Statistic-Driven Proton Transfer Affecting Nanoscopic Organization in an Ethylammonium Nitrate Ionic Liquid and 1,4-Diaminobutane Binary Mixture: A Steamy Pizza Model




Abstract

1. Introduction
2. Experimental Methods
3. Computational Methods
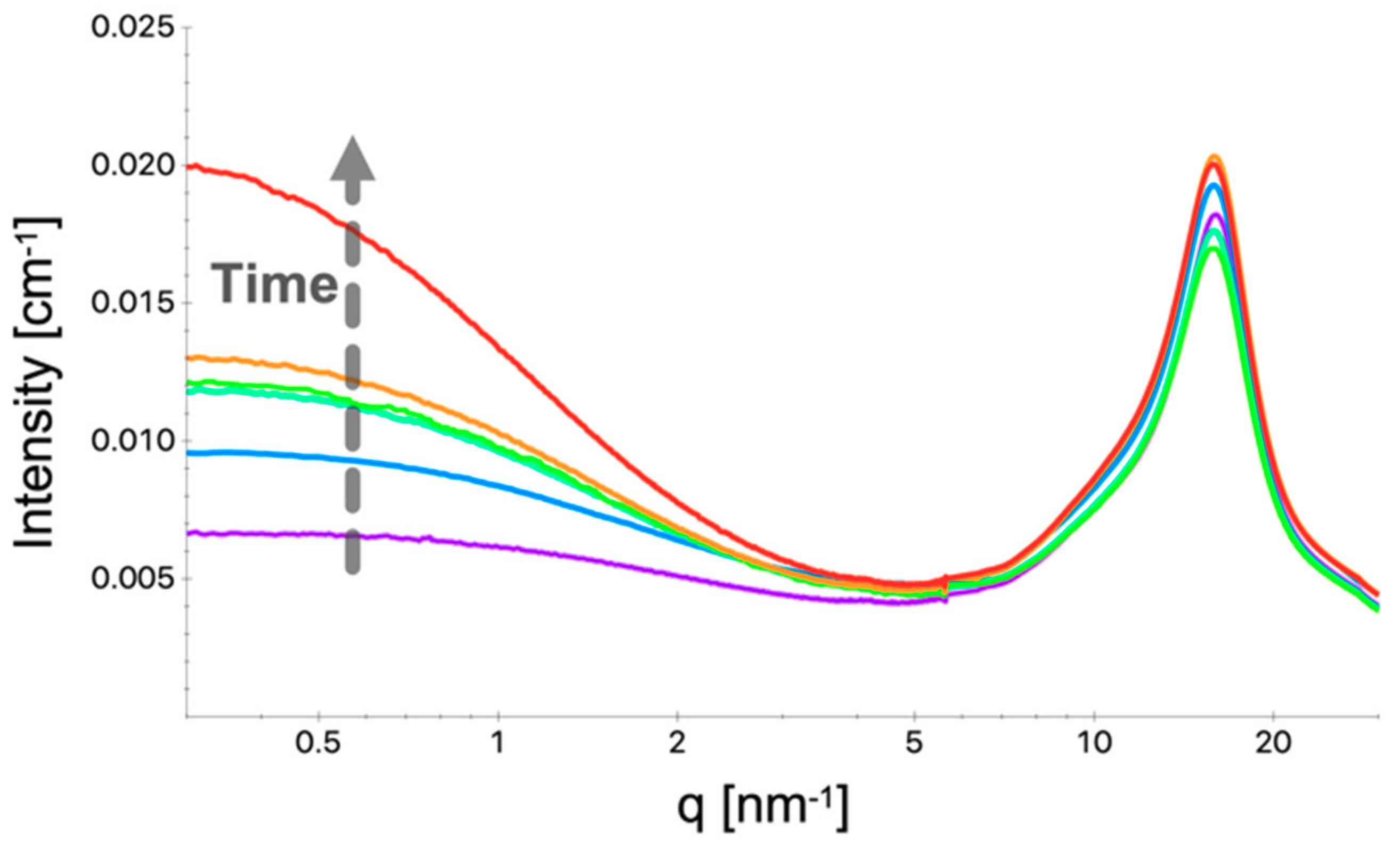
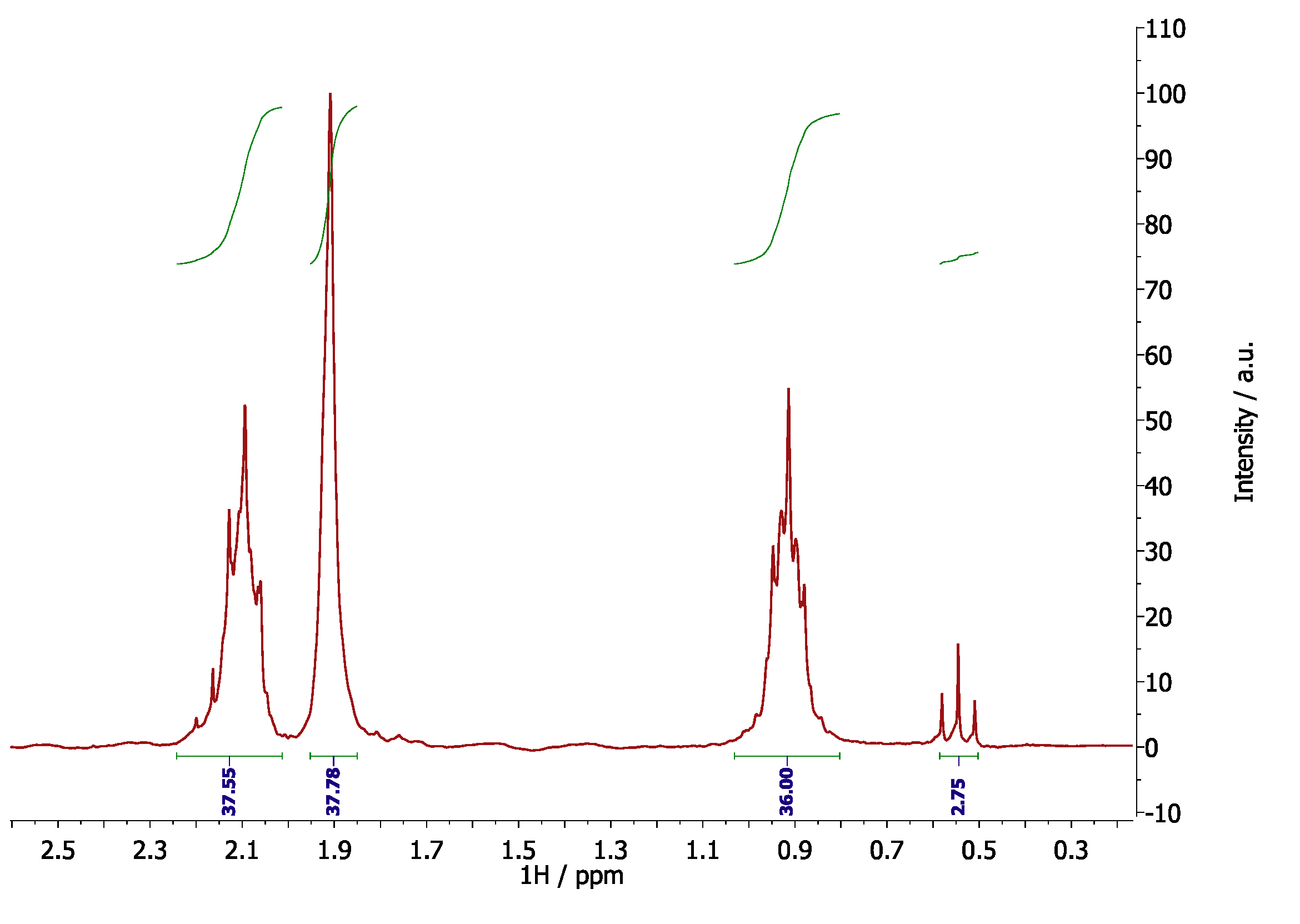
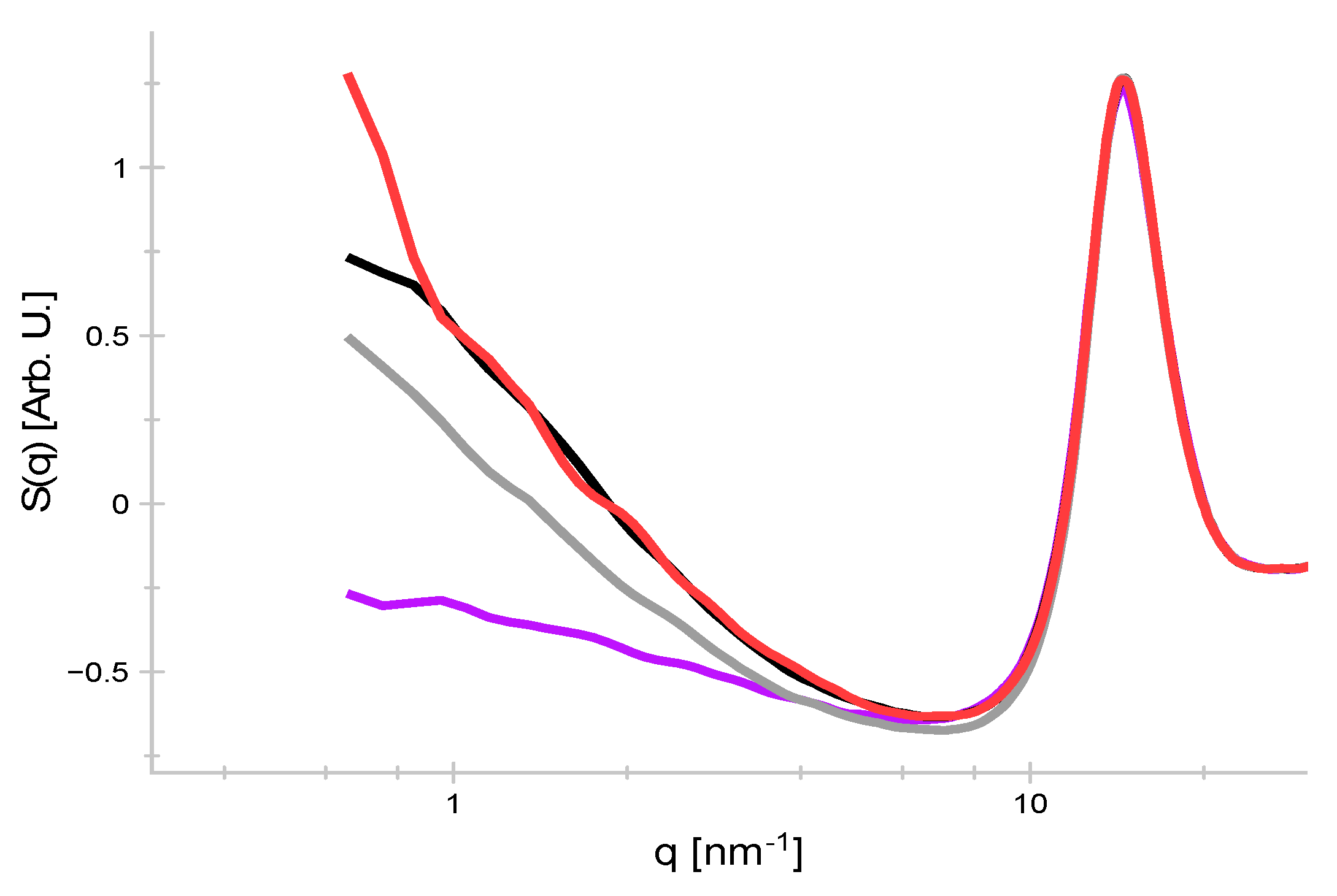
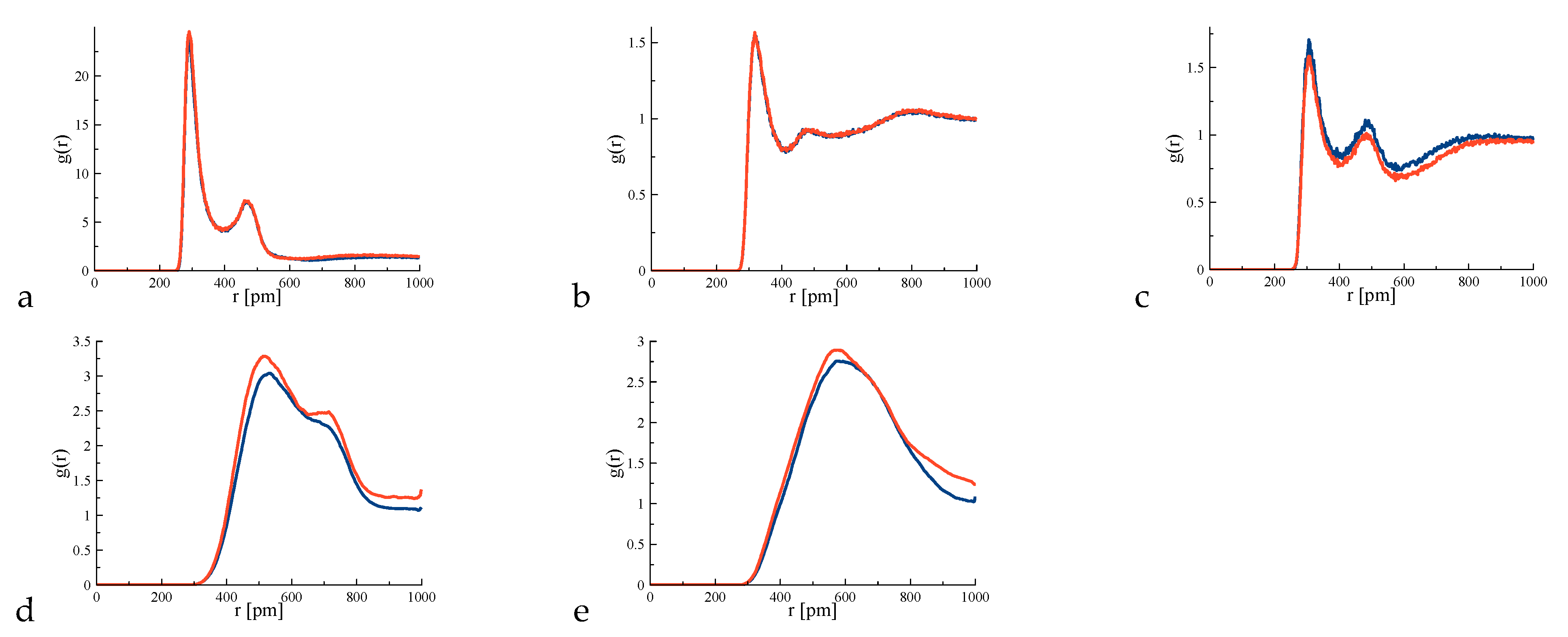
