Abstract
Since the first reports, the Ugi four-component reaction (U-4CR) has been recognized as a keystone transformation enabling the synthesis of peptide mimetics in a single step and with high atom economy. In recent decades, the U-4CR has been a source of inspiration for many chemists fascinated by the possibility of identifying new efficient organic reactions by simply changing one of the components or by coupling in tandem the multicomponent process with a huge variety of organic transformations. Herein we review the synthetic potentialities, the boundaries, and the applications of the U-4CR involving α-amino acids, where the presence of two functional groups—the amino and the carboxylic acids—allowed a 5-center 4-component Ugi-like reaction, a powerful tool to gain access to drug-like multi-functionalized scaffolds.
1. Introduction
Multicomponent reactions (MCRs) represent an efficient one-pot synthetic strategy to generate, from three or more reagents, a new product containing almost all portions of the starting materials. For their convergent nature, atom economy and efficiency, MCRs are considered valuable methodologies for both medicinal and organic chemists. In particular, Isocyanide-based Multicomponent Reactions (IMCRs) [1,2,3,4], have been proven to be an ideal tool to provide in one single step medium-complexity molecular skeletons, usually accessible only via a multistep approach. Most of MCR chemistry is performed with isocyanides and is related to the Ugi reaction [5,6], a four-component transformation (4CR) described in 1959 by Professor Ivar Ugi. In the reaction, an acid component, like a carboxylic acid, reacts with an oxo-component (a ketone or an aldehyde), a primary amine, and an isocyanide. In detail, as shown in Scheme 1, the first step is the condensation between the oxo-component 1 and the amine 2 to generate the Schiff base 6. Then, the acid component protonates the nitrogen atom of the Schiff base thus increasing its electrophilicity. Hence, the nucleophilic addition of the isocyanide 3 to the Schiff base produces the nitrilium ion 9, which rapidly reacts with the nucleophilic carboxylic acid anion 8. The α-adduct 10 so formed is finally converted into the Ugi product 5 through an intramolecular acylation, which resembles the Mumm-type rearrangement.
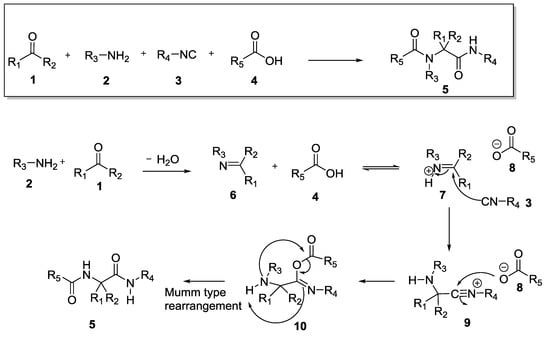
Scheme 1.
The Ugi 4-component reaction.
The replacement of one of the components of the Ugi reaction, or the incorporation of two or more functional groups into a single moiety, as well as the combination of the Ugi reaction with other chemical transformations have provided straightforward synthetic approaches to a large number of different scaffolds with a rich structural diversity. In particular, the presence of a primary amine and a carboxylic acid functional group both in natural and unnatural α-amino acids make them very useful synthons suitable for a three-component Ugi-like reaction (3CUR) [7].
In this review, we summarized all the Ugi reactions involving α-amino acids reported to date (March 2019). For the sake of simplicity, this review has been divided into four sections. In the second section, linear products will be gathered, while the third section will contain cyclic products obtained via the 3CUR. In the fourth section, we will focus our attention on products designed and synthesized for medicinal chemistry applications, while in the fifth section synthetic and biosynthetic reactions of α-amino acids in Ugi-like transformations for the obtainment of natural products will be taken into consideration. For better clarity, in some cases, specific examples have been reported, while in others, d the general reaction is reported. Nevertheless, for any given transformation, the number of reported examples and the range of yields are always reported, when available.
2. Linear Compounds
2.1. Ugi 5C-4CR Using α-Amino Acids, Aldehydes, Chloroaldehydes and Ketones
In 1996, Ugi et al. described the first Ugi 5C-4CR using an α-amino acid as a starting bi-functional material to yield 1,1′-iminodicarboxylic acid derivatives (97–99% yield, Scheme 2) [8,9,10,11,12,13,14]. In the proposed reaction mechanism (Scheme 2) the amine functional group of the α-amino acid condenses with the aldehyde 1 to give the imine 12. The subsequent α-addition of the isocyanide followed by the intramolecular interception of the nitrilium ion, forms an O-acylamide 13. The nucleophilic attack of the fourth component (the alcohol, i.e., the fifth reacting center) 14 at the carboxylic carbon atom and the subsequent rearrangement provides the 1,1′-iminodicarboxylic acid ester derivative 15. Notably, the side chain of tri-functional α-amino acids (L-Serine, L-Threonine, L-Tyrosine, L-Asparagine, L-Glutamine, L-Methionine) did not participate in the Ugi reaction.
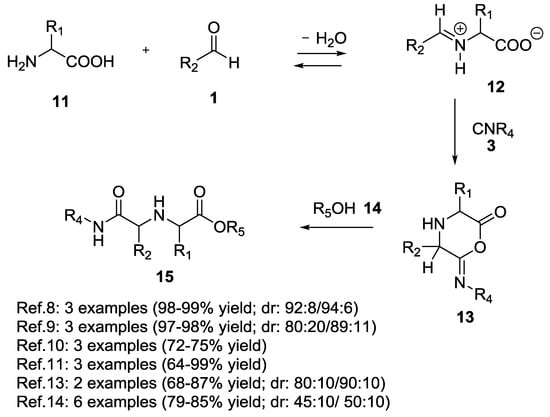
Scheme 2.
The Ugi 5-center-4-components reaction.
Ugi et al. also investigated the use of a halogen-substituted aldehydes (Figure 1), in particular chloroacetaldehyde with methylisocyanide in methanol. Interestingly, only traces (up to 10%) of the substitution on halogen with the secondary amine of U-5C-4CR product were observed. Yields and diasteromeric excesses were less than those observed using unsubstituted aldehydes.
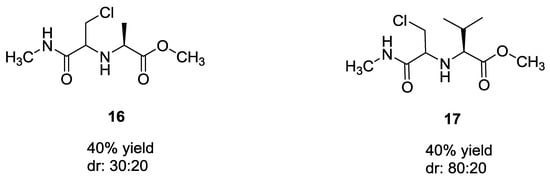
Figure 1.
Use of halogen-substituted aldehydes with α-amino acids.
The U-5C-4CR was conducted also by using ketones instead of aldehydes [15]. Due to the low reaction rate of ketones, the reaction time increased from approximately one day to several weeks (4 examples, 64–94% yield). The same authors reported the formation of cyclic analogues, which are reported in Section 3.
2.2. Tandem Ugi-asserini Reactions Involving α-Amino Acids
The use of lysine triggered some interesting studies regarding the possibility of combining more than one MCR in tandem. Indeed, this α-amino acid can react with two equivalents of aldehyde, an isocyanide, and a carboxylic acid to afford the linear product 23 in 22.8% yield (Scheme 3) along with the Passerini product 24 in 18.5% yield [16]. Excess of isocyanide, and carboxylic acid led to the Ugi Nine Center Seven Component Reaction (U-9C-7CR) product 25 in 8% yield.
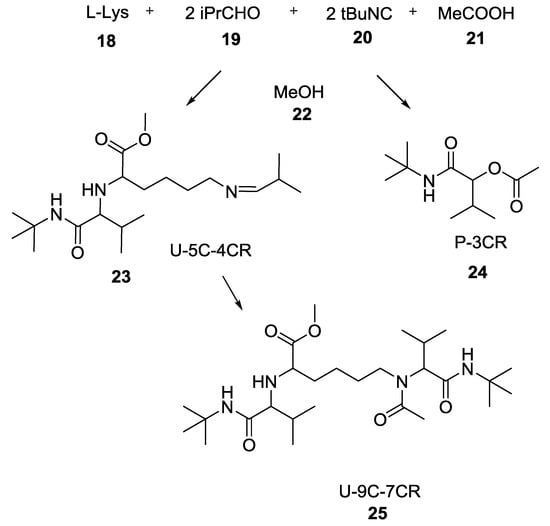
Scheme 3.
Use of L-lysine in the combination of MCRs.
Similarly, a tandem Ugi-Passerini reaction was observed when L- glutamic and L-aspartic acid were used (Scheme 4) [9]. In particular, the Ugi reaction using L-aspartic acid (26) with two equivalents of both aldehyde and isocyanide provided a linear compound 27 in contrast to from L-glutamic acid that gave cyclic diketopiperazine.
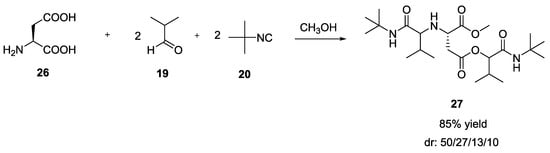
Scheme 4.
Use of L-aspartic acid in the combination of MCRs.
2.3. α-Amino Acids as Chiral Auxiliaries in the U-5C-4CR Reaction
Transtech Pharma patented the use of α-amino acids as chiral auxiliaries for the synthesis of enantiomerically pure N-alkyl-N-acyl-α-amino amides 15, which after the cleavage of both the chiral auxiliary amine and the hydrolysis of amide, and the subsequent protection of the amino group, provided N-protected α-amino acids 30 (Scheme 5) [17].
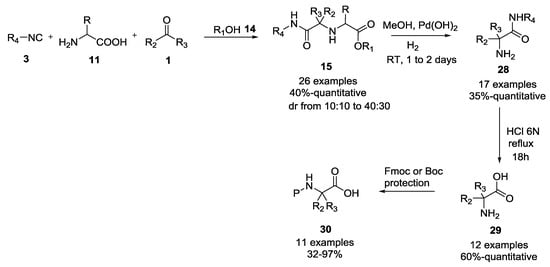
Scheme 5.
Use of α-amino acids as chiral auxiliaries in U-5C-4CR.
2.4. Diastereoselectivity in the U-5C-4CR Reaction
Sung, K. et al. investigated the steric effect of both aldehydes and α-amino acids on the diasteroselectivity of the Ugi reaction [18]. When bulky aldehydes (9-anthraldehydes, 2-ethylbutyraldehyde and isobutyraldehyde) were used in combination with L/D α-amino acids, the diasteroisomeric excess was 99%, while with less bulky aldehydes (such as benzaldehyde or n-butyraldehyde), the final products were obtained with a lower diasteroisomeric excess (43–79% de). Similarly, bulky enantiomerically pure L/D α-amino acids gave higher de (99%). The proposed mechanism showed that bulky substituents preferred to stay at equatorial positions of the six-membered Ugi intermediate to prevent serious 1,3-diaxal and ‘butan-gauche’ nonbonding interactions (Scheme 6).
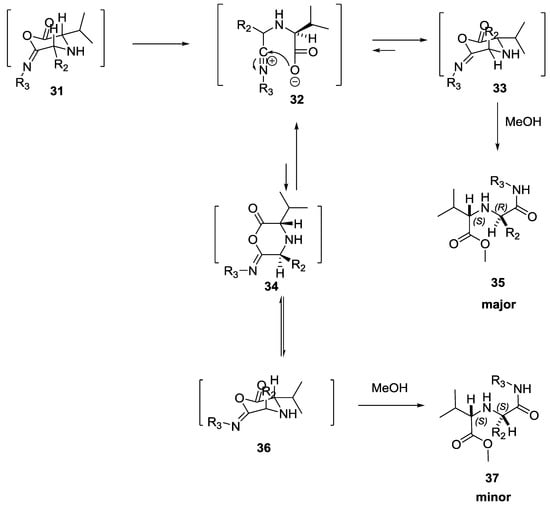
Scheme 6.
Use of bulky aldehydes in combination with L/D α-amino acids.
Further information about the possibility of a substantial diastereoselectivity in the U-5C-4CR came from a report by Chen X. et al. The Ugi reaction of α-amino acids such as L-valine and L-serine, aromatic aldehydes, and isocyanides was employed for the synthesis of 1,1′-iminodicarboxylic acid derivatives, which were then used as key intermediates to synthesize tetrahydroisoquinoline compounds (Scheme 7) [19]. L-valine gave tetrahydroisoquinolin-4-ol 42 in nine reaction steps with high stereoselectivity. The configuration of the cyclic compound 42 was hence determined by NOESY NMR. Substituent at C-3 and C-4 resulted in a cis configuration as showed by correlation between H-3 (δ = 2.73 ppm) and H-4 (δ = 4.57 ppm). Consequently, the absolute configuration of C-1 and C-4 in 42 was determined to be R and S, respectively. Subsequently, Ugi product 41 from (S)-valine had the S,R configuration. This result could support a general rule regarding the stereochemistry of the similar Ugi reactions harnessing primary α-amino acids.
