Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy


Abstract
1. Introduction
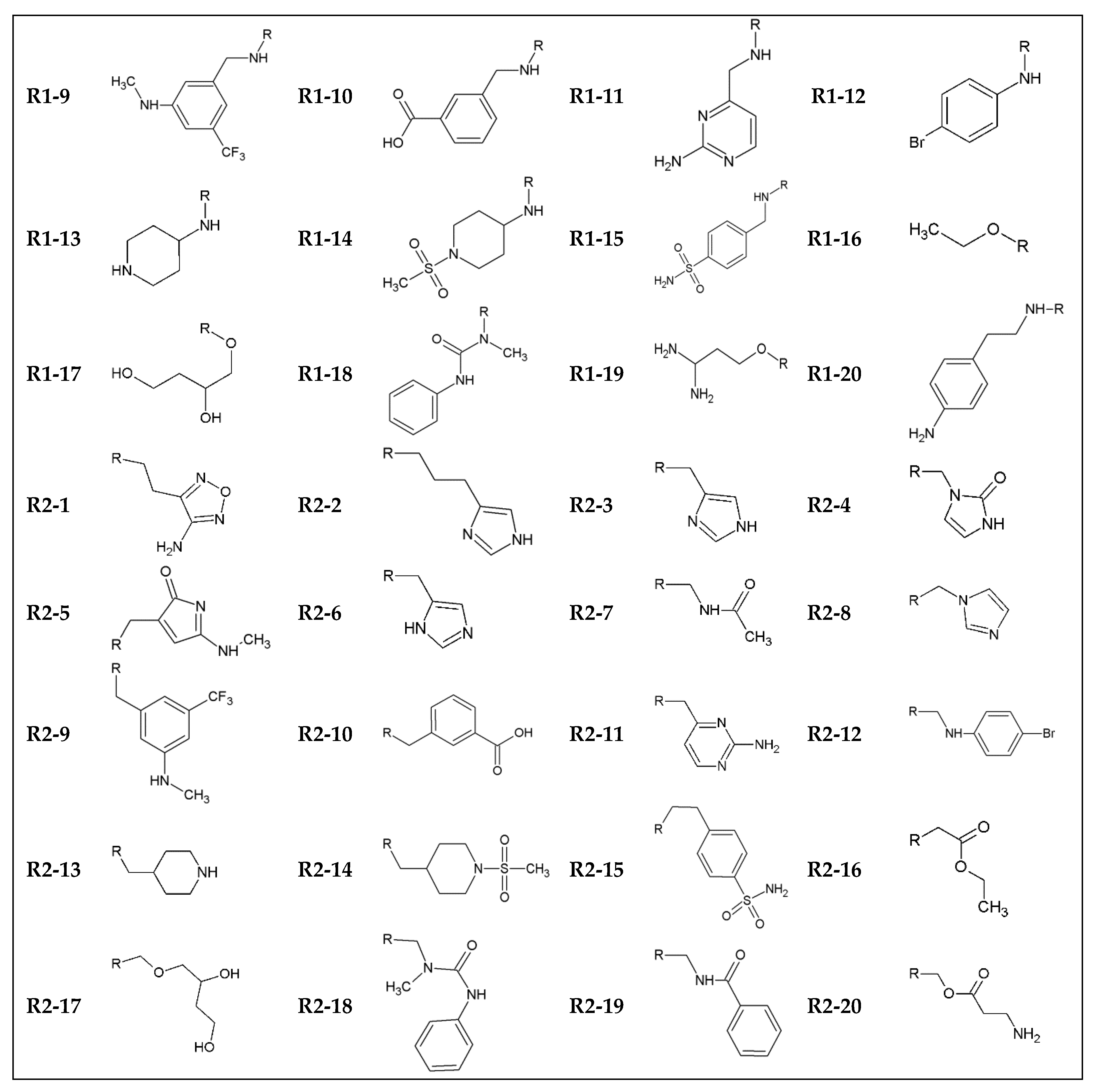
2. Methods
3. Results and Discussion
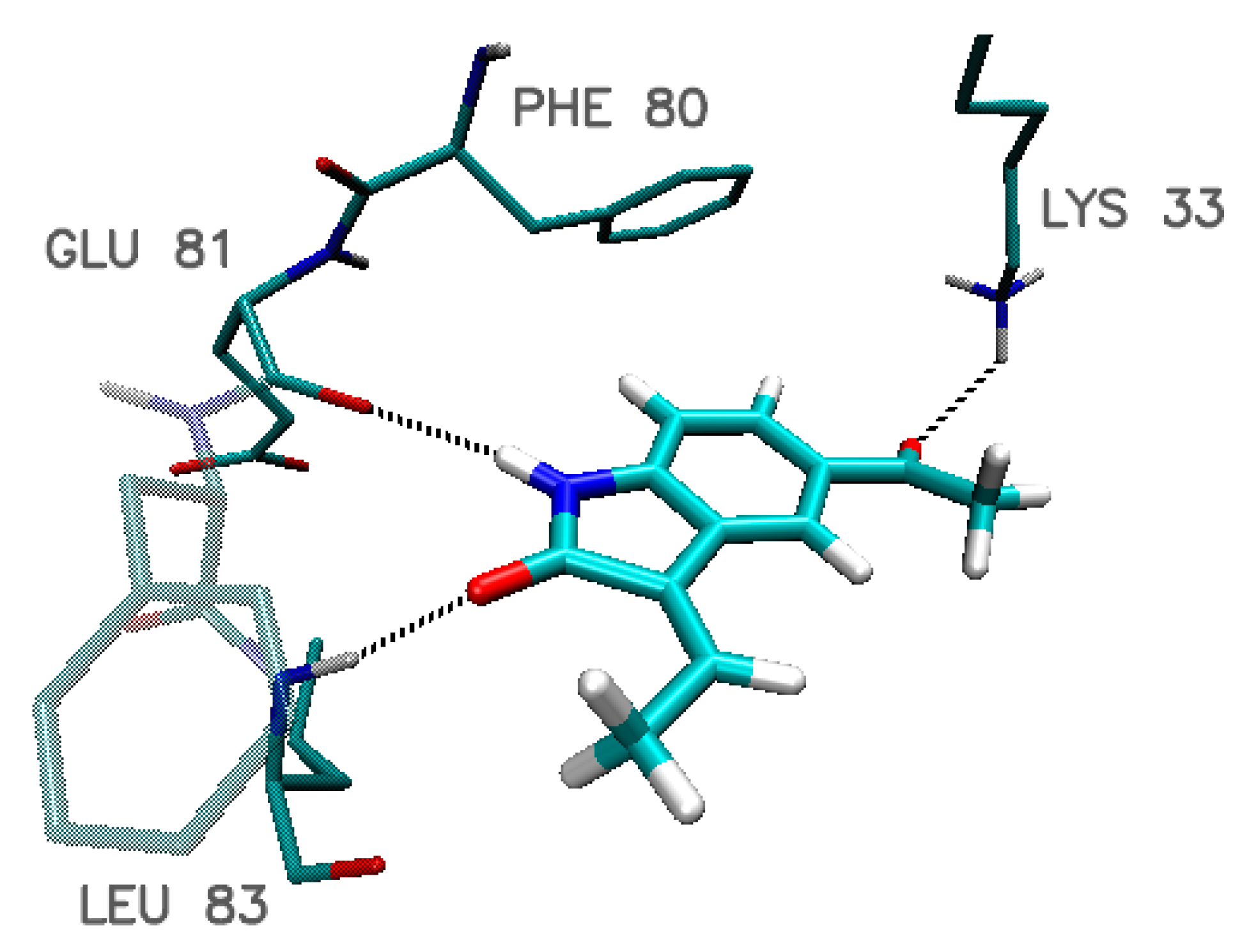
4. Molecular Dynamics of Chosen Complexes
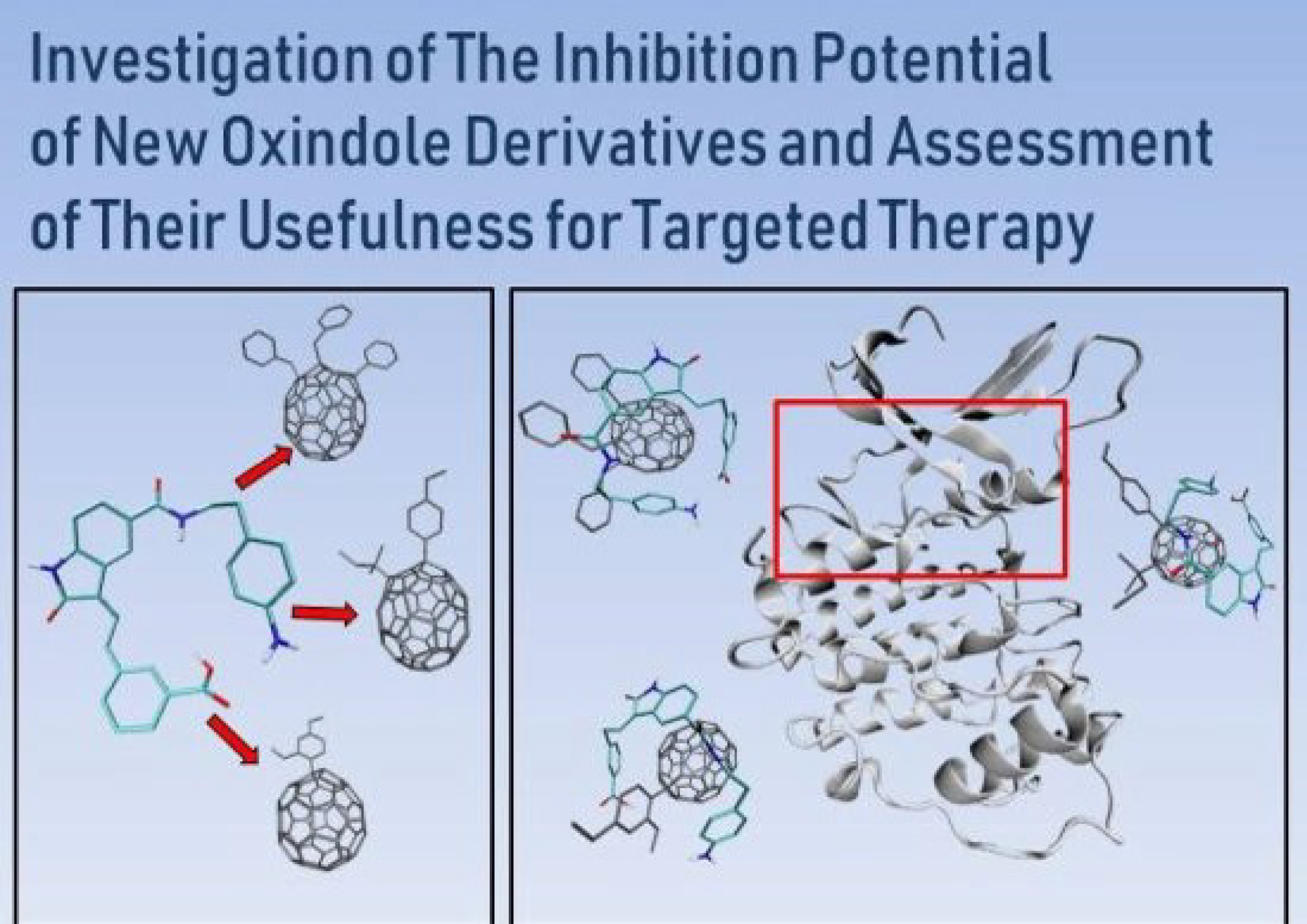
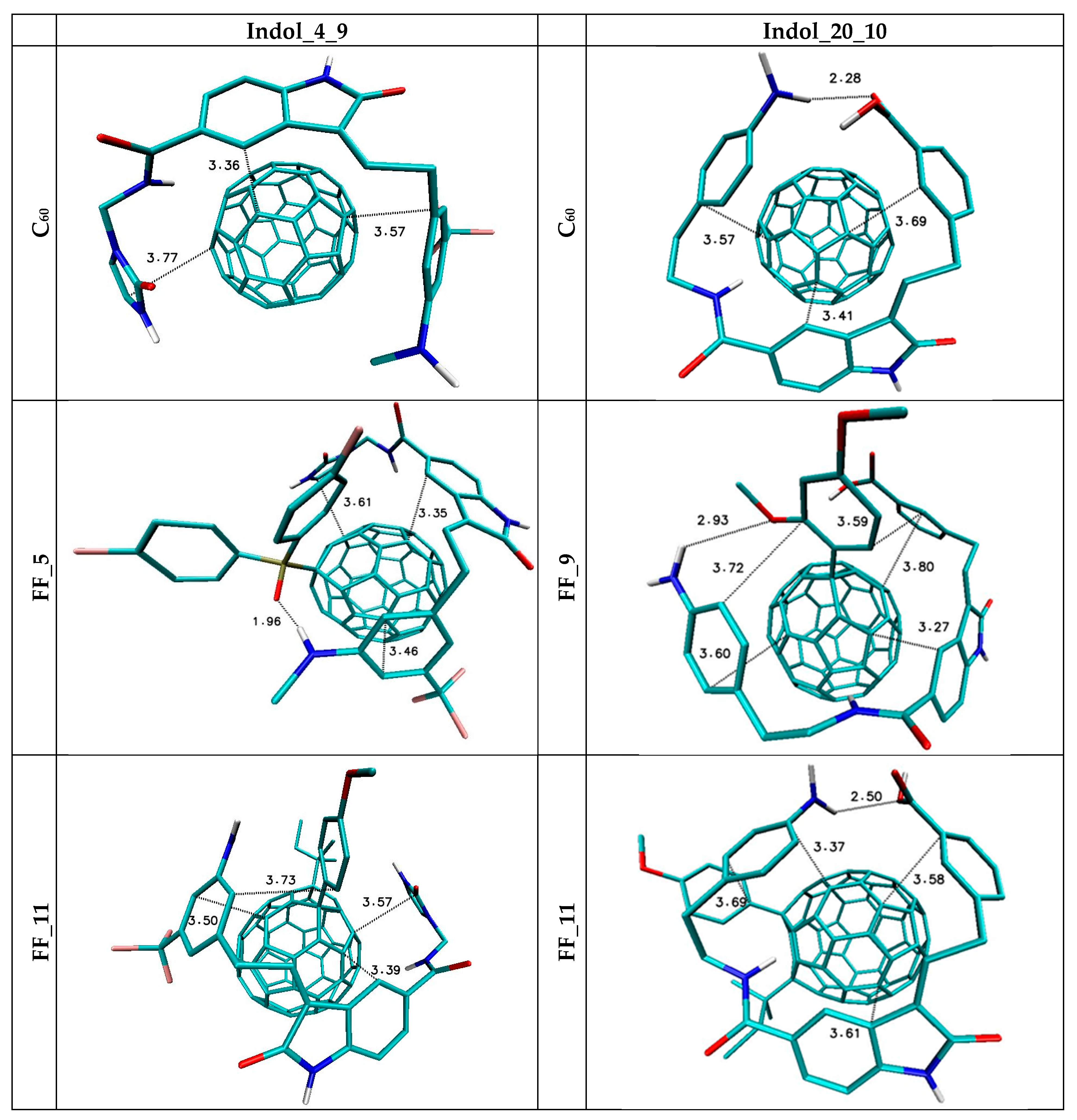
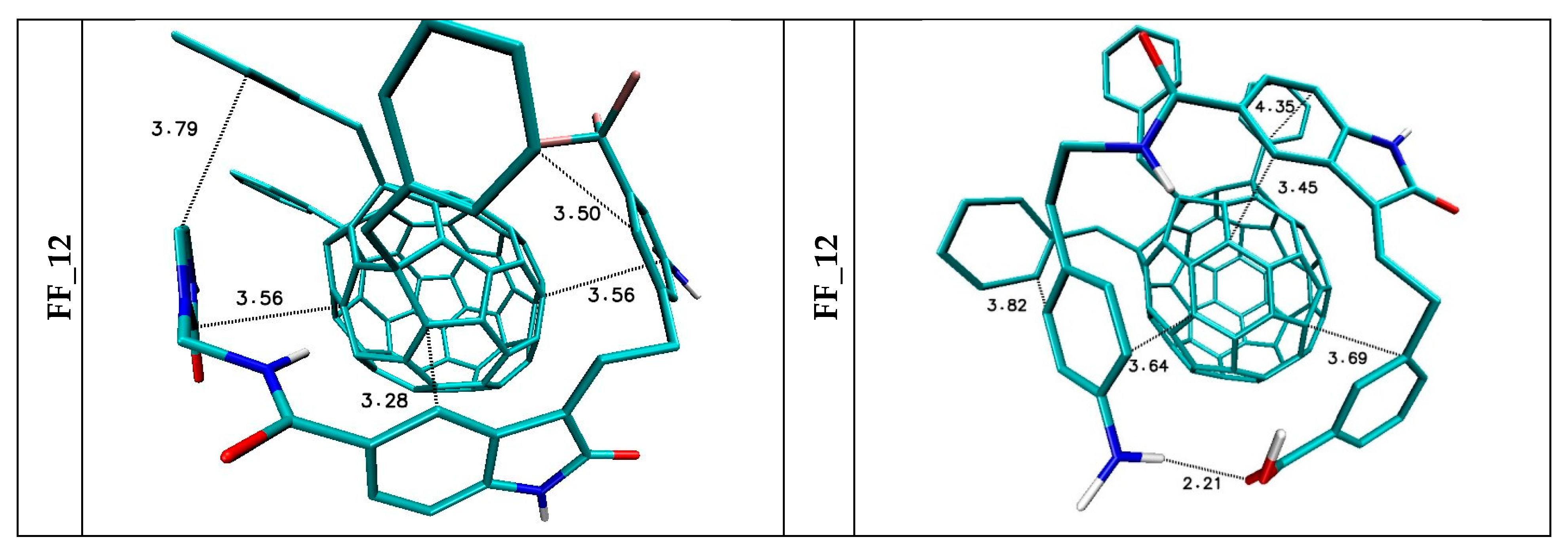
5. The Immobilization of Chosen Ligands with the Use of C60 Fullerene Derivatives
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | SMILES | ΔG (kcal/mol) | IC (nM) | Tox | LogP | MW (g/mol) | n HbA | n HbD |
|---|---|---|---|---|---|---|---|---|
| indol_1_10 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −11 | 8.65 | 0.8 | 2.056 | 419.4 | 10 | 5 |
| indol_1_11 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.66 | 15.35 | 0.93 | 0.074 | 392.38 | 11 | 6 |
| indol_1_12 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −10.48 | 20.80 | 0.9 | 2.85 | 469.3 | 9 | 5 |
| indol_1_13 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/CC1CCNCC1)/C(=O)N2 | −10.3 | 28.18 | 0.86 | 1.692 | 382.42 | 9 | 5 |
| indol_1_15 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/CCc1ccc(cc1)S(=O)(=O)N)/C(=O)N2 | −10.04 | 43.71 | 0.84 | 0.416 | 468.5 | 11 | 6 |
| indol_1_19 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −9.56 | 98.27 | 0.75 | 1.136 | 418.41 | 10 | 5 |
| indol_1_3 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −10.00 | 46.76 | 0.94 | 0.465 | 365.35 | 10 | 5 |
| indol_1_4 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/Cn1c(=O)[nH]cc1)/C(=O)N2 | −10.1 | 39.50 | 0.97 | −0.015 | 381.35 | 11 | 5 |
| indol_1_5 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.62 | 88.81 | 0.92 | −1.075 | 407.39 | 11 | 5 |
| indol_1_6 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −10.1 | 39.50 | 0.94 | 0.227 | 365.35 | 10 | 5 |
| indol_1_9 | c12c(cc(cc1)C(=O)NCc1nonc1N)/C(=C/Cc1cc(cc(c1)NC)C(F)(F)F)/C(=O)N2 | −11.08 | 7.56 | 0.89 | 3.527 | 472.43 | 9 | 5 |
| indol_4_1 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/CCc1nonc1N)/C(=O)N2 | −10.2 | 33.37 | 0.97 | 0.314 | 395.38 | 11 | 5 |
| indol_4_10 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −11.1 | 7.30 | 0.07 | 2.043 | 418.41 | 9 | 4 |
| indol_4_11 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.7 | 14.35 | 0.88 | −0.012 | 391.39 | 10 | 5 |
| indol_4_12 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −10.4 | 23.81 | 0.35 | 2.801 | 468.31 | 8 | 4 |
| indol_4_13 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/CC1CCNCC1)/C(=O)N2 | −10.4 | 23.81 | 0.17 | 1.408 | 381.44 | 8 | 4 |
| indol_4_15 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/CCc1ccc(cc1)S(=O)(=O)N)/C(=O)N2 | −10.72 | 13.87 | 0.01 | 0.445 | 467.51 | 10 | 5 |
| indol_4_19 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −9.28 | 157.64 | 0 | 0.855 | 417.43 | 9 | 4 |
| indol_4_3 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −10 | 46.76 | 0.76 | 0.405 | 364.37 | 9 | 4 |
| indol_4_5 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.66 | 83.01 | 0.43 | −1.317 | 406.4 | 10 | 4 |
| indol_4_6 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −10.02 | 45.21 | 0.76 | 0.031 | 364.37 | 9 | 4 |
| indol_4_9 | c12c(cc(cc1)C(=O)NCn1c(=O)[nH]cc1)/C(=C/Cc1cc(cc(c1)NC)C(F)(F)F)/C(=O)N2 | −11 | 8.65 | 0.32 | 3.579 | 471.44 | 8 | 4 |
| indol_9_1 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/CCc1nonc1N)/C(=O)N2 | −9.6 | 91.86 | 0.88 | 3.562 | 486.45 | 9 | 5 |
| indol_9_10 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −10.54 | 18.80 | 0.39 | 5.116 | 509.48 | 7 | 4 |
| indol_9_11 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.24 | 31.19 | 0.69 | 3.281 | 482.47 | 8 | 5 |
