(6) | 157 |
| C19–H19B⋯O4iii | 0.96 | 2.58 | 3.439(3) | 149 |
| 4a | ||||
| N1–H1⋯O2i | 0.86 | 2.28 | 2.923(3) | 131.6 |
| C6–H6⋯O2i | 0.98 | 2.57 | 3.313(3) | 133 |
| C18–H18⋯O4ii | 0.93 | 2.52 | 3.375(3) | 152 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kranjc, K.; Juranovič, A.; Kočevar, M.; Perdih, F. Supramolecular Diversity of Oxabicyclo[2.2.2]octenes Formed between Substituted 2H-Pyran-2-ones and Vinyl-Moiety-Containing Dienophiles. Symmetry 2020, 12, 1714. https://doi.org/10.3390/sym12101714
Kranjc K, Juranovič A, Kočevar M, Perdih F. Supramolecular Diversity of Oxabicyclo[2.2.2]octenes Formed between Substituted 2H-Pyran-2-ones and Vinyl-Moiety-Containing Dienophiles. Symmetry. 2020; 12(10):1714. https://doi.org/10.3390/sym12101714
Chicago/Turabian StyleKranjc, Krištof, Amadej Juranovič, Marijan Kočevar, and Franc Perdih. 2020. "Supramolecular Diversity of Oxabicyclo[2.2.2]octenes Formed between Substituted 2H-Pyran-2-ones and Vinyl-Moiety-Containing Dienophiles" Symmetry 12, no. 10: 1714. https://doi.org/10.3390/sym12101714
APA StyleKranjc, K., Juranovič, A., Kočevar, M., & Perdih, F. (2020). Supramolecular Diversity of Oxabicyclo[2.2.2]octenes Formed between Substituted 2H-Pyran-2-ones and Vinyl-Moiety-Containing Dienophiles. Symmetry, 12(10), 1714. https://doi.org/10.3390/sym12101714