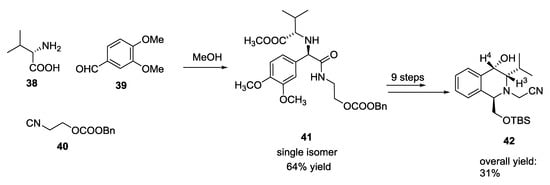
Scheme 7.
Synthesis of strained tetrahydroisoquinolines via a U-5C-4CR.
2.5. Use of TiCl4 as Lewis Acid in the U-5C-4CR Reaction
Ciufolini and coworkers screened two Brønsted acids and thirteen Lewis acids to extend the scope of Ugi reaction involving α-amino acids and aromatic aldehydes (Scheme 8) [20]. Brønsted acids proved to be ineffective and harmful promoters. For example, trifluoracetic acid showed no improvement of the reaction and methanesulfonic acid gave no desired compound, perhaps due to polymerization or degradation of the isocyanide. In contrast, Lewis acids had beneficial effects on both yields and rates. TiCl4 showed the best results (75–90% yield), which were not attributable to the HCl in situ release. The sense of diastereoinduction is (S,S), as confirmed by X-ray crystallography.
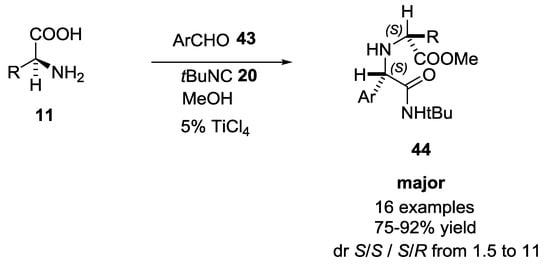
Scheme 8.
Titanium (IV) chloride catalyzed U-5C-4CR with aromatic aldehydes.
2.6. Ketones and Secondary α-Amino Acids in the U-5C-4CR Reaction
To expand the molecular diversity of Ugi 5C-4CR, various symmetrical and asymmetrical ketones were combined with secondary α-amino acids [21]. Also in this case, catalytic amounts of TiCl4 as Lewis acid proved to increase the reaction yield. The combination of bulky ketones with unbulky isocyanides gave the best yields, while unbulky ketones gave no significative difference in yields if combined with bulky or unbulky isocyanides (0–67% yield, Scheme 9). The diastereoselectivity of the U-5C-4CR may be sensitive to the nature of coupling reagents and reaction conditions. Following 2D-NMR studies on strained cyclic analogues, the authors noticed that the degree and the sense of diastereoinduction observed could not be easily rationalized.
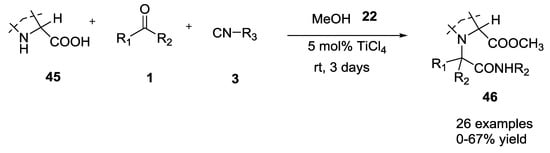
Scheme 9.
Use of ketones and secondary α-amino acids in U-5C-4CR.
In general, the stereochemical outcomes of this Ugi-5C-4CR variant depended on the structure of both the isocyanide and the ketone employed. These results indicated that it was not possible to draw general conclusions and that the mechanism and the diastereoselectivity of the Ugi-5C-4CR employing ketones is still a subject of debate.
2.7. Direct Conversion of Ugi Ester Scaffold to Amide
Dömling and co-workers investigated the one-pot conversion of the methyl ester function of the U-5C-4CR scaffold [22]. They described a one-pot amidation (24 examples, 15–82% yield, Scheme 10) from α-amino acid methyl esters under solventless conditions and at ambient temperature or using THF as solvent and several different amines, including aliphatic, heterocyclic, aromatic, and functionalized ones.
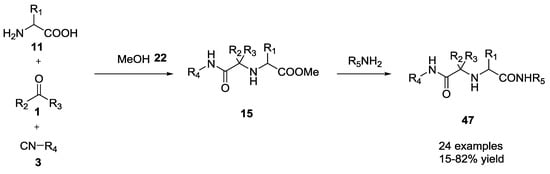
Scheme 10.
One-pot conversion of the methyl ester of U-5C-4CR products.
2.8. Selenium α-Amino Acids in the U-5C-4CR Reaction
The same group reported a variation of the Ugi reaction using bifunctional selenium α-amino acids as starting materials to generate derivatives including methyl selenocysteine 48 and selenomethionine 49 (11 examples, 48–95% yield, Scheme 11) [23].
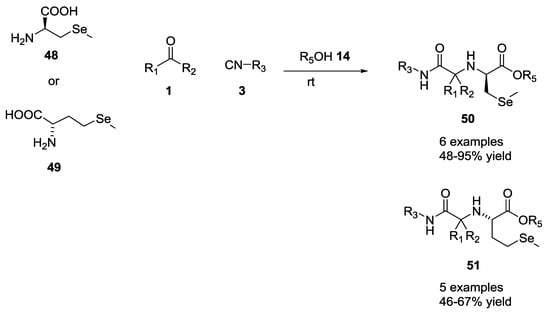
Scheme 11.
Use of selenium α-amino acids in U-5C-4CR.
On the other hand, the use of diselenocysteine in the Ugi 5C-4CR (Scheme 12) gave low yield of the desired compound 55, even when stirring the reaction for 24 h or refluxing for 12 h, most probably because of the poor solubility of the diselenide 54.
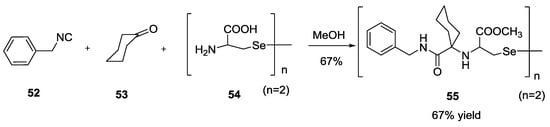
Scheme 12.
Use of diselenocysteine in Ugi 5C-4CR.
2.9. Isocyanocarbamates in the U-5C-4CR
Sello et al. reported the U-5C-4CR with isocyanocarbamates, α-amino acids and aldehydes [24]. It is worth noting that, when histidine was used as starting α-amino acid, the product was linear rather than cyclic as reported by Ugi (Figure 2). On the other hand, with glutamine, although a linear product was expected, the observation of a piperazinedione structure was indicative of a ring-closure due to intramolecular attack of the glutamine primary amide into the Ugi imino anhydride intermediate.
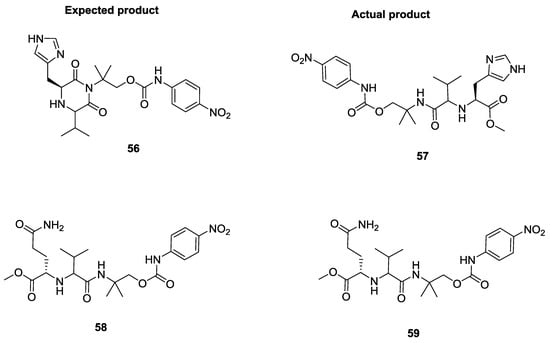
Figure 2.
U-5C-4CR with isocyanocarbamates and histidine and glutamine.
2.10. Silica Nanoparticles as Green Catalyst for U-5C-4CR
Recently, Esrafili and coworkers described an efficient and green one-pot synthesis of sulfonylamide derivatives (42–68% yield, Scheme 13) from L-α-amino acids, aromatic aldehydes and p-toluenesulfonylmethyl isocyanide in water/methanol using silica nanoparticles (SNP) as the catalyst [25]. In the absence of SNP, the reactions did not work efficiently, and products 61 were obtained in low yields.
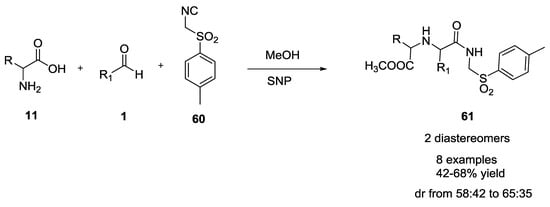
Scheme 13.
SNP catalyzed synthesis of sulfonylamide derivatives.
2.11. Use of DMAP-Based Aldehydes in U-5C-4CR
Dimethylaminopyridine (DMAP)-based aldehydes were used with α-amino acids and tert-butyl isocyanide in the Ugi reaction to produce diverse chiral DMAP derivatives [26]. 4-(dimethylamino)-2-pyridine-carboxaldehyde 62 (Scheme 14) afforded products with low diasteroselectivity compared to 4-(dimethylamino)-3-pyridine carboxaldehyde 65 (Scheme 15). Diastereomeric ratios and yields were obtained changing substrates concentration and α-amino acids used in the Ugi reaction.
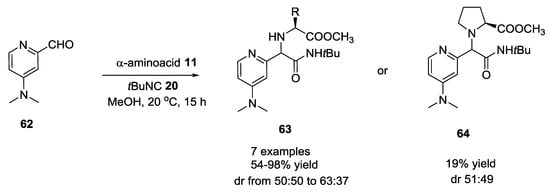
Scheme 14.
U-5C-4CR with 4-(dimethylamino)-2-pyridine-carboxaldehyde.
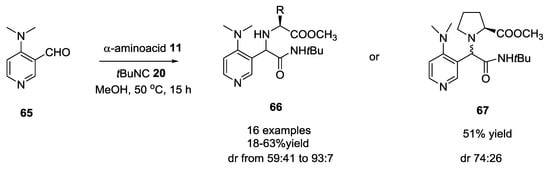
Scheme 15.
U-5C-4CR with 4-(dimethylamino)-3-pyridine carboxaldehyde.
The chiral diastereomerically pure DMAP derivatives were used for the kinetic resolution of secondary alcohols in presence of acetic anhydride and triethylamine in toluene [27].
3. Cyclic Compounds
3.1. Ugi-5C-4CRs and One-Pot Post-Condensation Modifications
One of the first reports regarding the obtaining of a diketopiperazine scaffold was by Ugi et al. in 1998. The formation of 2,6-piperazinediones was accomplished under weak basic conditions and in a one-pot reaction by using only ketones (43–70%, Scheme 16) [15]. Alternatively, the U-5C-4CR products 15 could be cyclized to 2,6-piperazindiones 68 by refluxing them in THF under basic conditions using potassium tert-butoxide (68–72% yield). However, stronger basic conditions and higher temperatures resulted in the racemization of the chiral centers.
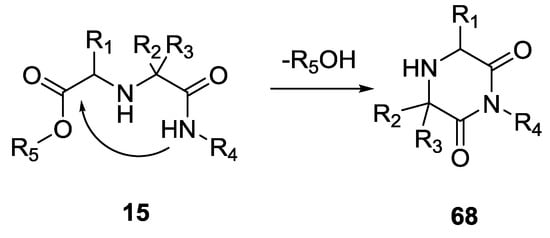
Scheme 16.
Intramolecular attack leading to 2,6-piperazinediones.
One exception had been reported, again by Ugi, a couple of years previously, when histidine 69 was used as starting α-amino acid with an aldehyde and an isocyanoester. In this case, the formation of intramolecular H-bonds between the -NH of imidazole ring and the carbonyl group of isocyanoester 71 allowed for the isolation of the diketopiperazine derivative 72 when the reaction was run at room temperature and under neutral conditions (Scheme 17).
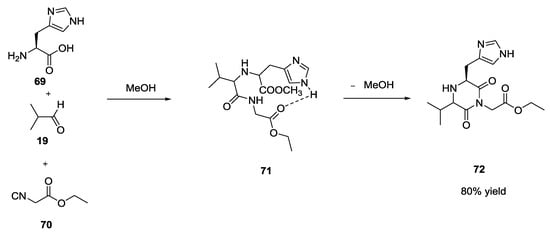
Scheme 17.
U-5C-4CR with histidine yielding diketopiperazine (72).
The sense of diastereoinduction, for reactions with unsymmetrical ketones, was studied by Turło et al. by converting the resulting Ugi adducts into the corresponding rigid 2,6-diketopiperazine derivatives (Scheme 18). In particular, in this study, the authors expanded the scope of the Ugi-5C-4CR to secondary α-amino acids, such as proline, and to ketones. The linear adducts then underwent a N-detert-butylation/cyclocondensation sequence leading to N-unsubstituted cyclic derivatives 75, 76, 80–82 by reacting them in BF3⋯2 CH3COOH. Direct cyclization with NaOH of 83 and 86 gave directly cyclic compounds 85 and 87. NOESY spectra of cyclic compounds showed that stereochemistry was not the same for all the synthesized compounds, but depended on the structure of both the isocyanide and the ketone inputs [21].