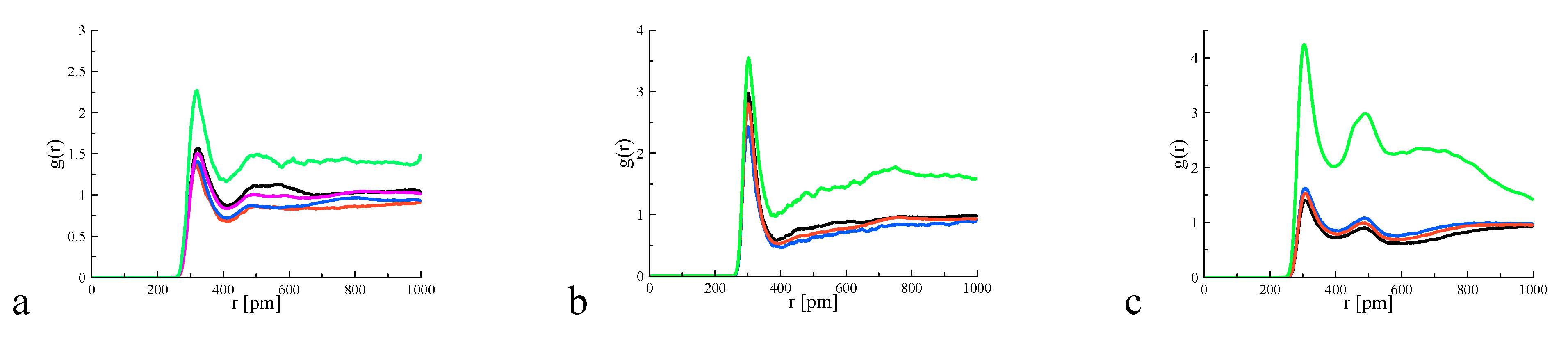
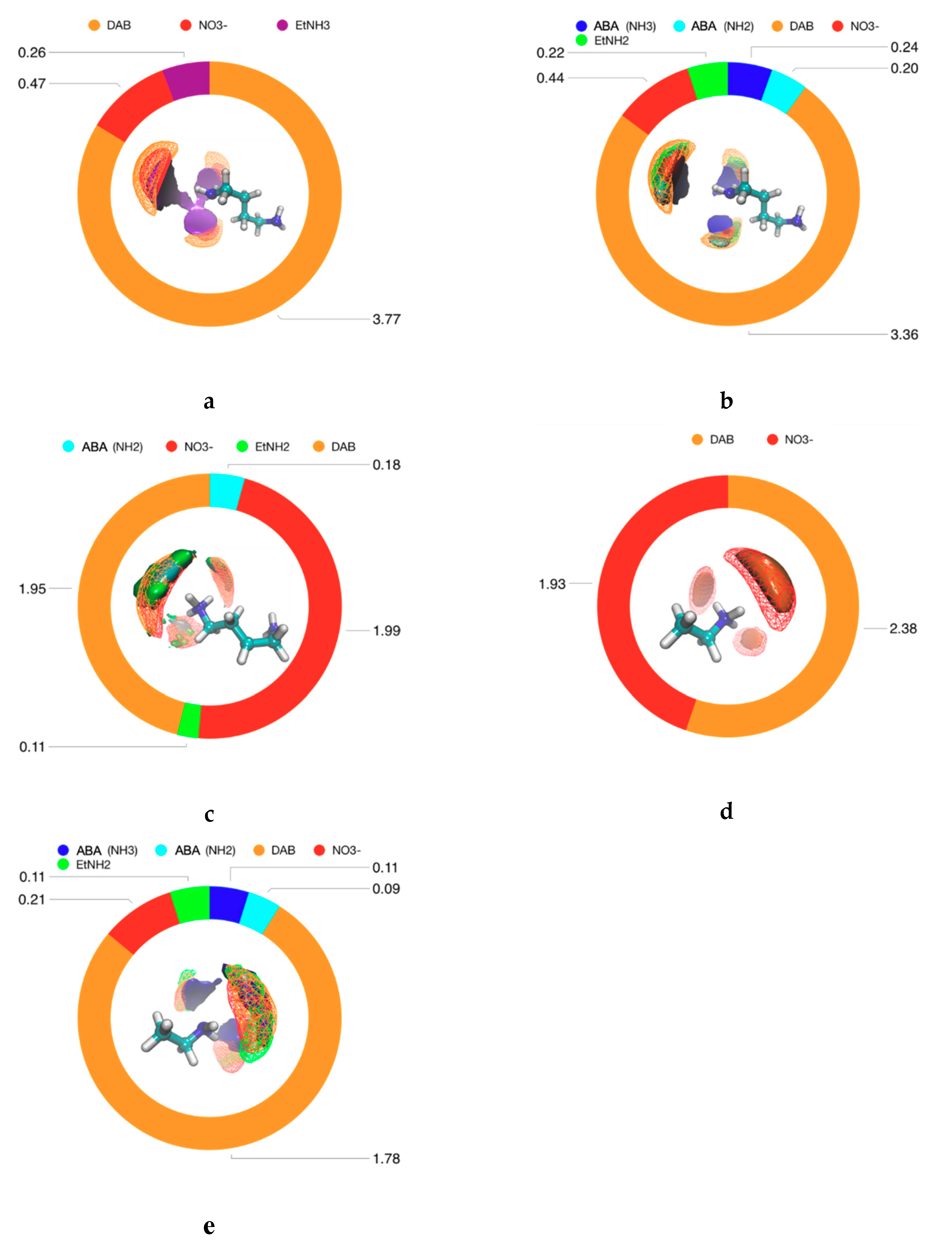
4. Results and Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li Chum, H.; Koch, V.R.; Miller, L.L.; Osteryoung, R.A. An Electrochemical Scrutiny of Organometallic Iron Complexes and Hexamethylbenzene in a Room Temperature Molten Salt. J. Am. Chem. Soc. 1975, 97, 3264–3265. [Google Scholar] [CrossRef]