| indol_9_12 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −9.72 | 75.01 | 0.59 | 5.9 | 559.39 | 6 | 4 |
| indol_9_13 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/CC1CCNCC1)/C(=O)N2 | −10.22 | 32.26 | 0.45 | 4.809 | 472.51 | 6 | 4 |
| indol_9_15 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/CCc1ccc(cc1)S(=O)(=O)N)/C(=O)N2 | −10.58 | 17.57 | 0.19 | 3.644 | 558.58 | 8 | 5 |
| indol_9_19 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −9.72 | 75.01 | 0.11 | 4.076 | 508.5 | 7 | 4 |
| indol_9_3 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −9.9 | 55.36 | 0.66 | 3.694 | 455.44 | 7 | 4 |
| indol_9_4 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/Cn1c(=O)[nH]cc1)/C(=O)N2 | −10.08 | 40.86 | 0.24 | 2.923 | 471.44 | 8 | 4 |
| indol_9_5 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.86 | 59.23 | 0.36 | 2.07 | 497.48 | 8 | 4 |
| indol_9_6 | c12c(cc(cc1)C(=O)NCc1cc(cc(c1)NC)C(F)(F)F)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −9.92 | 53.52 | 0.67 | 3.455 | 455.44 | 7 | 4 |
| indol_10_1 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/CCc1nonc1N)/C(=O)N2 | −9.54 | 101.65 | 0.79 | 2.235 | 433.42 | 10 | 5 |
| indol_10_11 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.7 | 14.35 | 0.4 | 1.66 | 429.44 | 9 | 5 |
| indol_10_12 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −10.16 | 35.70 | 0.51 | 4.497 | 506.36 | 7 | 4 |
| indol_10_13 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/CC1CCNCC1)/C(=O)N2 | −10.36 | 25.47 | 0.28 | 3.377 | 419.48 | 7 | 4 |
| indol_10_15 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/CCc1ccc(cc1)S(=O)(=O)N)/C(=O)N2 | −10.58 | 17.57 | 0.45 | 2.318 | 505.55 | 9 | 5 |
| indol_10_19 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −9.3 | 152.41 | 0.03 | 2.936 | 455.47 | 8 | 4 |
| indol_10_3 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −10.18 | 34.51 | 0.46 | 2.176 | 402.41 | 8 | 4 |
| indol_10_4 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/Cn1c(=O)[nH]cc1)/C(=O)N2 | −10.44 | 22.25 | 0.08 | 1.582 | 418.41 | 9 | 4 |
| indol_10_5 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.98 | 48.37 | 0.15 | 0.749 | 444.45 | 9 | 4 |
| indol_10_6 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −10.16 | 35.70 | 0.45 | 1.903 | 402.41 | 8 | 4 |
| indol_10_9 | c12c(cc(cc1)C(=O)NCc1cc(ccc1)C(=O)O)/C(=C/Cc1cc(cc(c1)NC)C(F)(F)F)/C(=O)N2 | −10.88 | 10.59 | 0.46 | 5.274 | 509.48 | 7 | 4 |
| indol_15_1 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/CCc1nonc1N)/C(=O)N2 | −10.08 | 40.86 | 0.87 | −1.335 | 468.5 | 11 | 6 |
| indol_15_10 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −10.78 | 12.54 | 0.49 | 0.998 | 491.52 | 9 | 5 |
| indol_15_11 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.48 | 20.80 | 0.24 | −0.85 | 464.51 | 10 | 6 |
| indol_15_12 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −10.4 | 23.81 | 0.3 | 1.952 | 541.43 | 8 | 5 |
| indol_15_13 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/CC1CCNCC1)/C(=O)N2 | −10.08 | 40.86 | 0.14 | 0.662 | 454.55 | 8 | 5 |
| indol_15_19 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −10.28 | 29.15 | 0.01 | 0.095 | 490.54 | 9 | 5 |
| indol_15_3 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −10 | 46.76 | 0.29 | −0.86 | 437.48 | 9 | 5 |
| indol_15_4 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/Cn1c(=O)[nH]cc1)/C(=O)N2 | −10.2 | 33.37 | 0.01 | −1.221 | 453.48 | 10 | 5 |
| indol_15_5 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.1 | 213.61 | 0.02 | −2.226 | 479.52 | 10 | 5 |
| indol_15_6 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −10.02 | 45.21 | 0.27 | −0.753 | 437.48 | 9 | 5 |
| indol_15_9 | c12c(cc(cc1)C(=O)NCc1ccc(cc1)S(=O)(=O)N)/C(=C/Cc1cc(cc(c1)NC)C(F)(F)F)/C(=O)N2 | −9.36 | 137.73 | 0.25 | 2.576 | 544.55 | 8 | 5 |