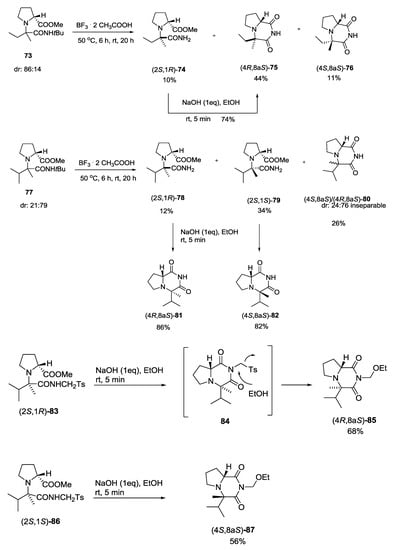
Scheme 18.
Diketopiperazine derivatives useful to study diastero-induction of the U-5C-4CR.
The use of siloxycyclopropanes in the Ugi-5C-4CR was described by Hans-Ulrich Reissig and coworkers to form highly substituted pyrrolidinone derivatives (Scheme 19) [28]. Siloxycyclopropanes 88 were used as direct precursor of β-formyl esters with tert-butyl isocyanide and α-amino acids yielding the linear coupling products 89 (46–82% yield). These compounds were cyclized to pyrrolidinone derivatives 90 (16–98% yield) by heating them in toluene at reflux temperature.
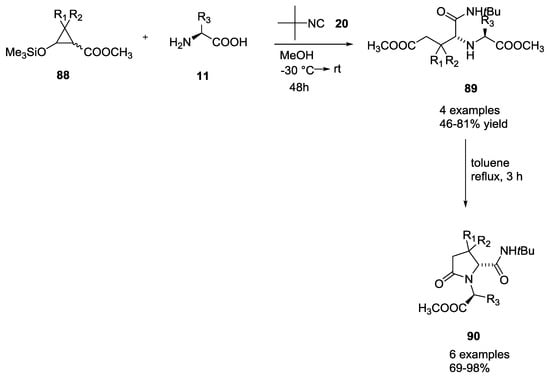
Scheme 19.
Reaction of siloxycyclopropanes with α-amino acids and isocyanides.
When a chiral syloxycyclopropane was employed, the reaction resulted in 4 diastereoisomers; while non-chiral syloxycyclopropanes produced only 2 diastereoisomers (Scheme 20).
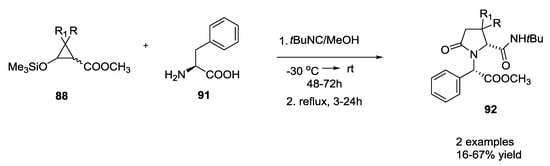
Scheme 20.
Reaction of chiral siloxycyclopropanes with α-amino acids and isocyanides.
Precursors of γ-ketoesters such as 93 seemed to be inefficient in Ugi reactions, providing only 16% of the desired adduct 94 (Scheme 21).
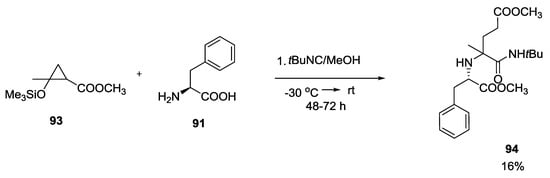
Scheme 21.
Reaction of precursors of γ-ketoesters with α-amino acids and isocyanides.
Similarly, 1-trifluoromethyl-2-(trimethylsilyloxy)cyclopropanecarboxylate 95, isonitriles and α-amino acids (such as glycine 97 and phenylalanine 91) provided CF3-substituted γ-lactam (Scheme 22) [29]. In particular, the reaction with glycine at reflux temperature gave the cyclic compound 99 directly, without isolation of the linear compound 98. On the contrary, when the U-5C-4CR was performed with phenylalanine, the reaction mixture was heating for 5 days to cyclize to γ-lactam 101.
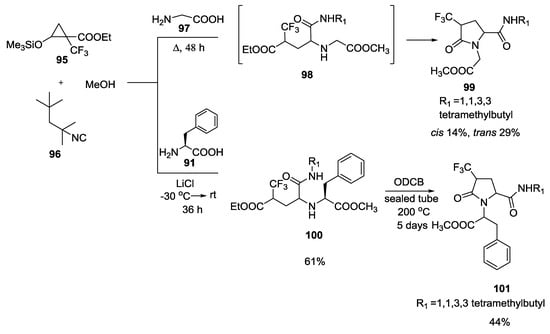
Scheme 22.
U-5C-4CR with 1-trifluoromethyl-2-(trimethylsilyloxy)-cyclopropane-carboxylate, isonitriles and α-amino acids.
The U-5C-4CR can be extremely powerful in getting access to molecular diversity and complexity when the linear adduct is further manipulated in a post-MCR transformation. A few examples have been reported herein. For example, highly functionalized constrained nitrogen-heterocycles were reported by Gracias et al. as examples of post-Ugi reaction manipulation [30]. Allyl glycine 103, o-bromo-benzaldehyde 102 and benzyl isocyanide 52 in methanol yielded the Ugi cyclic adduct 104, which was quickly converted into the aminoester 103. Finally, a microwave-assisted Heck cyclization generated N-containing heterocycle (106) (90% yield, Scheme 23).
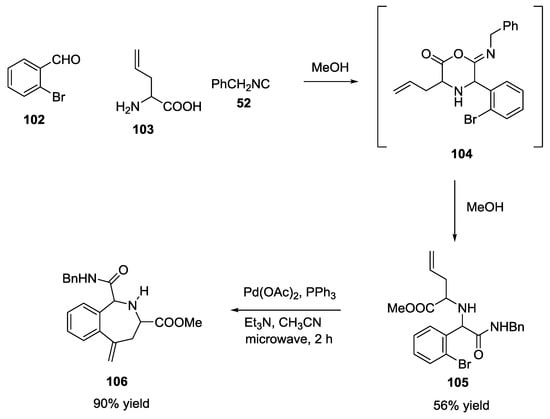
Scheme 23.
U-5C-4CR and Heck post-MCR modifications.
Chiral highly functionalized dihydroisoquinolines and isoindoles were synthesized by Dyker and coworkers using U-5C-4CR followed by a gold-catalyzed hydroamination [31,32]. Dyker et al. used L-valine as the chiral amine component, and benzaldehydes with an alkyne moiety to generate highly functionalized secondary amines converted in dihydroisoquinolines 111 and 112 by a 6-endo-dig cyclization (23% and 35% yield) or in isoindoles 110 by a 5-exo-dig cyclization and subsequent aromatization such as in 113 and 114 (38% and 49%, Scheme 24). A further tandem Diels Alder reaction was carried on isoindole derivatives using acetylene dicarboxylic acid dimethyl ester 115.
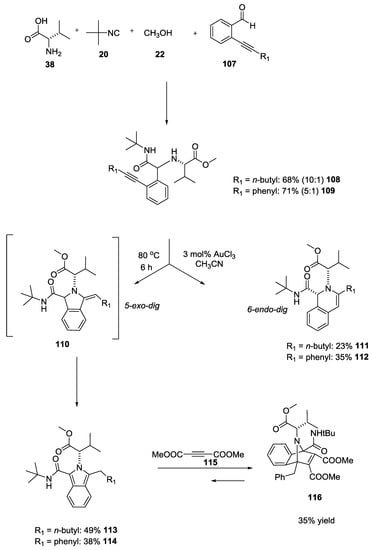
Scheme 24.
U-5C-4CR and post-MCR modifications leading to dihydroisoquinolines and isoindoles.
The Pictet–Spengler reaction was recently applied in tandem as Ugi post-condensation transformation to yield complex polycyclic scaffold (Scheme 25). In these cases, the U-5C-4-CR adducts were reacted in one pot, without any purification, for the subsequent Pictet–Spengler cyclization [33,34,35]. This procedure was successfully carried out on differently functionalized Ugi scaffolds in order to obtain a wide range of multi-functionalized heterocycles. Isoindolone scaffold 119 was obtained by introducing methyl 2-formylbenzoate as the oxo-component and by monitoring the reaction via supercritical fluid chromatography-mass spectrometry (SFC-MS). Primary amines provided diastereomeric ratios of 70:30, and only the major diastereomer was precipitated during the reaction. In contrast, secondary amines gave four stereoisomers in equal ratios, suggesting racemization of the amino acid (Scheme 25).
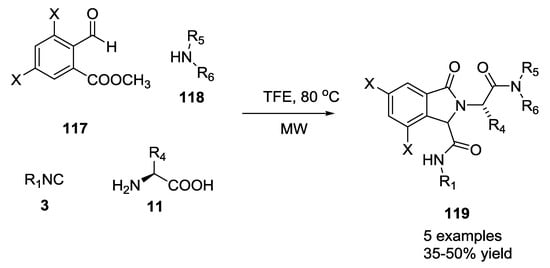
Scheme 25.
Isoindolones via U-5C-4CR and tandem Pictet–Spengler cyclization.
When β-ketoester, such as 2-oxocyclohexane carboxylic acid ethyl ester, was employed with 3 equivalents of cesium carbonate, pyrrolidinedione scaffold 121 was obtained either as a single diastereomer or mixture of two diastereomers easily isolated using preparative TLC plates (Scheme 26).
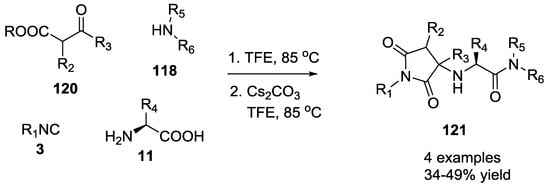
Scheme 26.
Pyrrolidinediones via U-5C-4CR and tandem Pictet–Spengler cyclization.
Reaction involving L-triptophan as a starting α-amino acid provided the strained tricyclic 3,9-diazabicyclo[3.3.1]nonanes 124 in a one-pot two-step reaction, employing formic acid as the catalyst (Scheme 27).
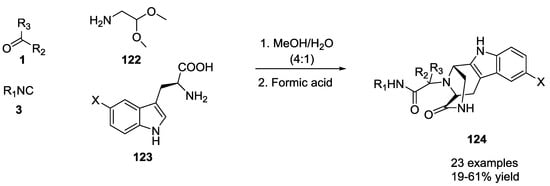
Scheme 27.
3,9-Diazabicyclo[3.3.1]nonanes via U-5C-4CR and tandem Pictet–Spengler cyclization.
Only U-5C-4C products bearing an electron-donating group on the meta position of the phenyl ring, such as 3-methoxy-phenylalanine and 3,4 dimethoxy-phenylalanine, were able to form isoquinolines derivatives 125 (Scheme 28).
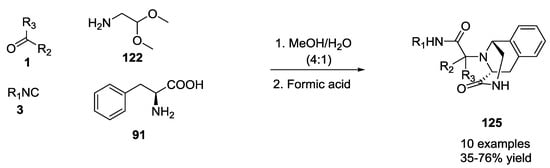
Scheme 28.
Isoquinolines via U-5C-4CR and tandem Pictet–Spengler cyclization.
The use of electron-rich aromatic α-amino acids such as phenylalanine also gave bicyclic tetrahydroimidazo-[1,2-a] pyrazine-2,6(3H,5H)-diones 126 with the formation of only trans diastereomers (Scheme 29).
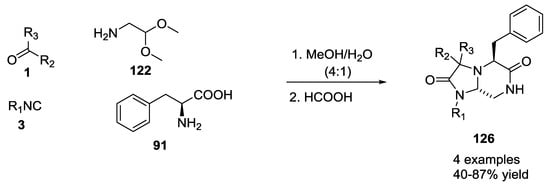
Scheme 29.
Tetrahydroimidazo-[1,2-a] pyrazine-2,6(3H,5H)-diones via U-5C-4CR and tandem Pictet–Spengler cyclization.
L-leucine, cyclohexanone, benzyl isocyanide and morpholine gave an Ugi product whose formation was monitored through SFC-MS, subsequently reacted in the presence of one equivalent of potassium carbonate to provide β-lactam derivatives 130 (Scheme 30).
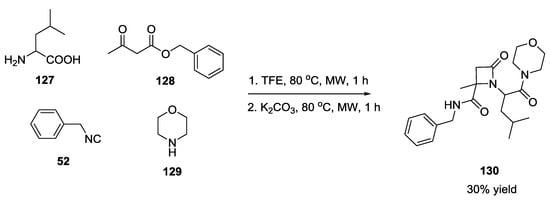
Scheme 30.
β-lactam derivatives via U-5C-4CR and tandem Pictet–Spengler cyclization.