| indol_18_1 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/CCc1nonc1N)/C(=O)N2 | −9.88 | 57.26 | 0.94 | 0.754 | 432.44 | 10 | 4 |
| indol_18_10 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −10.88 | 10.59 | 0.03 | 2.979 | 455.47 | 8 | 3 |
| indol_18_11 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.52 | 19.44 | 0.7 | 1.089 | 428.45 | 9 | 4 |
| indol_18_12 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −10.54 | 18.80 | 0.28 | 3.852 | 505.37 | 7 | 3 |
| indol_18_13 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/CC1CCNCC1)/C(=O)N2 | −9.62 | 88.81 | 0.11 | 2.613 | 418.5 | 7 | 3 |
| indol_18_15 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/CCc1ccc(cc1)S(=O)(=O)N)/C(=O)N2 | −10.44 | 22.25 | 0 | 1.649 | 504.57 | 9 | 4 |
| indol_18_19 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −9.16 | 193.03 | 0 | 1.9 | 454.49 | 8 | 3 |
| indol_18_3 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −9.86 | 59.23 | 0.57 | 1.49 | 401.43 | 8 | 3 |
| indol_18_4 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/Cn1c(=O)[nH]cc1)/C(=O)N2 | −10.06 | 42.26 | 0.06 | 0.688 | 417.43 | 9 | 3 |
| indol_18_5 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.76 | 70.12 | 0.12 | 0.137 | 443.46 | 9 | 3 |
| indol_18_6 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −10.07 | 41.20 | 0.56 | 1.264 | 401.43 | 8 | 3 |
| indol_18_9 | c12c(cc(cc1)C(=O)N(C(=O)Nc1ccccc1)C)/C(=C/Cc1cc(cc(c1)NC)C(F)(F)F)/C(=O)N2 | −10.34 | 26.34 | 0.21 | 4.55 | 508.5 | 7 | 3 |
| indol_20_1 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −10.4 | 23.81 | 0.39 | 1.261 | 441.49 | 7 | 5 |
| indol_20_10 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cc1cc(ccc1)C(=O)O)/C(=O)N2 | −11.2 | 6.17 | 0.39 | 3.606 | 441.49 | 7 | 5 |
| indol_20_11 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cc1ccnc(n1)N)/C(=O)N2 | −10.7 | 14.35 | 0.46 | 1.814 | 414.47 | 8 | 6 |
| indol_20_12 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/CNc1ccc(cc1)Br)/C(=O)N2 | −10.4 | 23.81 | 0.54 | 4.528 | 491.39 | 6 | 5 |
| indol_20_13 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/CC1CCNCC1)/C(=O)N2 | −10.6 | 16.99 | 0.4 | 3.151 | 404.51 | 6 | 5 |
| indol_20_15 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/CCc1ccc(cc1)S(=O)(=O)N)/C(=O)N2 | −10.8 | 12.12 | 0.19 | 2.184 | 490.58 | 8 | 6 |
| indol_20_19 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/CNC(=O)c1ccccc1)/C(=O)N2 | −9.9 | 55.36 | 0.1 | 2.488 | 440.5 | 7 | 5 |
| indol_20_3 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cc1nc[nH]c1)/C(=O)N2 | −10.3 | 28.18 | 0.49 | 2.065 | 387.44 | 7 | 5 |
| indol_20_4 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cn1c(=O)[nH]cc1)/C(=O)N2 | −10.4 | 23.81 | 0.11 | 1.461 | 403.44 | 8 | 5 |
| indol_20_5 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/CC1=CC(=NC1=O)NC)/C(=O)N2 | −9.88 | 57.26 | 0.16 | 0.231 | 429.48 | 8 | 5 |
| indol_20_6 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cc1cnc[nH]1)/C(=O)N2 | −10.28 | 29.15 | 0.49 | 1.849 | 387.44 | 7 | 5 |
| indol_20_9 | c12c(cc(cc1)C(=O)NCCc1ccc(cc1)N)/C(=C/Cc1cc(cc(c1)NC)C(F)(F)F)/C(=O)N2 | −10.88 | 10.59 | 0.5 | 5.415 | 494.52 | 6 | 5 |
| FF_1 | CID_11332103 |
| C67H14F3O4P | |
| 1-(diethoxyphosphorylmethyl)-7-(2,2,2-trifluoroethoxy)(C60-Ih)[5,6]fullerene | |
| FF_2 | CID_11468612 |
| C65H13O3P | |
| 9-(diethoxyphosphorylmethyl)-1H-(C60-Ih)[5,6]fullerene | |
| FF_3 | CID_16146387 |
| C67H16O2Si | |
| methyl 9-(2-trimethylsilylethyl)(C60-Ih)[5,6]fullerene-1-carboxylate | |
| FF_4 | CID_16150529 |
| C70H20N2O2 | |
| 1-N,1-N,9-N,9-N-tetraethyl(C60-Ih)[5,6]fullerene-1,9-dicarboxamide | |
| FF_5 | CID_16156307 |
| C72H9F2OP | |
| 9-bis(4-fluorophenyl)phosphoryl-1H-(C60-Ih)[5,6]fullerene | |
| FF_6 | CID_53469305 |
| C64H11O3P | |
| 9-diethoxyphosphoryl-1H-(C60-Ih)[5,6]fullerene | |
| FF_7 | CID_71618962 |
| C68H10O2 | |
| 9-(3,5-dimethoxyphenyl)-1H-(C60-Ih)[5,6]fullerene | |
| FF_8 | CID_71619055 |
| C68H10O2 | |
| 9-(2,6-dimethoxyphenyl)-1H-(C60-Ih)[5,6]fullerene | |
| FF_9 | CID_71619159 |
| C68H10O2 | |
| 9-(2,4-dimethoxyphenyl)-1H-(C60-Ih)[5,6]fullerene | |
| FF_10 | CID_101218232 |
| C63H4ClF3O | |
| 1-(chloromethyl)-7-(2,2,2-trifluoroethoxy)(C60-Ih)[5,6]fullerene | |
| FF_11 | CID_101218236 |
| C69H9Cl3O | |
| 1-(4-methoxyphenyl)-7-(1,1,2-trichloroethyl)(C60-Ih)[5,6]fullerene | |
| FF_12 | CID_101266715 |
| C80H22 | |
| 12,15-dibenzyl-9-phenyl-6,18-dihydro-1H-(C60-Ih)[5,6]fullerene | |
| FF_13 | CID_101382121 |
| C62F6 | |
| 1,9-bis(trifluoromethyl)(C60-Ih)[5,6]fullerene |
References
- Besson, A.; Dowdy, S.F. Roberts JM CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Barbacid M Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Child, E.S.; Hendrychová, T.; McCague, K.; Futreal, A.; Otyepka, M.; Mann, D.J. A cancer-derived mutation in the PSTAIRE helix of cyclin-dependent kinase 2 alters the stability of cyclin binding. Biochim. Biophys. Acta. Mol. Cell Res. 2010, 1803, 858–864. [Google Scholar] [CrossRef]
- Echalier, A.; Hole, A.J.; Lolli, G.; Endicott, J.A.; Noble, M.E. An Inhibitor’s-Eye View of the ATP-Binding Site of CDKs in Different Regulatory States. ACS Chem. Biol. 2014, 9, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Canavese, M.; Santo, L.; Raje, N. Cyclin dependent kinases in cancer. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef]
- LEE, M.Y.; Liu, Y.W.; Chen, M.H.; Wu, J.Y.; Ho, H.Y.; Wang, Q.F.; Chuang, J.J. Indirubin-3′-monoxime promotes autophagic and apoptotic death in JM1 human acute lymphoblastic leukemia cells and K562 human chronic myelogenous leukemia cells. Oncol. Rep. 2013, 29, 2072–2078. [Google Scholar] [CrossRef]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.; Lawrie, A.; Tunnah, P.; Niederberger, E. Indirubin the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef]
- Shin, E.K.; Kim, J.K. Indirubin derivative E804 inhibits angiogenesis. BMC Cancer 2012, 12, 164. [Google Scholar] [CrossRef]
- Marko, D.; Schätzle, S.; Friedel, A.; Genzlinger, A.; Zankl, H.; Meijer, L.; Eisenbrand, G. Inhibition of cyclin-dependent kinase 1 (CDK1) by indirubin derivatives in human tumour cells. Br. J. Cancer 2012, 84, 283–289. [Google Scholar] [CrossRef]
- Martin, L.; Magnaudeix, A.; Wilson, C.M.; Yardin, C.; Terro, F. The new indirubin derivative inhibitors of glycogen synthase kinase-3, 6-BIDECO and 6-BIMYEO, prevent tau phosphorylation and apoptosis induced by the inhibition of protein phosphatase-2A by okadaic acid in cultured neurons. J. Neurosci. Res. 2011, 89, 1802–1811. [Google Scholar] [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Eisenbrand, G. Indirubins Inhibit Glycogen Synthase Kinase-3beta and CDK5/P25, Two Protein Kinases Involved in Abnormal Tau Phosphorylation in Alzheimer’s Disease. A PROPERTY COMMON TO MOST CYCLIN-DEPENDENT KINASE INHIBITORS? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef]
- Beauchard, A.; Ferandin, Y.; Frère, S.; Lozach, O.; Blairvacq, M.; Meijer, L.; Besson, T. Synthesis of novel 5-substituted indirubins as protein kinases inhibitors. Bioorg. Med. Chem. 2006, 14, 6434–6443. [Google Scholar] [CrossRef]
- Dermatakis, A.; Luk, K.C.; DePinto, W. Synthesis of potent oxindole CDK2 inhibitors. Bioorg. Med. Chem. 2003, 11, 1873–1881. [Google Scholar] [CrossRef]
- Luk, K.C.; Simcox, M.E.; Schutt, A.; Rowan, K.; Thompson, T.; Chen, Y.; Dermatakis, A. A new series of potent oxindole inhibitors of CDK2. Bioorg. Med. Chem. Lett. 2004, 14, 913–917. [Google Scholar] [CrossRef]