3.2. Exploiting α-Amino Acids in the Combination of Tandem MCRs
In theory, thanks to the presence of both the amino and the carboxylic acid functionality on the α-amino acids, and to the use of two equivalents of aldehyde and isocyanide, it could be possible to combine more than one MCR in the same flask. For example, glycine 97, 2 equivalents of propionaldehyde 131, 2 equivalents of methyl isocyanide 132, and 1 equivalent of sodium azide 133 in methanol and in the presence of Dowex 50 allowed for a tandem Ugi-5C-4CR/ Ugi-azide 4CR, resulting in the formation in a single step of both tetrazole and 2,6-diketopiperazine rings 134 (Scheme 31) [16].
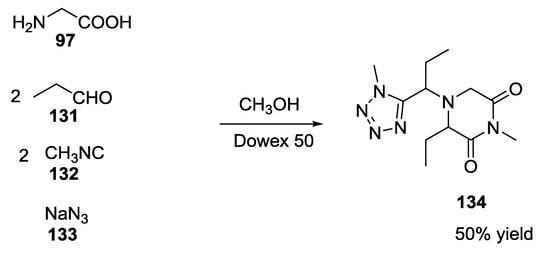
Scheme 31.
Formation of tetrazole derivatives via tandem MCRs.
To enable a wide variability in the functionalization of the final product, the reaction could also be performed in two steps: equimolar amounts of α-amino acid 97, aldehyde 131 and isocyanide 132 were one-pot mixed to give the Ugi-5C-4CR adduct 135, which was not isolated, but reacted directly with a different aldehyde 19, a different isocyanide 136 and sodium azide 133 to give a 1,1′-iminodicarboxylic acid derivative 137 (Scheme 32). It is worth noting that in this one-pot two-step approach, no cyclization was observed, and the linear adduct 137 was obtained.
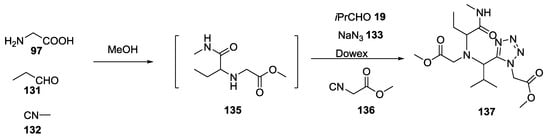
Scheme 32.
Formation of tetrazole derivatives via one-pot two-step tandem MCRs.
The possibility of performing more than one MCR in tandem, exploiting the amino and the carboxylic acid functionalities in two different reactions, has recently been investigated further. It was shown, indeed, that unprotected natural and unnatural α-amino acids, β/γ/ω-amino acids 138 with different aldehydes 1, isocyanides 3 and sodium azide 133 furnished tetrazolo peptidomimetics 139 in good yields as diastereomeric mixtures (Scheme 33) [36]. Two sequential Ugi tetrazole/Ugi reactions were also performed to provide more complex structures 141.
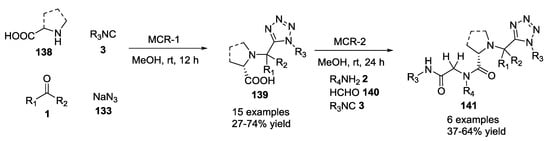
Scheme 33.
Tetrazoles via tandem Ugi-tetrazole/U-4CR.
The Ugi tetrazole reaction also worked with dipeptides (Gly Gly) 143 and tripeptides (Gly Gly Gly) 144 (Scheme 34).
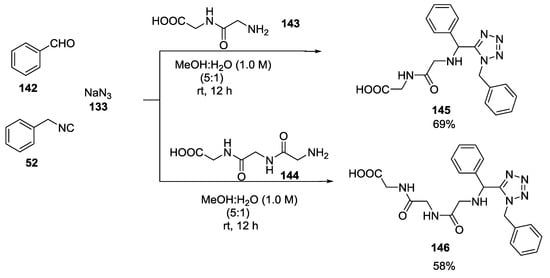
Scheme 34.
Ugi-tetrazole reaction employing dipeptides and tripeptides.
The application of two or more one-pot tandem MCRs was also made possible by the use of equimolar amounts of sodium glycinate 149, isocyanide 147, and acetone 148 as the oxo-component and silver acetate as Lewis acid catalyst (Scheme 35) [37]. This reaction inserted a carboxylic acid function in the 2H-2-imidazoline 150, which was then protonated and used in another Ugi 4-CR using i-PrCHO 19, n-propylamine or benzylamine 2 and tert-butylisocyanide 20 to give compounds 151 and 152.
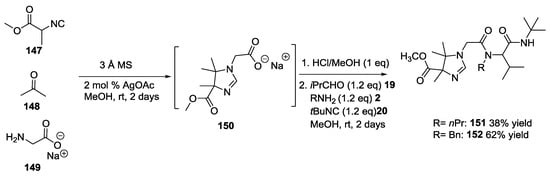
Scheme 35.
Two-step one-pot tandem U-5C-4CR/U-4CR.
Notably, this reaction was conducted in a one-pot sequence in which three different MCRs were combined to give the first example of an 8CR (Scheme 36). Extraordinarily, in this reaction, 5 new C-N bonds, and 4 new C-C bonds were formed in one pot.
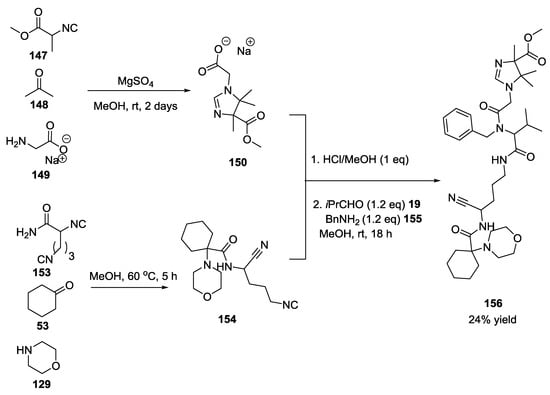
Scheme 36.
One-pot tandem U-5C-4CR/U-4CR.
On the other hand, as reported by Martens et al., when the α-amino acid was used in its hydrochloride form, the presence of the HCl served as the amine-protecting group, with only the carboxylic acid being able to be involved in an Ugi-like 4-CR [38,39]. In this reaction, the spiro derivatives of two 3-thiazolidines and one 3-oxazoline as imine component 157 were combined with glycine, β-alanine and γ-aminobutyric acid as hydrochloride salts 158. Cyclohexylisocyanide, ethyl isocyanoacetate, tert-butyl isocyanoacetate, and tert-butyl isocyanide were employed to give a small library of oligopeptide analogues 159 (20–85%, Scheme 37).
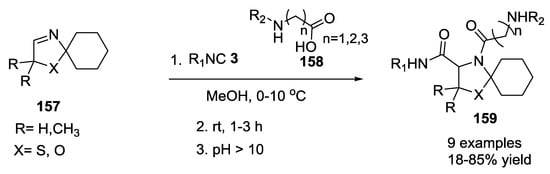
Scheme 37.
Spiro derivatives of two 3-thiazolidines and one 3-oxazoline as imine components.
3.3. Use of tri-Functional α-Amino Acids in Ugi-5C-4CRs
The use of α-amino acids with nucleophilic functional groups in their side chains such as amino-, sulfhydryl-, and hydroxyl-, enabled the synthesis of lactams, thiolactons, and lactones, respectively.
L-lysine 18, for example, formed a ε-lactam 161 from the O-acylamide intermediate 160 via nucleophilic attack of the side chain amine to the ester (Scheme 38) [9].
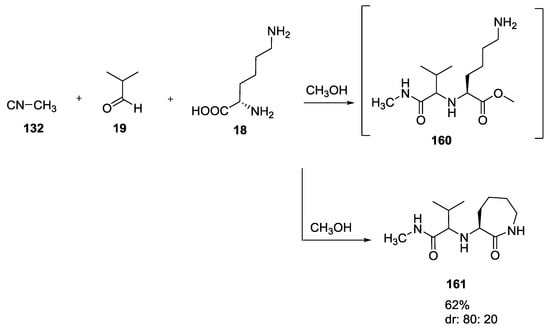
Scheme 38.
U-5C-4CR of L-lysine forming an ε-lactam.
A thiolactone scaffold 164 was generated during an Ugi 5C-4CR reaction using the α-amino acid homocysteine 162 with aldehydes and isocyanides in trifluorethanol as reported by Dömling et al. (Scheme 39) [40,41]. Trifluorethanol was preferred as a solvent, as it did not react with the 6-membered α-adduct of the Ugi reaction, and because it favored the intramolecular reaction with the nucleophilic side chain of homocysteine.
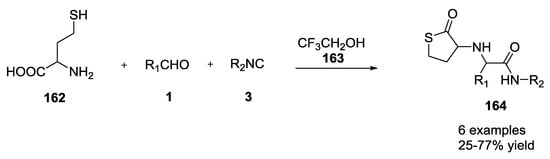
Scheme 39.
U-5C-4CR of homocysteine forming thiolactones.
Kim et al. illustrated an efficient synthesis for N-carbamoylmethyl-α-aminobutyrolactones 166 (42–97%, Scheme 40) starting from L-homoserine (165), aldeydes or ketones 1, and isocyanides 3 in 2,2,2-trifluoroethanol (163) [42]. It is worth noting that the reaction with hindered aldehydes proceeded with high diastereoselectivity, probably due to steric factors.
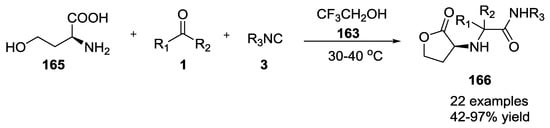
Scheme 40.
U-5C-4CR of homoserine affording α-aminobutyrolactones.
When methanol was used as a solvent, the cyclic compound was obtained along with the ring-opened compound coming from methanol attack on the carboxylate carbon of the imino-anhydride intermediate 167 (Scheme 41).
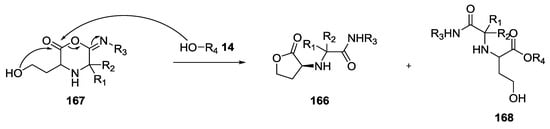
Scheme 41.
Alternative formation of ring-opened compound 168.
3.4. Combination of Other Bifunctional Starting Materials with α-Amino Acids
Kim et al. were able to demonstrate that commercially available glycolaldehyde dimer could be used efficiently in the Ugi condensation with α-amino acids to generate 3-substituted morpholin-2-one-5-carboxamide derivatives 170 (32–90% yield, Scheme 42) [43].
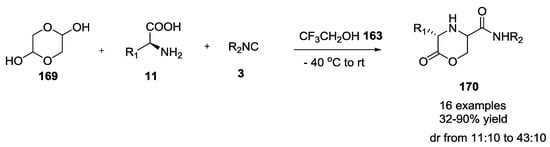
Scheme 42.
Use of glicolaldehyde dimer in the U-5C-4CR.
When cyclic α-amino acids were used, other unique heterobicyclic compounds 172 (37–90% yield, Scheme 43) were produced in moderate to good yields.
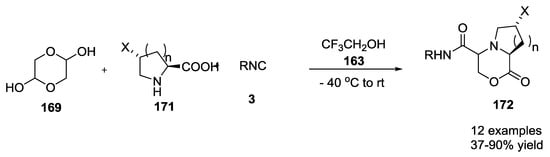
Scheme 43.
Use of glicolaldehyde dimer and cyclic α-amino acids in the U-5C-4CR.
The reaction mechanism forecasting the addition of the intramolecular hydroxyl group to the carboxylate carbon, resulting in the formation of 3-substituted morpholin-2-one-5-carboxamide derivatives 170 (Scheme 44).
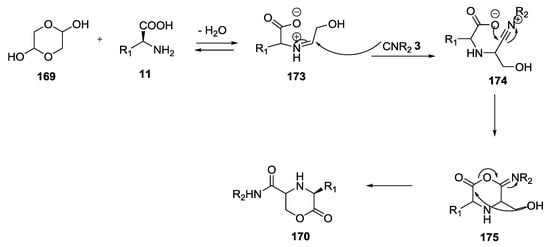
Scheme 44.
Reaction mechanism for the formation of morpholinones 170.
The basic idea of the interception of the iminoanydride Ugi adduct by a hydroxyl group was also applied to the synthesis of macrolactones 178. Cyclic chiral hemiacetals 176 and α-amino acids 11 combined with aliphatic, dipeptidic, glucosidic and lipidic isocyanides 3 in an Ugi 5C-3CR provided polysubstituted nine-membered ring lactones 178 (Scheme 45) [44]. The reaction led to poor diastereoselectivity in the formation of the new stereogenic center, but the complexity of the generated structures was remarkable.
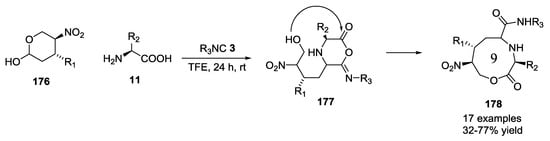
Scheme 45.
Formation of nine-membered lactones via U-5C-3CR.