- Czeleń Molecular dynamics study on inhibition mechanism of CDK-2 and GSK-3β by CHEMBL272026 molecule. Struct. Chem. 2016, 27, 1807–1818. [CrossRef]
- Czeleń, P. Inhibition mechanism of CDK-2 and GSK-3β by a sulfamoylphenyl derivative of indoline—A molecular dynamics study. J. Mol. Model. 2017, 23, 230. [Google Scholar] [CrossRef]
- Czeleń, P.; Szefler, B. The Immobilization of Oxindole Derivatives with Use of Cube Rhombellane Homeomorphs. Symmetry 2019, 11, 900. [Google Scholar]
- Szefler, B.; Czeleń, P. Docking of Cisplatin on Fullerene Derivatives and Some Cube Rhombellane Functionalized Homeomorphs. Symmetry 2019, 11, 874. [Google Scholar] [CrossRef]
- Szefler, B.; Czeleń, P.; Diudea, M.V. Docking of indolizine derivatives on cube rhombellane functionalized homeomorphs. Stud. Univ. Babes-Bolyai Chem. 2018, 63, 7–18. [Google Scholar] [CrossRef]
- Morgen, M.; Bloom, C.; Beyerinck, R.; Bello, A.; Song, W.; Wilkinson, K.; Shamblin, S. Polymeric Nanoparticles for Increased Oral Bioavailability and Rapid Absorption Using Celecoxib as a Model of a Low-Solubility, High-Permeability Drug. Pharm. Res. 2012, 29, 427–440. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J.A. Drug delivery and nanoparticles:applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, L.; Sun, Y. Nanotechnology applied to overcome tumor drug resistance. J. Control. Release 2012, 162, 45–55. [Google Scholar] [CrossRef]
- Turov, V.V.; Chehun, V.F.; Barvinchenko, V.N.; Krupskaya, T.V.; Prylutskyy, Y.I.; Scharff, P.; Ritter, U. Low-temperature 1H-NMR spectroscopic study of doxorubicin influence on the hydrated properties of nanosilica modified by DNA. J. Mater. Sci. Mater. Med. 2011, 22, 525–532. [Google Scholar] [CrossRef]
- Szefler, B. Nanotechnology, from quantum mechanical calculations up to drug delivery. Int. J. Nanomed. 2018, 13, 6143–6176. [Google Scholar] [CrossRef]
- Cataldo, F.; Da Ros, T. Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes; Springer: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Panchuk, R.R.; Prylutska, S.V.; Chumak, V.V.; Skorokhyd, N.R.; Lehka, L.V.; Evstigneev, M.P.; Ritter, U. Application of C60 Fullerene-Doxorubicin Complex for Tumor Cell Treatment In Vitro and In Vivo. J. Biomed. Nanotechnol. 2015, 11, 1139–1152. [Google Scholar] [CrossRef]
- Andrievsky, G.; Klochkov, V.; Derevyanchenko, L. Is the C60 Fullerene Molecule Toxic? Fuller. Nanotub. Carbon Nanostruct. 2005, 13, 363–376. [Google Scholar] [CrossRef]
- Prylutska, S.; Bilyy, R.; Overchuk, M.; Bychko, A.; Andreichenko, K.; Stoika, R.; Ritter, U. Water-soluble pristine fullerenes C60 increase the specific conductivity and capacity of lipid model membrane and form the channels in cellular plasma membrane. J. Biomed. Nanotechnol. 2012, 8, 522–527. [Google Scholar] [CrossRef]
- Qiao, R.; Roberts, A.P.; Mount, A.S.; Klaine, S.J.; Ke, P.C. Translocation of C60 and Its Derivatives Across a Lipid Bilayer. Nano Lett. 2007, 7, 614–619. [Google Scholar] [CrossRef]
- TURBOMOLE 7.0. Available online: http://www.turbomole.com/.
- Potemkin, V.; Grishina, M. Principles for 3D/4D QSAR classification of drugs. Drug. Discov. Today 2008, 13, 952–959. [Google Scholar] [CrossRef]
- Potemkin, V.A.; Grishina, M.A. A new paradigm for pattern recognition of drugs. J. Comput. Aided Mol. Des. 2008, 22, 489–505. [Google Scholar] [CrossRef]
- Chemosophia. Available online: http://www.chemosophia.com/ (accessed on 1 March 2017).
- Eckert, F.; Klamt, A. Fast solvent screening via quantum chemistry: COSMO-RS approach. AIChE J. 2002, 48, 369–385. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 1 May 2019).