Dömling et al. also described a variation of the previous reaction involving glycolaldehyde dimer, to form thiomorpholines, such as 180 and 181 (16–68%, Scheme 46), using α-amino acids 11, mercaptoacetaldehyde 179, and isocyanides 3 in trifluoroethanol as solvent [41,45]. Products were generally formed as a mixture of diastereomers that could be separated by silica gel chromatography. Cyclic α-amino acids, however, afforded mostly one diastereomer (Figure 3).
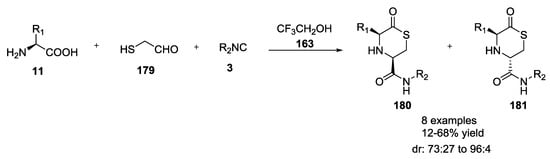
Scheme 46.
Formation of thiomorpholines.
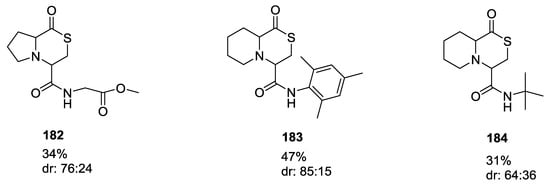
Figure 3.
Thiomorpholines formed with cyclic α-amino acids.
Novel glycopeptide structures were obtained by the extension of the Ugi reaction to an unprotected disaccharide with α-amino acids and isocyanides without any catalyst or reagent [46]. Boiling methanol, catalytic amounts of tertiary amines, and excess carbohydrate reduced reaction times and increased yields. D-ribose 185 was used with D/L α-amino acids 186 and ethyl isocyanoacetate 70 to provide a seven membered lactone such as 187 and 188 (Scheme 47). The α-amino acid isoelectric point influenced the reaction; neutral α-amino acids showed best results, while acidic or basic α-amino acids did not react. L/D-configured α-amino acids produced 1,2-syn- or 1,2-anti-configured seven-membered lactones, respectively, with a diastereoselectivity controlled by the steric demand of the α-amino acids employed. The configuration of the carbohydrates dictates the installation of the configuration at the carbon atom C-1 of the former carbohydrates. Disaccharides (maltose 189, Scheme 48) or dipeptides (β-aspartame 192, Scheme 49) were reacted under the same conditions.
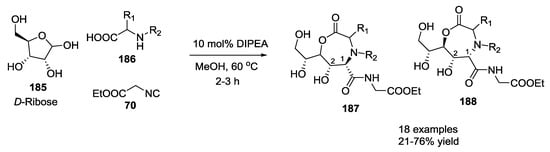
Scheme 47.
Use of D-ribose, D/L α-amino acids and ethyl isocyanoacetate.
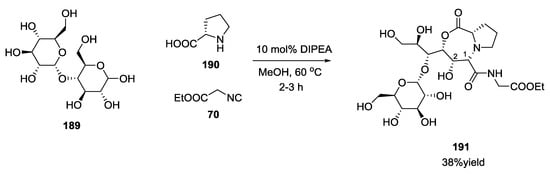
Scheme 48.
Reaction of disaccharides and L-proline.
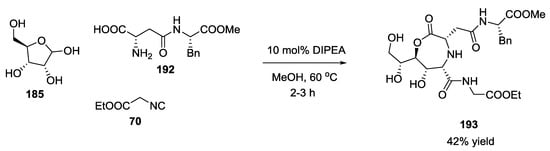
Scheme 49.
Reaction of dipeptides and D-ribose.
The reaction scope was explored with different pentoses and hexoses 194, L-proline 190 or D-proline 195, and ethyl isocyanoacetate 70 or toluenesulfonylmethyl isocyanide 60 (Scheme 50).
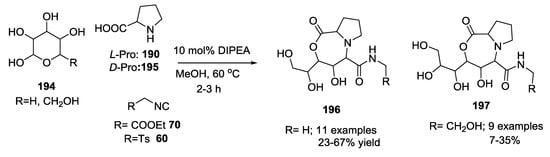
Scheme 50.
Use of hexoses, L/D-proline, and ethyl isocyanoacetate.
Subsequently, this methodology was expanded to hydroxy ketones and dihydroxyketones with unprotected α-amino acids (Scheme 51) [47]. Reaction of hydroxyketones 198 and dihydroxyketones 200 with L/D α-amino acids 11 generated 2-oxo-morpholines 199 and 201 in good to high yields. Stereoselectivity is dictated by the nature of α-amino acids employed. Syn-configured oxomorpholines were detected preferentially.
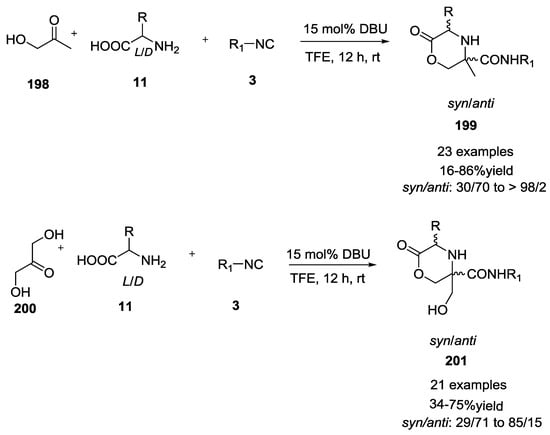
Scheme 51.
Use of hydroxy- and dihydroxy-ketones.
Reaction of L-erythrulose (ketotetrose) 202 with L-valine (38) or D-valine 206 and tert-butyl isocyanide 20 in the presence of 1,5-diazabiciclo(5.4.0)undec-7-ene (DBU) gave different results depending on the α-amino acid stereochemistry (Scheme 52). In particular, the use of L-valine produced two different oxomorpholines 203 and 204 and a seven-membered lactone 205, in contrast to D-valine, which gave a mixture of syn- and anti-configured 2-oxomorpholines 207.
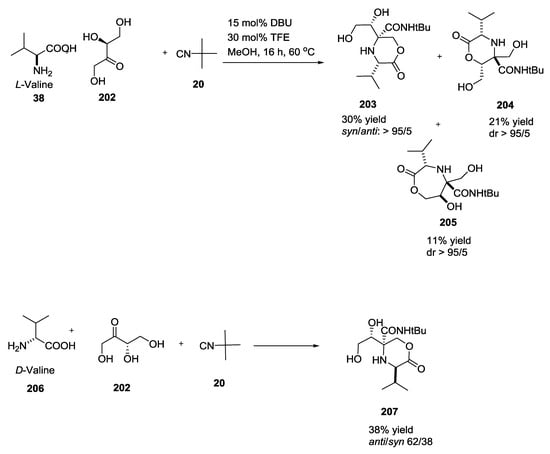
Scheme 52.
Reaction of L-erythrulose (ketotetrose) with L/D-valine and tert-butyl isocyanide.
Ketohexoses showed different results in yield (Scheme 53). Only 60% of the yield of the L-valine series were observed when D-valine was used.
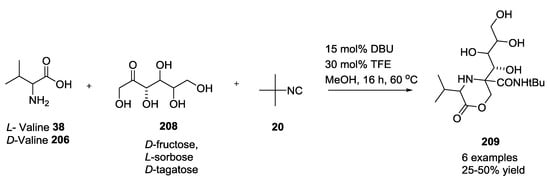
Scheme 53.
Use of Ketohexoses with L/D-valine and tert-butyl isocyanide.
L-amino acids were used by Yudin A. et al. as starting material with isocyanides and aziridine aldehydes for piperazinone synthesis [48,49,50]. Piperazinone products 211 and 212 were obtained in all cases as a single diasteroisomers without formation of linear product (Scheme 54). A detailed analysis through X-ray crystallography and NOESY NMR studies revealed that diastereoselectivity of piperazinones was dependent by the α-amino acid employed [51]. Primary α-amino acids gave a trans orientation to the new amino acid stereocenter 212; in contrast, proline- and N-substituted α-amino acids formed cis product 211 (Figure 4). Achiral α-amino acids led to piperazinones with low diastereoselectivity.
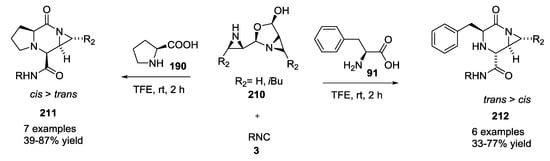
Scheme 54.
Use of aziridine aldehydes, isocyanides, and α-amino acids.
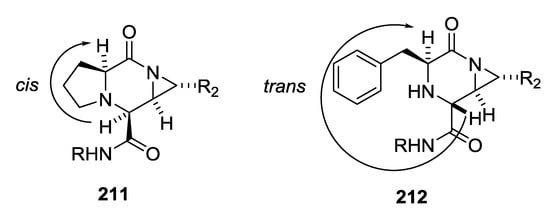
Figure 4.
Cis and trans configurations of Yudin piperazinones.
The authors also investigated the role of the isocyanide in diastereoselectivity. Bulky isocyanides increased stereoselectivity, while electron-withdrawing groups on isocyanide decreased diastereoselectivity. The same trends were observed using bulky or unsubstituted aziridine aldehyde dimers. They also compared the reactivity and the stereoselectivity of diastereomeric cis and trans aziridine aldehyde dimers (Scheme 55). Using the trans methyl aziridine aldehyde dimer 213, a moderate yield (up to 37%) was observed. In contrast, the cis methyl aziridine aldehyde dimer 215, showed excellent diastereoselectivities, and moderate to good yields.
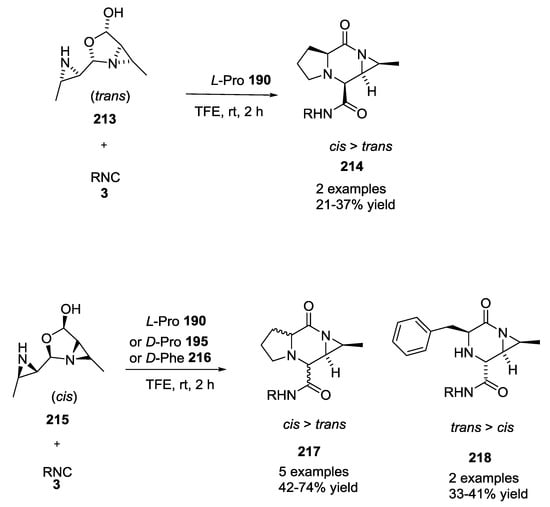
Scheme 55.
Comparison between cis and trans aziridine aldehyde dimers.
New synchronized synthesis of peptide-based macrocycles from three different components using a digital microfluidic platform was also presented [52]. The authors carried out the synthesis of a nine-membered macrocycle with an aziridine moiety in a fast, automated and well-controlled way. The system featured ten reagent reservoirs and eighty-eight actuation electrodes dedicated to dispensing, merging, and mixing droplets of reagents and products.
Solvatochromic fluorescent isocyanides 220 were also used in combination with aziridine aldehyde dimer 210 and α-amino acids 219 in the synthesis of environment-sensitive probes to obtain peptide macrocycles equipped with a fluorescent tag such as 221 (Scheme 56) [53]. Fluorophore macrocyclic peptides increased the mitochondria-localization compared to the linear one and could be used as irreversible probes of enzyme activity.
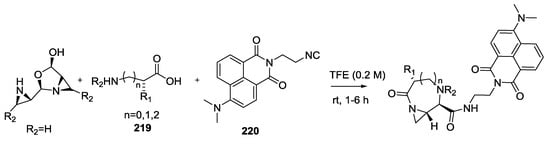
Scheme 56.
Use of fluorescent isocyanides to get macrocycles equipped with a fluorescent tag.
An environmentally friendly methodology for the synthesis of 6-alkyl/acyl phenanthridines such as 223 and 224 was recently reported by using unprotected α-amino acids as a source of stable alkyl/acyl radicals under metal free conditions with optimized reaction conditions (potassium carbonate and potassium persulfate as base and oxidant, respectively, Scheme 57) [54]. According to the proposed mechanism (Scheme 58), the homolytic cleavage of potassium persulfate generated sulfate radical anions 225 that gave single electron oxidative decarboxylation of α-amino acid anion 226. This species was oxidized to iminium 227 and converted into aldehyde 1. The sulfate radical anion abstracted the aldehyde hydrogen atom and this radical 228 could follow two different pathways. Direct decarboxylation could provide R radical 229 that reacted with isocyanide 230 to give an imidoyl radical 231. This radical gave an intramolecular cyclization 232 and a subsequent oxidation to form the alkyl phenanthridine 223.
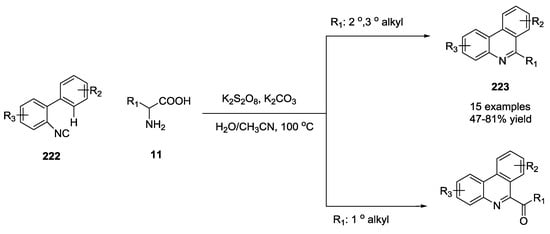
Scheme 57.
Synthesis of phenathridines by using isocyanobiphenyls and α-amino acids.
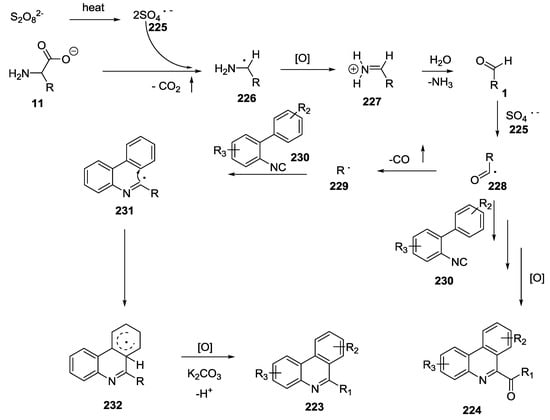
Scheme 58.
Proposed reaction mechanism for the formation of phenathridines.
The aldehyde radical could also react directly with isocyanide 230 to provide acylated phenanthridines 224.
Uyeda et al. reported the use of Pt/TiO2 catalysts for light-induced dehydrogenation of methanol to formaldehyde 140 [55]. Upon excitation with UV light, TiO2 is capable, when coupled with an efficient proton reduction catalyst such as Pt metal, of oxidizing alcohol substrates to aldehyde under mild dehydrogenation and less energetic visible/near-UV light illumination. H2 was the only stoichiometric byproduct (Scheme 59).
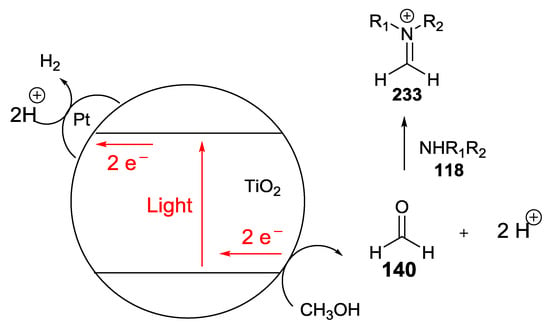
Scheme 59.
Light-induced dehydrogenative imine transformations using Pt/TiO2.
In this report, a 100W Hg lamp was used for MeOH dehydrogenation to give formaldehyde 140 that was used in a Ugi reaction with unprotected L-proline 190, 2,6-dimethylphenylisonitrile 234 in acetic acid to provide methyl ester product 235 in 54% yield (Scheme 60).
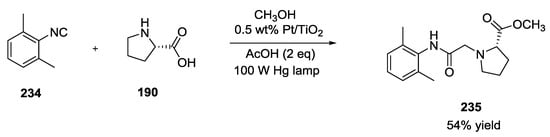
Scheme 60.
Ugi reaction enabled by the photocatalytic dehydrogenation of MeOH.
4. Ugi Compounds in Medicinal Chemistry
The Ugi reaction, including the U-5C-4CR, has been extensively exploited for combinatorial diversity-oriented syntheses in the field of medicinal chemistry.
Peptide mimetic inhibitors of the P. falciparum M1 alanylaminopeptidase (APN), a key enzyme involved in malaria infection, were obtained by Ugi 5C-4C reaction. Gazarini et al. described an increase in both the number and the diversity of their 1,1′-iminodicarboxylic acid analogues produced by such multicomponent approach.
Previously [14] and newly synthesized [56] 1,1′-iminodicarboxylic acid analogues 236 were tested for PfA-M1 inhibition and for their in vitro antimalarial activity on the growth of P. falciparum erythrocytic stages (3D7 and FcB1 strains) (Scheme 61).
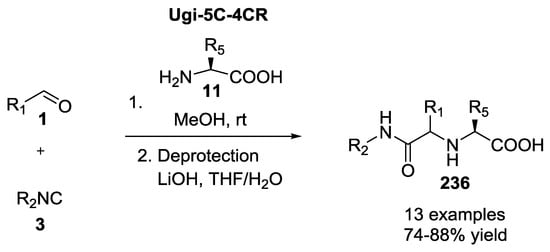
Scheme 61.
Peptidomimetic inhibitors of the P. falciparum M1 alanyl-amino-peptidase.
The p53 protein has a key role in protecting cells with oncogenic mutations. It mediates growth arrest, senescence, and apoptosis in response to cellular damage. In normal cells, p53 is present in very low levels because of its MDM-2 mediated degradation. So MDM2 represents the principal antagonist of p53 by limiting the p53 tumor suppressor function. It is structurally and biologically well understood that the key for p53–MDM2 interaction is a triad of p53 α-amino acids that inserts itself into the MDM2 cleft: Phe19, Trp23, and Leu26. For this reason, the design of molecules that inhibit the interaction of p53 and MDM2 could provide new therapeutic strategies for cancer.
Inhibitor KK271 (239) of the MDM2–p53 interaction based on the 6-chloroindole scaffold was synthesized through an U-5C-4CR using L-leucine 237, ethyl 6-chloro-3-formyl-1H-indole-2-carboxylate 238 and benzyl isocyanide 52 (Scheme 62) [57].
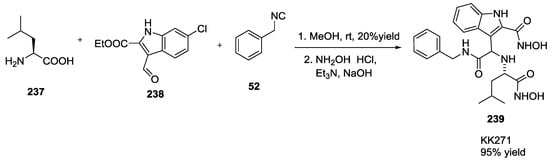
Scheme 62.
Inhibitor KK271 of the MDM2–p53 interaction.
6-Chloroindole-2-hydroxamic acid of KK271 fitted into the Trp23 pocket of MDM2, the isobutyl element filled the Phe19 pocket, and the benzyl moiety was bound within the Leu26 pocket. Analysis of the KK271-MDM2 crystal complex revealed the similarity with the native MDM2-p53 structure and the possibility for further modifications on the central, peptidic core in order to improve the drug likeness.
High-voltage-activated Ca2+ channels are involved in contraction, secretion, neurotransmitter release, and gene expression and, as shown in many reports, T-type Ca2+ channel blockers are crucial for treatment of epilepsy and neuropathic pain.
Morpholin-2-one-5-carboxamide derivatives, such as 240 and 241, synthesized via an Ugi-5C-4CR described by Kim et al. were tested as a novel class of potent and selective T-type Ca2+ channel blockers (Scheme 63) [58]. They were preliminarily screened against CaV3.2 T-type Ca2+ channels expressed in Xenopus oocytes. Compounds exhibiting more than 45% inhibition were re-evaluated for the blocking effects on CaV3.1 channels expressed in HEK293 cells. Usually, 3,5-cis adducts 240 showed higher activity than their trans analogues 241 and selective effects on T-type channels compared with N-type channels.
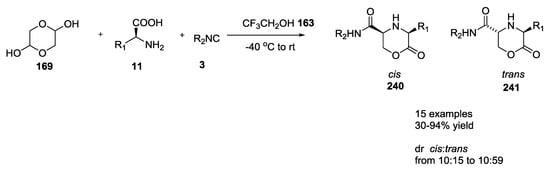
Scheme 63.
Morpholin-2-one-5-carboxamides as T-type Ca2+ channel blockers.
Substantial need for new, more effective and safer anticonvulsant drugs with lower side effects and drug–drug interactions, and with the challenge of being disease modifying, prompted Turlo et al. to Test 2,6 diketopiperazines previously synthesized via U-5C-4CR in various animal models of epilepsy.
2,6-Diketopiperazines were synthesized, starting from non-polar α-amino acid 186 (L-valine, L-leucine, L-isoleucine, L-phenylalanine, L-phenylglycine), benzaldehyde 142, tert-butyl isocyanide 20, and methanol in the presence of a catalytic amount of Iron (III) chloride (Scheme 64) [59]. Tert-butyl cleavage by use of BF3·CH3COOH and base-induced intramolecular cyclocondensation gave the final products 244 and 245, which displayed a good anticonvulsant activity in various animal models of epilepsy. Structure–activity relationship studies highlighted all the requirements for the anticonvulsant activity: proper stereochemistry on the stereogenic centers (S,S), the presence of an imide moiety and a benzene ring attached to 2,6-DKP scaffold. They also analyzed less sterically constrained monocyclic piperazines by removing the second condensed ring derived from L-proline or L-homoproline, thereby better fitting into the putative receptor(s). Synthesized compounds showed weak to good anticonvulsant activities in maximal electroshock seizure tests.
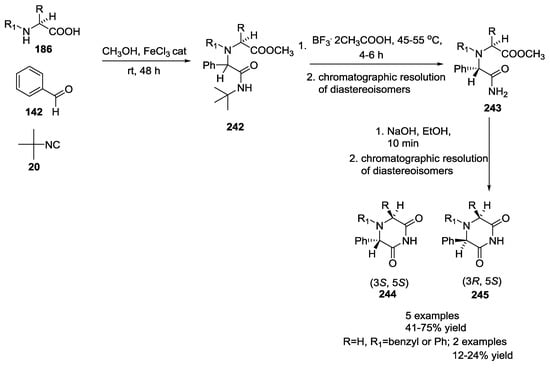
Scheme 64.
2,6-Diketopiperazines with anticonvulsant activity.
A similar approach was used by Glaxo to synthesize linear compounds subsequently cyclized into diketopiperazines that had high affinity as antagonists of the oxytocin receptors on the uterus of rats and humans [60]. DKPs exhibited also high affinity at the human recombinant oxytocin receptor in CHO cells.
5. UGI-5C-4CR in the Synthesis of Natural Products
The development of new synthetic strategies of terpene isocyanides found in marine organisms has represented a crucial investigation over recent decades.
Boneratamides A–C were isolated from the marine sponge Axinyssa aplysinoides by Andersen and co-workers in 2004. A retrosynthetic approach showed a simple synthetic strategy through the use of three building blocks (axisonitrile-3 246), a carbonyl component, either acetone or acetaldehyde 148 or 247, and glutamic acid 248 (Scheme 65) [61,62].
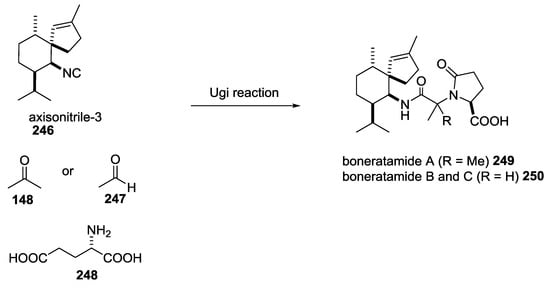
Scheme 65.
U-5C-4CR leading to boneratamides A–C.
Hypothetical biosyntheses of the right-side portion of boneratamide A 252 and 253 (Scheme 66) and exigurin 283 (Scheme 67) were proposed by Yoshiyasu and coworkers according to an U-5C-4CR. Based on these proposals, a biomimetic approach was designed and applied to the synthesis of these molecular frameworks.
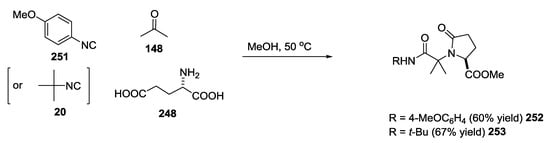
Scheme 66.
Biomimetic approach for the synthesis of boneratamide A.
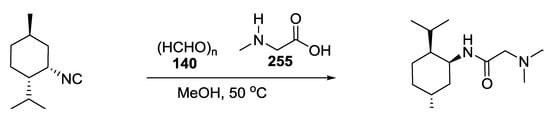
Scheme 67.
Biomimetic approach for the synthesis of exigurin (right side).
To exclude mixtures of γ-lactams, boneratamide B–C right-side portion syntheses were conducted through a stepwise synthetic route (Scheme 68).
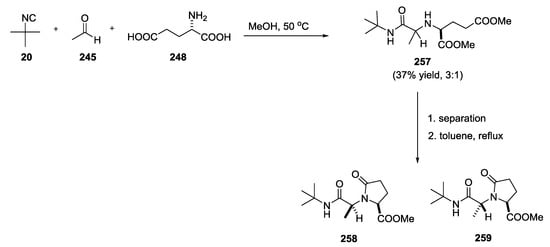
Scheme 68.
Stepwise synthetic route to boneratamide B–C.
Synthesis of the despirocyclic boneratamide A analogue 263 was also explored by preparing the isocyanide 262 from (+)-menthol 260 via azide formation 261, hydrogenation to amine, formamide formation and final dehydration (Scheme 69).