- Bartashevich, E.V.; Potemkin, V.A.; Grishina, M.A.; Belik, A.V. A Method for Multiconformational Modeling of the Three-Dimensional Shape of a Molecule. J. Struct. Chem. 2002, 43, 1033–1039. [Google Scholar] [CrossRef]
- Rosita, G.; Manuel, C.; Franco, M.; Cinzia, N.; Donatella, F.; Emiliano, L.; Roberta, G. Permethrin and its metabolites affect Cu/Zn superoxide conformation: Fluorescence and in silico evidences. Mol. Biosyst. 2015, 11, 208–217. [Google Scholar] [CrossRef]
- Mangiaterra, G.; Laudadio, E.; Cometti, M. Inhibitors of multidrug efflux pumps of Pseudomonas aeruginosa from natural sources: An in silico high-throughput virtual screening and in vitro validation. Med. Chem. Res. 2017, 26, 414–430. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Adelman, S.A. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: Oakland, CA, USA, 2014. [Google Scholar]








| Name | Binding Affinity (kcal/mol) | Increase of Binding Affinity (%) | Name | Binding Affinity (kcal/mol) | Increase of Binding Affinity (%) |
|---|---|---|---|---|---|
| Indol_9_0 | −9.6 | 15.7 | Indol_0_10 | −10.3 | 24.1 |
| Indol_15_0 | −9.5 | 14.5 | Indol_0_9 | −10.2 | 22.9 |
| Indol_10_0 | −9.4 | 13.3 | Indol_0_15 | −10.1 | 21.7 |
| Indol_20_0 | −9.3 | 12.0 | Indol_0_11 | −9.8 | 18.1 |
| Indol_1_0 | −9.3 | 12.0 | Indol_0_1 | −9.66 | 16.4 |
| Indol_4_0 | −9.24 | 11.3 | Indol_0_13 | −9.6 | 15.7 |
| Indol_18_0 | −9.22 | 11.1 | Indol_0_12 | −9.58 | 15.4 |
| Indol_11_0 | −9 | 8.4 | Indol_0_4 | −9.3 | 12.0 |
| Indol_8_0 | −8.9 | 7.2 | Indol_0_6 | −9.3 | 12.0 |
| Indol_6_0 | −8.9 | 7.2 | Indol_0_19 | −9.3 | 12.0 |
| Indol_3_0 | −8.9 | 7.2 | Indol_0_3 | −9.2 | 10.8 |
| Indol_13_0 | −8.9 | 7.2 | Indol_0_5 | −9.2 | 10.8 |
| Indol_14_0 | −8.86 | 6.7 | Indol_0_8 | −9 | 8.4 |
| Indol_2_0 | −8.8 | 6.0 | Indol_0_14 | −9 | 8.4 |
| Indol_12_0 | −8.76 | 5.5 | Indol_0_2 | −8.96 | 8.0 |
| Indol_5_0 | −8.74 | 5.3 | Indol_0_18 | −8.82 | 6.3 |
| Indol_19_0 | −8.2 | −1.2 | Indol_0_7 | −8.8 | 6.0 |
| Indol_17_0 | −8.2 | −1.2 | Indol_0_16 | −8.5 | 2.4 |
| Indol_7_0 | −8.02 | −3.4 | Indol_0_20 | −8.3 | 0.0 |
| Indol_16_0 | −7.92 | −4.6 | Indol_0_17 | −8.24 | −0.7 |
| Name | LogP | Toxicity | Binding Affinity (kcal/mol) | Increase of Binding Affinity (%) | Inhibition Constant (nM) | |
|---|---|---|---|---|---|---|
| 1. | indol_20_10 | 3.61 | 0.39 | −11.20 | 34.94 | 6.17 |
| 2. | indol_4_10 | 2.04 | 0.07 | −11.10 | 33.73 | 7.30 |
| 3. | indol_4_9 | 3.58 | 0.32 | −11.00 | 32.53 | 8.65 |
| 4. | indol_9_15 | 3.64 | 0.19 | −10.88 | 31.08 | 10.59 |
| 5. | indol_20_15 | 2.18 | 0.19 | −10.80 | 30.12 | 12.12 |
| 6. | indol_18_12 | 3.85 | 0.28 | −10.54 | 26.99 | 18.80 |
| 7. | indol_18_15 | 1.65 | 0.00 | −10.44 | 25.78 | 22.25 |
| 8. | indol_10_4 | 1.58 | 0.08 | −10.44 | 25.78 | 22.25 |
| 9. | indol_18_10 | 2.98 | 0.03 | −10.40 | 25.30 | 23.81 |
| 10. | indol_20_4 | 1.46 | 0.11 | −10.40 | 25.30 | 23.81 |
| 11. | indol_4_13 | 1.41 | 0.17 | −10.40 | 25.30 | 23.81 |
| 12. | indol_15_12 | 1.95 | 0.30 | −10.40 | 25.30 | 23.81 |
| 13. | indol_4_12 | 2.80 | 0.35 | −10.40 | 25.30 | 23.81 |
| 14. | indol_20_1 | 1.26 | 0.39 | −10.40 | 25.30 | 23.81 |
| 15. | indol_10_13 | 3.38 | 0.28 | −10.36 | 24.82 | 25.47 |
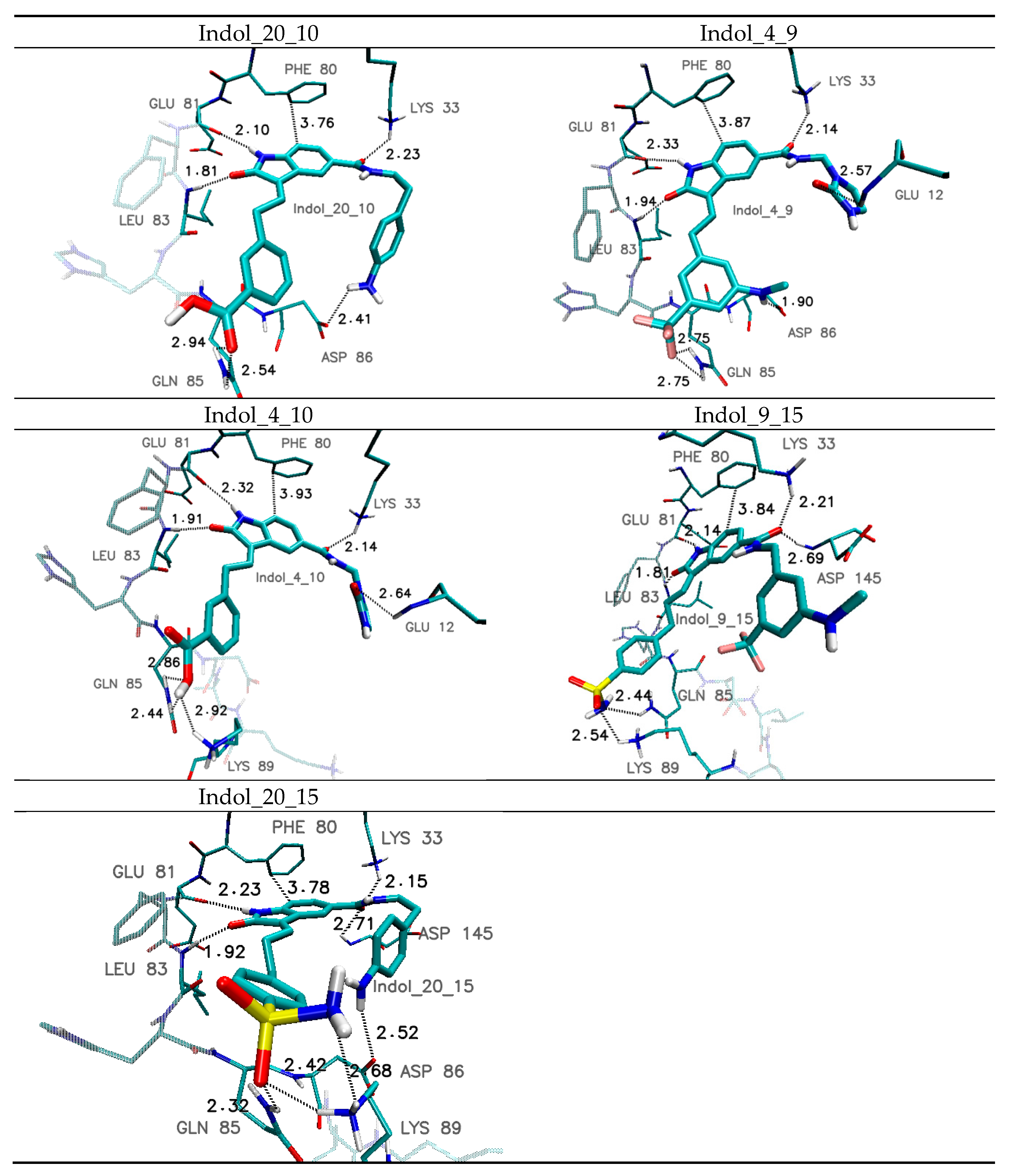
| Amino Acid | Hydrogen-Bond Length/Distance Between Aromatic Systems * (Å) | ||||