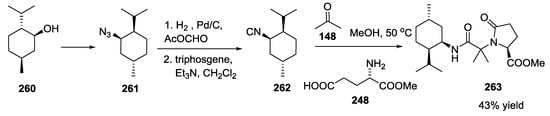
Scheme 69.
Synthesis of the despirocyclic boneratamide A.
Halichonadin G is a natural marine product isolated in 2011 by Kobayashi and co-workers from the sponge Halichondria sp. Yoshiyasu and coworkers hypothesized the natural synthesis of Halichonadin G from a natural precursor, i.e., Halichonadin C, considering a putative “ugiase” that might promote a Ugi reaction (Scheme 70) [63]. In contrast to Boneratamide A–C, the synthesis of Halichonadin G using N-unprotected α-amino acids was unsuccessful, giving only undesired products. However, the use of N-benzylglycine allowed biomimetic U-5C-4CR, a menthyl analogue of Halichonadin G.
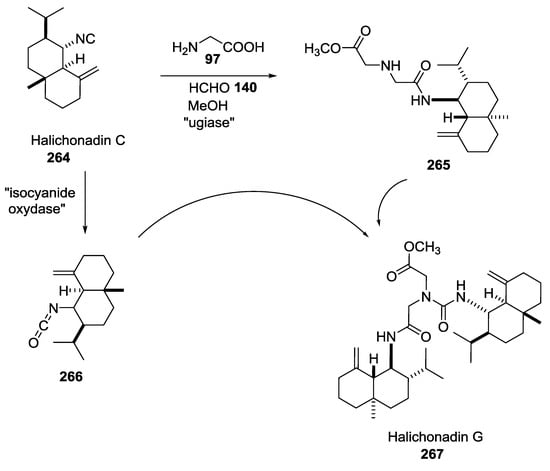
Scheme 70.
Natural synthesis of Halichonadin G promoted by a putative “ugiase”.
The syntheses of the right-hand portion of Halichonadin Q and the central part of Halichonadin M have also been reported [64].
The Kobayashi group isolated Halichonadins M–Q, from the light-brown marine sponge Halichondria sp. collected at Unten Port in Okinawa Island. Like other terpenes previously described, the natural synthesis could start from Halichonadin C isocyanide 264 by a putative “ugiase” (Scheme 71).
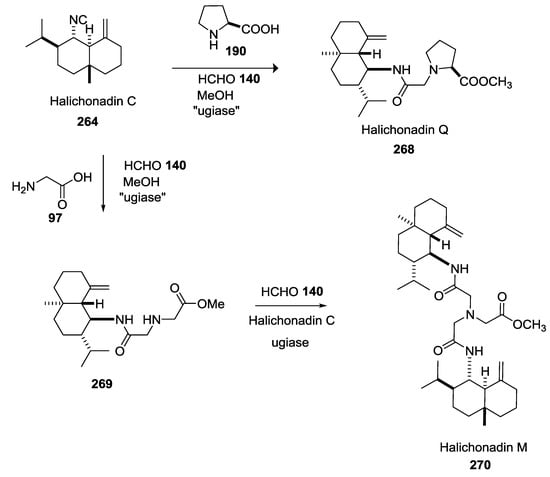
Scheme 71.
Plausible biosynthetic pathway of Halichonadin M.
A biomimetic one-pot process could also be applied for the synthesis of these terpenes 271 and 272 in a simple and remarkable manner (Scheme 72).
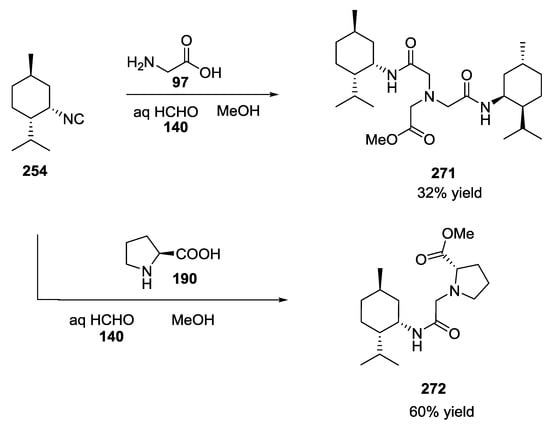
Scheme 72.
Biomimetic one-pot process for the synthesis of halichonadin analogues.
Analogously, the syntheses of despiro analogues of exigurin 274 (Scheme 73) and boneratamide B–C (Scheme 74) were explored using the same U-5C-4CR [65].
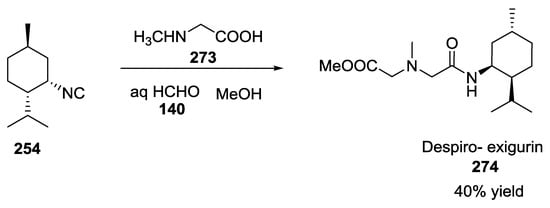
Scheme 73.
Synthesis of despiro analogue of exigurin.
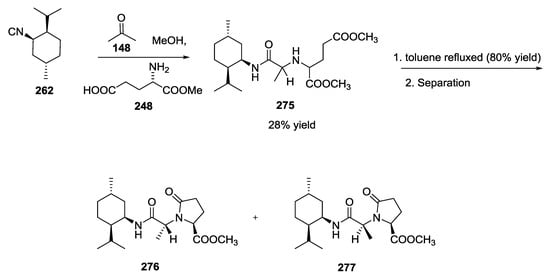
Scheme 74.
Synthesis of boneratamide B–C analogues.
6. Conclusions
As highlighted by this review, the U-5C-4CR has been exploited for the synthesis of a wide range of organic compounds, including peptide mimetics, heterocycles, and natural compounds. Notably, its potentialities have also been exploited by Yudin et al. in the macrocyclization of peptides, which is often a difficult task for organic chemists facing long-standing and unsolved problems affecting yields and purity of cyclic peptides. Despite being a 70-year-old reaction, we believe that the potentialities of the U-5C-4CR have still to be fully discovered, and we hope that this collection of literature reports will be useful in renewing the never-ending interest in such efficient processes and triggering new relevant applications in all the fields of chemistry.
Author Contributions
Conceptualization, S.P., M.G., G.C.T.; Writing-original draft preparation, S.P., I.A.A., U.G.; Writing-review and editing, S.P.; M.G.; G.C.T.; E.N.
Funding
This research received no external funding.
Acknowledgments
Financial support from Università degli Studi “Federico II” Napoli, Italy and Università del Piemonte Orientale, Novara is acknowledged. S.P., I.A.A., and M.G. gratefully acknowledge MFAG 18793.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Ugi, I.; Werner, B.; Dömling, A. The chemistry of isocyanides, their multicomponent reactions and their libraries. Molecules 2003, 8, 53–66. [Google Scholar] [CrossRef]
- Dömling, A. Recent advances in isocyanide-based multicomponent chemistry. Curr. Opin. Chem. Biol. 2002, 6, 306–313. [Google Scholar] [CrossRef]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef] [PubMed]
- Giustiniano, M.; Novellino, E.; Tron, G.C. Nitrile N-oxides and nitrile imines as new fuels for the discovery of novel isocyanide-based multicomponent reactions. Synthesis 2016, 48, 2721–2731. [Google Scholar] [CrossRef]
- Mercalli, V.; Massarotti, A.; Varese, M.; Giustiniano, M.; Meneghetti, F.; Novellino, E.; Tron, G.C. Multicomponent reaction of Z-chlorooximes, isocyanides, and hydroxylamines as hypernucleophilic traps. A one-pot route to aminodioximes and their transformation into 5-amino-1,2,4-oxadiazoles by mitsunobu–beckmann rearrangement. J. Org. Chem. 2015, 80, 9652–9661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, S.; Liu, J.; Xie, Z.; Luan, S.; Xiao, C.; Tao, Y.; Wang, X. Ugi reaction of natural amino acids: A general route toward facile synthesis of polypeptoids for bioapplications. ACS Macro Lett. 2016, 5, 1049–1054. [Google Scholar] [CrossRef]
- Demharter, A.; Hörl, W.; Herdtweck, E.; Ugi, I. Synthesis of chiral 1,1′-iminodicarboxylic acid derivatives from α-amino acids, aldehydes, isocyanides, and alcohols by the diastereoselective five-center–four-component reaction. Angew. Chem. Int. Ed. 1996, 35, 173–175. [Google Scholar] [CrossRef]
- Ugi, I.; Demharter, A.; Hörl, W.; Schmid, T. Ugi reactions with trifunctional α-amino acids, aldehydes, isocyanides and alcohols. Tetrahedron 1996, 52, 11657–11664. [Google Scholar] [CrossRef]
- Silva, E.H.B.; Emery, F.S.; Ponte, G.D.; Donate, P.M. Synthesis of some functionalized peptomers via Ugi four-component reaction. Synth. Commun. 2015, 45, 1761–1767. [Google Scholar] [CrossRef]
- Vazquez, M.P.; Morshed, M.M.; Hickey, J.L.; Poupart, M.-A.; Yang, G.; Gilard, J.; Kafal, A.P.; Roughton, A.L. Fragment Synthesis of Cyclic Peptides. Publication No. WO2017079821A1, 18 May 2017. [Google Scholar]
- Mollica, A.; Pelliccia, S.; Famiglini, V.; Stefanucci, A.; Macedonio, G.; Chiavaroli, A.; Orlando, G.; Brunetti, L.; Ferrante, C.; Pieretti, S.; et al. Exploring the first Rimonabant analog-opioid peptide hybrid compound, as bivalent ligand for CB1 and opioid receptors. J. Enzyme Inhib. Med. Chem. 2017, 32, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Sollis, S.L. Short and novel stereospecific synthesis of trisubstituted 2,5-diketopiperazines. J. Org. Chem. 2005, 70, 4735–4740. [Google Scholar] [CrossRef] [PubMed]
- Méndez, Y.; Pérez-Labrada, K.; González-Bacerio, J.; Valdés, G.; De Los Chávez, M.Á.; Osuna, J.; Charli, J.-L.; Pascual, I.; Rivera, D.G. Combinatorial multicomponent access to natural-products-inspired peptidomimetics: Discovery of selective inhibitors of microbial metallo-aminopeptidases. ChemMedChem 2014, 9, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Ugi, I.; Hörl, W.; Hanusch-Kompa, C.; Schmid, T.; Herdtweck, E. MCR 6: Chiral 2,6-piperazinediones via Ugi reactions with a-amino acids, carbonyl compounds, isocyanides and alcohols. Heterocycles 1998, 47, 965–975. [Google Scholar] [CrossRef]
- Ugi, I.K.; Ebert, B.; Hörl, W. Formation of 1,1′-iminodicarboxylic acid derivatives, 2,6-diketo-piperazine and dibenzodiazocine-2,6-dione by variations of multicomponent reactions. Chemosphere 2001, 43, 75–81. [Google Scholar] [CrossRef]
- Mjalli, A.M.M. A method for the Synthesis of Compounds of Formula 1 and Derivatives Thereof. Publication No. WO2000043352A1, 27 July 2000. [Google Scholar]
- Sung, K.; Chen, F.-L.; Chung, M.-J. Application of MCR: Facile one-pot diastereoselective syntheses of novel chiral α,α-iminodiacetic acid analogues. Mol. Divers. 2003, 6, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, L.; Chen, R.; Ni, D.; Xia, L. An approach to the synthesis of enantiopure tetrahydroisoquinoline via a key asymmetric Ugi reaction. Synlett 2013, 24, 241–245. [Google Scholar] [CrossRef][Green Version]
- Godet, T.; Bonvin, Y.; Vincent, G.; Merle, D.; Thozet, A.; Ciufolini, M.A. Titanium catalysis in the Ugi reaction of α amino acids with aromatic aldehydes. Org. Lett. 2004, 6, 3281–3284. [Google Scholar] [CrossRef]
- Dawidowski, M.; Sobczak, S.; Wilczek, M.; Kulesza, A.; Turło, J. Expanding the substrate scope of Ugi five-center, four-component reaction U-5C-4CR): Ketones as coupling partners for secondary amino acids. Mol. Divers. 2014, 18, 61–77. [Google Scholar] [CrossRef]
- Wang, W.; Dömling, A. Efficient synthesis of arrays of amino acid derived Ugi products with subsequent amidation. J. Comb. Chem. 2009, 11, 403–409. [Google Scholar] [CrossRef]
- Liu, H.; Dömling, A. One-pot synthesis of highly functionalized seleno amino acid derivatives. Chem. Biol. Drug Des. 2009, 74, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Totaro, K.A.; Okandeji, B.O.; Sello, J.K. Use of a multicomponent reaction for chemoselective derivatization of multiple classes of metabolites. ChemBioChem 2012, 13, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Edjlali, L.; Vessally, E.; Jafari, Z.; Esrafili, M.D. A novel multicomponent reaction between amino acids, aromatic aldehydes and p-toluenesulfonylmethyl isocyanide: An efficient and green one-pot synthesis using nanosilica reaction between amino acids, aromatic aldehydes and ptoluenesulfonylmethyl isocyanide: An efficient and green one-pot synthesis using nanosilica. Green Chem. Lett. Rev. 2016, 9, 13–19. [Google Scholar]
- Mandai, H.; Irie, S.; Mitsudo, K.; Suga, S. Studies on the synthesis of DMAP derivatives by diastereoselective Ugi reactions. Molecules 2011, 16, 8815–8832. [Google Scholar] [CrossRef]
- Mandai, H.; Irie, S.; Akehi, M.; Yuri, K.; Yoden, M.; Mitsudo, K.; Suga, S. Kinetic Resolution of Secondary alcohols by chiral DMAP derivatives prepared by the Ugi Multicomponent Reaction. Heterocycles 2013, 87, 329–340. [Google Scholar] [CrossRef]
- Zimmer, R.; Ziemer, A.; Gruner, M.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Siloxycyclopropanes in Ugi four-component reaction: A new method for the synthesis of highly substituted pyrrolidinone derivatives. Synthesis 2001, 11, 1649–1658. [Google Scholar] [CrossRef]
- Gladow, D.; Senf, D.; Wiecko, J.; Lentz, D.; Zimmer, R.; Reissig, H.-U. New trifluoromethyl-substituted heterocycles by multicomponent reactions of siloxycyclopropanes. Chem. Heterocycl. Compd. 2017, 53, 416–421. [Google Scholar] [CrossRef]
- Gracias, V.; Moore, J.D.; Djuric, S.W. Sequential Ugi/Heck cyclization strategies for the facile construction of highly functionalized N-heterocyclic scaffolds. Tetrahedron Lett. 2004, 45, 417–420. [Google Scholar] [CrossRef]
- Famiglini, V.; Coluccia, A.; Brancale, A.; Pelliccia, S.; La Regina, G.; Silvestri, R. Arylsulfone-based HIV-1 non-nucleoside reverse transcriptase inhibitors. Future Med. Chem. 2013, 5, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Kadzimirsz, D.; Hildebrandt, D.; Merz, K.; Dyker, G. Isoindoles and dihydroisoquinolines by gold-catalyzed intramolecular hydroamination of alkynes. Chem. Commun. 2006, 6, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Khoury, K.; Sinha, M.K.; Nagashima, T.; Herdtweck, E.; Dömling, A. Efficient assembly of iminodicarboxamides by a “truly” four-component reaction. Angew. Chem. Int. Ed. 2012, 51, 10280–10283. [Google Scholar] [CrossRef]
- Sinha, M.K.; Khoury, K.; Herdtweck, E.; Dömling, A. Various cyclization scaffolds by a truly Ugi 4-CR. Org. Biomol. Chem. 2013, 11, 4792–4796. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.K.; Khoury, K.; Herdtweck, E.; Dömling, A. Tricycles by a new Ugi variation and pictet–spengler reaction in one pot. Chem. Eur. J. 2013, 19, 8048–8052. [Google Scholar] [CrossRef] [PubMed]
- Madhavachary, R.; Wang, Q.; Dömling, A. With unprotected amino acids to tetrazolo Peptidomimetics. Chem. Commun. 2017, 53, 8549–8552. [Google Scholar] [CrossRef]
- Elders, N.; Van der Born, D.; Hendrickx, L.J.D.; Timmer, B.J.J.; Krause, A.; Janssen, E.; de Kanter, F.J.J.; Ruijter, E.; Orru, R.V.A. The efficient one-pot reaction of up to eight components by the union of multicomponent reactions. Angew. Chem. Int. Ed. 2009, 48, 5856–5859. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, J.; Martens, J. Vereinfachte Peptidsynthese mit Schutzgruppenfreien Aminosäure- Hydrochloriden nach dem Prinzip der Vierkomponenten- Kondensation. Synthesis 1992, 9, 837–838. [Google Scholar] [CrossRef]
- Hatam, M.; Tehranfar, D.; Martens, J. Single-Step Synthesis of Racemic Di- and Tripeptides derived from unnatural β-hydroxy and β-mercapto α-amino acids by the Ugi Reaction. Synthesis 1994, 6, 619–623. [Google Scholar] [CrossRef]
- Beck, B.; Srivastava, S.; Dömling, A. New End-on Thiolactone Scaffold by an isocyanide-based Multicomponent Reaction. Heterocycles 2007, 73, 177–182. [Google Scholar]
- Beck, B.; Srivastava, S.; Khoury, K.; Herdtweck, E.; Dömling, A. One-pot multicomponent synthesis of two novel thiolactone scaffolds. Mol. Divers. 2010, 14, 479–491. [Google Scholar] [CrossRef]
- Park, S.J.; Keum, G.; Kang, S.B.; Koh, H.Y.; Kim, Y.; Lee, D.H. A facile synthesis of N-carbamoylmethyl- α -aminobutyrolactones by the Ugi multicomponent condensation reaction. Tetrahedron Lett. 1998, 39, 7109–7112. [Google Scholar] [CrossRef]
- Kim, Y.B.; Choi, E.H.; Keum, G.; Kang, S.B.; Lee, D.H.; Koh, H.Y.; Kim, Y. An efficient synthesis of morpholin-2-one derivatives using glycolaldehyde dimer by the ugi multicomponent reaction. Org. Lett. 2001, 3, 4149–4152. [Google Scholar] [CrossRef] [PubMed]
- Torre, A.F.D.L.; Rivera, D.G.; Concepción, O.; Echemendia, R.; Correa, A.G.; Paixão, M.W. Multicomponent synthesis of cyclic depsipeptide mimics by Ugi reaction including cyclic hemiacetals derived from asymmetric organocatalysis. J. Org. Chem. 2016, 81, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Beck, B.; Herdtweck, E.; Khoury, K.; Dömling, A. A novel Δ-thiolactone scaffold by a versatile intramolecular multicomponent reaction. Heterocycles 2009, 77, 731–738. [Google Scholar]
- Voigt, B.; Linke, M.; Mahrwald, R. Multicomponent cascade reactions of unprotected carbohydrates and amino acids. Org. Lett. 2015, 17, 2606–2609. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Trung, M.N.; Mahrwald, R. Multicomponent cascade reactions of unprotected ketoses and amino acids—Access to a defined configured quaternary stereogenic center. J. Org. Chem. 2015, 80, 10849–10865. [Google Scholar] [CrossRef] [PubMed]
- Yudin, A.; Hili, R. Cyclic Amino Acid Molecules and Methods of Preparing the Same. Publication No. WO2010105363A1, 23 September 2010. [Google Scholar]
- Heine, N.B.; Kaldas, S.J.; Belding, L.; Shmatova, O.; Dudding, T.; Nenajdenko, V.G.; Studer, A.; Yudin, A.K. Synthesis of chiral piperazinones using amphoteric aziridine aldehyde dimers and functionalized isocyanides. J. Org. Chem. 2016, 81, 5209–5216. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, S.; Adachi, S.; Rotstein, B.H.; Hickey, J.L.; Scully, C.C.G.; Denis, J.D.S.; Courtemanche, R.; Yu, J.C.Y.; Chung, B.K.W.; Yudin, A.K. Stereocontrolled disruption of the Ugi reaction toward the production of chiral piperazinones: Substrate scope and process development. J. Org. Chem. 2014, 79, 9948–9957. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Rai, V.; Ryan Hili, R.; Yudin, A.K. Synthesis of peptide macrocycles using unprotected amino aldehydes. Nat. Protoc. 2010, 5, 1813–1822. [Google Scholar] [CrossRef]
- Jebrail, M.J.; Ng, A.H.C.; Rai, V.; Hili, R.; Yudin, A.K.; Wheeler, A.R. Synchronized synthesis of peptide-based macrocycles by digital microfluidics. Angew. Chem. 2010, 122, 8807–8811. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Mourtada, R.; Kelley, S.O.; Yudin, A.K. Solvatochromic reagents for multicomponent reactions and their utility in the development of cell-permeable macrocyclic peptide vectors. Chem. Eur. J. 2011, 17, 12257–12261. [Google Scholar] [CrossRef]
- Lu, S.-C.; Li, H.-S.; Gong, Y.-L.; Wang, X.-L.; Li, F.-R.; Li, F.; Duan, G.-Y.; Xu, S. Use of unprotected amino acids in metal-free tandem radical cyclization reactions: Divergent synthesis of 6-alkyl/acyl phenanthridines. RSC Adv. 2017, 7, 55891–55896. [Google Scholar] [CrossRef]
- Adolph, C.M.; Werth, J.; Selvaraj, R.; Wegener, E.C.; Uyeda, C. Dehydrogenative Transformations of Imines Using a Heterogeneous Photocatalyst. J. Org. Chem. 2017, 82, 5959–5965. [Google Scholar] [CrossRef] [PubMed]
- González-Bacerio, J.; Maluf, S.E.C.; Méndez, Y.; Pascual, I.; Florent, I.; Melo, P.M.; Budu, A.; Ferreira, J.C.; Moreno, E.; Carmona, A.K.; et al. KBE009: An antimalarial bestatin-like inhibitor of the Plasmodium falciparum M1 aminopeptidase discovered in an Ugi multicomponent reaction-derived peptidomimetic library. Bioorg. Med. Chem. 2017, 25, 4628–4636. [Google Scholar] [CrossRef] [PubMed]
- Bista, M.; Wolf, S.; Khoury, K.; Kowalska, K.; Huang, Y.; Wrona, E.; Arciniega, M.; Popowicz, G.M.; Holak, T.A.; Dömling, A. Transient protein states in designing inhibitors of the MDM2-p53 interaction. Structure 2013, 21, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Ku, I.W.; Cho, S.; Doddareddy, M.R.; Jang, M.S.; Keum, G.; Lee, J.-H.; Chung, B.Y.; Kim, Y.; Rhim, H.; Kang, S.B. Morpholin-2-one derivatives as novel selective T-type Ca2 channel blockers. Bioorg. Med. Chem. Lett. 2006, 16, 5244–5248. [Google Scholar] [CrossRef]
- Dawidowski, M.; Turło, J. Multicomponent synthesis and anticonvulsant activity of monocyclic 2,6-diketopiperazine derivatives. Med. Chem. Res. 2014, 23, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D.; Hatley, R.J.; Hickey, D.M.B.; Liddle, J.; Livermore, D.G.H.; Mason, A.M.; Miller, N.D.; Nerozzi, F.; Sollis, S.L.; Szardenings, A.K.; et al. Substituted Diketopiperazines as Oxytocin Antagonists. Publication No. WO2003053443A1, 3 July 2003. [Google Scholar]
- Ichikawa, Y.; Saito, K.; Nishimori, A.; Kotsuki, H.; Nakano, K. A biomimetic approach to terpenes isolated from marine sponges: A Ugi coupling reaction in a hypothetical biosynthesis. Synlett 2013, 24, 757–761. [Google Scholar] [CrossRef][Green Version]
- Chianese, G.; Yu, H.-B.; Yang, F.; Sirignano, C.; Luciano, P.; Han, B.-N.; Khan, S.; Lin, H.-W.; Taglialatela-Scafati, O. PPAR modulating polyketides from a chinese plakortis simplex and clues on the origin of their chemodiversity. J. Org. Chem. 2016, 81, 5135–5143. [Google Scholar] [CrossRef]
- Saito, K.; Nishimori, A.; Mimura, R.; Nakano, K.; Kotsuki, H.; Masuda, T.; Ichikawa, Y. A biomimetic approach to the synthesis of the core structure of the marine sponge terpene halichonadin G. Eur. J. Org. Chem. 2013, 31, 7041–7043. [Google Scholar] [CrossRef]
- Mimura, R.; Kitamori, A.; Ikeda, A.; Masuda, T.; Nakano, K.; Kotsuki, H.; Ichikawa, Y. Biomimetic approaches employing the Ugi five-center four- component reaction for synthesis of the right-hand portion of halichonadin Q and the central part of halichonadin M. Synthesis 2015, 47, 3043–3048. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Saito, K.; Mimura, R.; Kitamori, A.; Matsukawa, A.; Ikeda, A.; Masuda, T.; Kotsuki, H.; Nakano, K. A Biomimetic Approach to the synthesis of terpene-amino acid conjugates. The Ugi reaction in the hypothetical biosynthesis of marine natural products. Heterocycles 2016, 92, 1040–1053. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).