|---|---|---|---|---|---|
| Indol_4_9 | Indol_4_10 | Indol_9_15 | Indol_20_10 | Indol_20_15 | |
| Glu 12 | 2.57 | 2.64 | ---- | ---- | ---- |
| Lys 33 | 2.14 | 2.14 | 2.21 | 2.23 | 2.15 |
| Leu 83 | 1.94 | 1.91 | 1.81 | 1.81 | 1.92 |
| Glu 81 | 2.33 | 2.32 | 2.14 | 2.10 | 2.23 |
| Gln 85 | 2.75 | 2.44 2.86 | 2.44 | 2.54 2.94 | 2.32 |
| Asp 86 | 1.90 | ---- | ---- | 2.41 | 2.52 |
| Lys 89 | ---- | 2.92 | 2.54 | ---- | 2.42 2.68 |
| Asp146 | 2.86 | 2.86 | 2.69 | 2.61 | 2.71 |
| Phe 80 * | 3.87 | 3.87 | 3.84 | 3.76 | 3.84 |
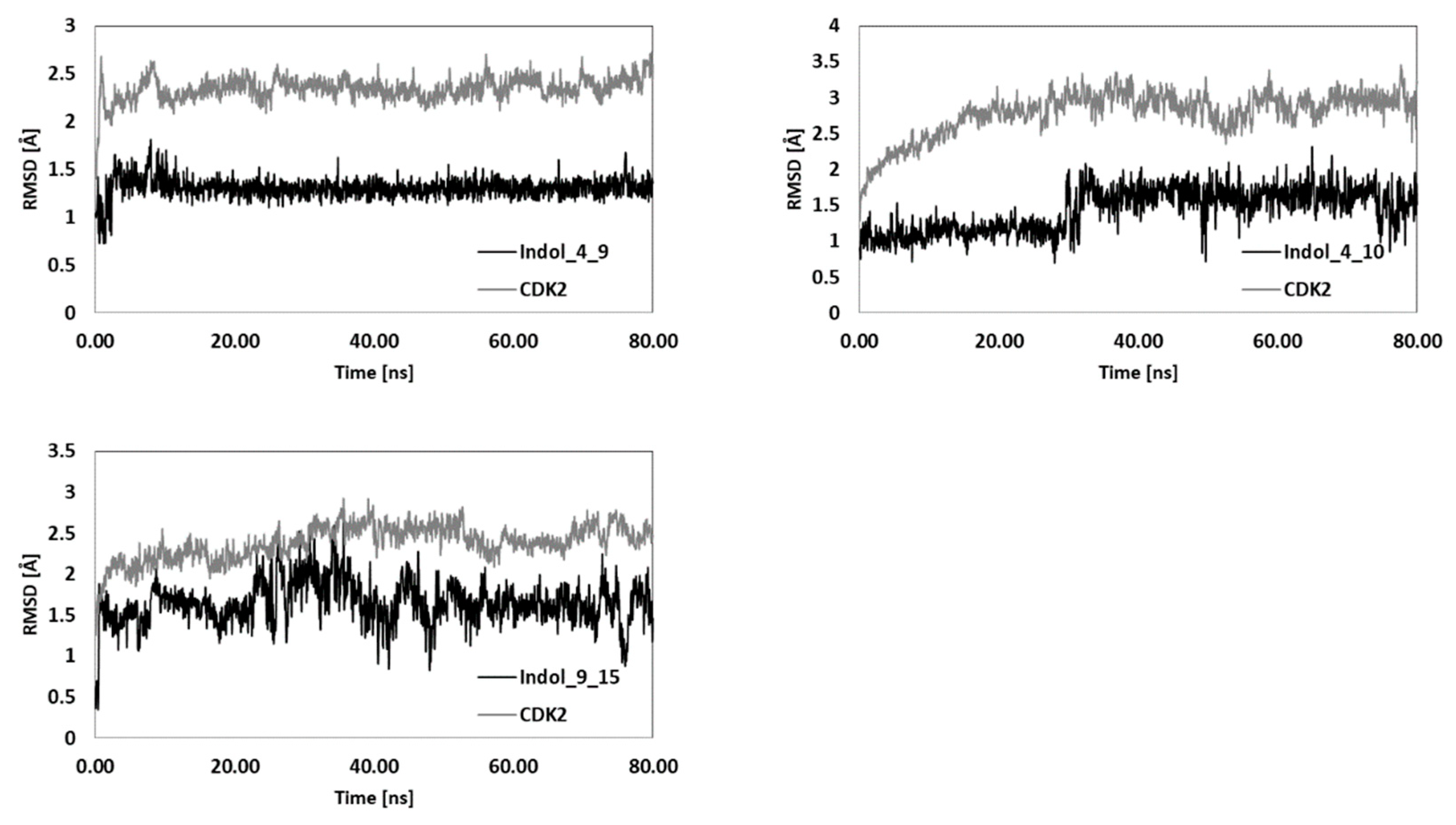
| Indol_4_9 | Indol_4_10 | Indol_9_15 | Indol_20_10 | Indol_20_15 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CDK2 | LIG | CDK2 | LIG | CDK2 | LIG | CDK2 | LIG | CDK2 | LIG | |
| RMSD | 2.36 | 1.30 | 2.91 | 1.54 | 2.46 | 1.68 | 2.72 | 0.93 | 2.49 | 1.46 |
| SD | 0.10 | 0.08 | 0.17 | 0.26 | 0.15 | 0.27 | 0.13 | 0.19 | 0.14 | 0.29 |
| Hydrogen Bond | Population % | |||||||
|---|---|---|---|---|---|---|---|---|
| ∑ | 1.5 Å | 1.75 Å | 2.0 Å | 2.25 Å | 2.5 Å | 2.75 Å | 3.0 Å | |
| Indol_4_9 | ||||||||
| Ligand (O2) … (HN) LEU 83 | 100.0 | 0.0 | 24.1 | 58.9 | 14.8 | 2.1 | 0.1 | 0.0 |
| Ligand (H3) … (O) GLU 81 | 100.0 | 0.0 | 14.1 | 58.3 | 24.1 | 3.3 | 0.3 | 0.0 |
| Ligand (O3) … (H) LYS 33 | 98.7 | 0.0 | 24.4 | 44.4 | 16.0 | 8.5 | 2.9 | 2.4 |
| Ligand (H1) … (O) ASP 86 | 98.3 | 0.0 | 10.2 | 37.1 | 29.4 | 13.7 | 5.5 | 2.4 |
| Ligand (H4) … (O) GLU 12 | 45.4 | 0.0 | 0.1 | 1.4 | 3.6 | 7.9 | 12.7 | 19.7 |
| Indol_4_10 | ||||||||
| Ligand (O2) … (HN) LEU 83 | 100.0 | 0.1 | 31.1 | 55.1 | 11.4 | 2.1 | 0.2 | 0.0 |
| Ligand (H3) … (O) GLU 81 | 100.0 | 0.0 | 19.5 | 57.2 | 19.1 | 3.3 | 0.9 | 0.0 |
| Ligand (O3) … (H) LYS 33 | 82.9 | 0.1 | 26.9 | 35.8 | 12.1 | 5.6 | 1.2 | 1.3 |
| Ligand (H2) … (O) GLU 8 | 69.1 | 15.4 | 43.6 | 8.4 | 1.0 | 0.4 | 0.1 | 0.2 |
| Ligand (H4) … (O) GLU 12 | 46.4 | 0.0 | 10.0 | 21.1 | 8.5 | 3.3 | 2.3 | 1.3 |
| Indol_9_15 | ||||||||
| Ligand (O3) … (HN) LEU 83 | 100.0 | 0.1 | 24.5 | 55.2 | 17.0 | 2.3 | 0.7 | 0.3 |
| Ligand (H5) … (O) GLU 81 | 100.0 | 0.1 | 27.1 | 57.8 | 12.9 | 1.9 | 0.3 | 0.0 |
| Ligand (O4) … (H) LYS 33 | 94.3 | 0.2 | 44.3 | 40.5 | 7.3 | 1.5 | 0.3 | 0.3 |
| Ligand (F) … (H) GLY 13 | 31.4 | 0.0 | 0.0 | 0.3 | 1.0 | 3.8 | 9.6 | 16.8 |
| Indol_20_10 | ||||||||
| Ligand (O1) … (HN) LEU 83 | 100 | 0.1 | 25.7 | 57.3 | 14.8 | 2.3 | 0.0 | 0.0 |
| Ligand (H5) … (O) GLU 81 | 100 | 0.0 | 27.9 | 56.6 | 13.5 | 1.9 | 0.2 | 0.0 |
| Ligand (O4) … (H) LYS 33 | 92.5 | 0.1 | 38.9 | 35.3 | 13.8 | 2.0 | 1.0 | 1.4 |
| Ligand (NHX) … (O) ASP 86 | 50.7 | 0.2 | 18.8 | 22.1 | 5.2 | 1.6 | 1.1 | 1.7 |
| Ligand (H4) … (O) GLU 8 | 85.7 | 21.3 | 54.2 | 8.8 | 1.0 | 0.3 | 0.1 | 0.1 |
| Ligand (NHX) … (O) ILE 10 | 61.3 | 0.0 | 10.9 | 25.0 | 11.2 | 4.5 | 4.9 | 4.8 |
| Indol_20_15 | ||||||||
| Ligand (O3) … (HN) LEU 83 | 100.0 | 0.1 | 23.2 | 59.4 | 15.0 | 2.3 | 0.1 | 0.0 |
| Ligand (H6) … (O) GLU 81 | 100.0 | 0.0 | 24.3 | 61.4 | 13.2 | 0.9 | 0.1 | 0.0 |
| Ligand (O4) … (H) LYS 33 | 89.6 | 0.3 | 38.4 | 39.4 | 8.2 | 2.5 | 0.6 | 0.2 |
| Ligand (H3) … (O) ASP 86 | 48.0 | 0.0 | 11.4 | 20.0 | 9.0 | 3.3 | 2.6 | 1.7 |
| Ligand (H1) … (O) HIE 84 | 56.7 | 0.1 | 20.9 | 25.1 | 6.8 | 1.6 | 0.9 | 1.2 |
| Ligand (O1/2) … (H) LYS 89 | 73.6 | 0.1 | 6.7 | 23.5 | 17.6 | 11.8 | 8.5 | 5.4 |
| Indol_4_9 | Indol_4_10 | Indol_9_15 | Indol_20_10 | Indol_20_15 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔE | SD | ΔE | SD | ΔE | SD | ΔE | SD | ΔE | SD | |
| EVDWAALS | −49.11 | 2.69 | −46.55 | 4.11 | −52.15 | 3.91 | −49.56 | 5.35 | −48.56 | 7.74 |
| EEL | −51.37 | 5.47 | −48.50 | 11.03 | −51.08 | 9.87 | −48.47 | 8.36 | −47.83 | 12.99 |
| EPB | 64.27 | 6.44 | 65.24 | 8.06 | 77.96 | 8.56 | 63.82 | 5.35 | 70.35 | 11.03 |
| ECAVITY | −4.47 | 1.04 | −4.65 | 1.70 | −4.22 | 2.20 | −5.11 | 0.19 | −6.98 | 3.60 |
| ΔH | −40.67 | 8.81 | −34.47 | 9.02 | −29.48 | 10.32 | −39.31 | 8.31 | −33.02 | 7.22 |
| Nanostructure Name | ΔG (kcal/mol) | Binding Constant (Kmax) | Difference of Kmax Relative to C60 (%) | |||
|---|---|---|---|---|---|---|
| MAX | MIN | AVERAGE | SD | |||
| FF_1 | −8.1 | −7.6 | −7.78 | 0.12 | 865722.5 | 657.9 |
| FF_2 | −7.9 | −7.6 | −7.76 | 0.07 | 617698.8 | 440.8 |
| FF_3 | −8.1 | −7.8 | −7.92 | 0.10 | 865722.5 | 657.9 |
| FF_4 | −7.9 | −7.7 | −7.76 | 0.07 | 617698.8 | 440.8 |
| FF_5 | −8.4 | −8 | −8.14 | 0.13 | 1436420.7 | 1157.5 |
| FF_6 | −7.9 | −7.5 | −7.74 | 0.10 | 617698.8 | 440.8 |
| FF_7 | −8 | −7.8 | −7.91 | 0.07 | 731270.0 | 540.2 |
| FF_8 | −7.9 | −7.7 | −7.79 | 0.04 | 617698.8 | 440.8 |
| FF_9 | −8.1 | −7.7 | −7.94 | 0.13 | 865722.5 | 657.9 |
| FF_10 | −7.6 | −7.3 | −7.47 | 0.08 | 372283.6 | 225.9 |
| FF_11 | −8.3 | −7.7 | −7.92 | 0.15 | 1213334.8 | 962.2 |
| FF_12 | −8.7 | −8.2 | −8.37 | 0.11 | 2383332.1 | 1986.5 |
| FF_13 | −8 | −7.6 | −7.80 | 0.13 | 731270.0 | 540.2 |
| C60 | −6.9 | −6.8 | −6.84 | 0.05 | 114226.6 | 0.0 |
| Nanostructure Name | ΔG (kcal/mol) | Binding Constant (Kmax) | Difference of Kmax Relative to C60 (%) | |||
|---|---|---|---|---|---|---|
| MAX | MIN | AVERAGE | SD | |||
| FF_1 | −8.60 | −8.10 | −8.31 | 0.12 | 2013184.5 | 962.2 |
| FF_2 | −8.40 | −7.80 | −8.14 | 0.19 | 1436420.7 | 657.9 |
| FF_3 | −8.90 | −8.30 | −8.56 | 0.15 | 3340307.9 | 1662.4 |
| FF_4 | −8.30 | −8.00 | −8.15 | 0.09 | 1213334.8 | 540.2 |
| FF_5 | −8.90 | −8.60 | −8.71 | 0.10 | 3340307.9 | 1662.4 |
| FF_6 | −8.50 | −8.10 | −8.24 | 0.10 | 1700523.4 | 797.2 |
| FF_7 | −8.70 | −8.50 | −8.63 | 0.07 | 2383332.1 | 1157.5 |
| FF_8 | −8.70 | −8.30 | −8.51 | 0.11 | 2383332.1 | 1157.5 |
| FF_9 | −8.90 | −8.30 | −8.61 | 0.17 | 3340307.9 | 1662.4 |
| FF_10 | −8.00 | −7.70 | −7.89 | 0.12 | 731270.0 | 285.8 |
| FF_11 | −9.20 | −7.80 | −8.31 | 0.35 | 5542292.2 | 2824.3 |
| FF_12 | −9.20 | −8.60 | −8.86 | 0.19 | 5542292.2 | 2824.3 |
| FF_13 | −8.60 | −8.20 | −8.36 | 0.11 | 2013184.5 | 962.2 |
| C60 | −7.20 | −7.10 | −7.14 | 0.05 | 189526.5 | 0.0 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czeleń, P. Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy. Symmetry 2019, 11, 974. https://doi.org/10.3390/sym11080974
Czeleń P. Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy. Symmetry. 2019; 11(8):974. https://doi.org/10.3390/sym11080974
Chicago/Turabian StyleCzeleń, Przemysław. 2019. "Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy" Symmetry 11, no. 8: 974. https://doi.org/10.3390/sym11080974
APA StyleCzeleń, P. (2019). Investigation of the Inhibition Potential of New Oxindole Derivatives and Assessment of Their Usefulness for Targeted Therapy. Symmetry, 11(8), 974. https://doi.org/10.3390/sym11080974