- Wilkes, J.S.; Levisky, J.A.; Wilson, R.A.; Hussey, C.L. Dialkylimidazolium Chloroaluminate Melts: A New Class of Room-Temperature Ionic Liquids for Electrochemistry, Spectroscopy, and Synthesis. Inorg. Chem. 1982, 21, 1263–1264. [Google Scholar] [CrossRef]
- Friedel, A.; Crafts, J.; Ador, E. Synthese der Benzoësäure und des Benzophenons mit Hülfe des Chlorkohlenoxyds. Ber. Der Dtsch. Chem. Ges. 1877, 10, 1854–1858. [Google Scholar] [CrossRef]
- Walden, P. Ueber die Molekulargr¨osse und elektrische Leitf¨ahigkeit einiger geschmolzenen Salze. Bull. I’Acad. Imp. Des. Sci. St.-Petersbg. 1914, 8, 405–422. [Google Scholar]
- De Santis, S.; Masci, G.; Casciotta, F.; Caminiti, R.; Scarpellini, E.; Campetella, M.; Gontrani, L. Cholinium-amino acid based ionic liquids: A new method of synthesis and physico-chemical characterization. Phys. Chem. Chem. Phys. 2015, 17, 20687–20698. [Google Scholar] [CrossRef]
- Ohno, H. Functional Design of Ionic Liquids. Bull. Chem. Soc. Jpn. 2006, 79, 1665–1680. [Google Scholar] [CrossRef]
- Totolin, V.; Minami, I.; Gabler, C.; Dörr, N. Halogen-free borate ionic liquids as novel lubricants for tribological applications. Tribol. Int. 2013, 67, 191–198. [Google Scholar] [CrossRef]
- Kuhlmann, E.; Himmler, S.; Giebelhaus, H.; Wasserscheid, P. Imidazolium dialkylphosphates - A class of versatile, halogen-free and hydrolytically stable ionic liquids. Green Chem. 2007, 9, 233–242. [Google Scholar] [CrossRef]
- Wasserscheid, P.; Van Hal, R.; Bösmann, A. 1-n-butyl-3-methylimidazolium ([bmim]) octylsulfate–An even “greener” ionic liquid. Green Chem. 2002, 4, 400–404. [Google Scholar] [CrossRef]
- Thuy Pham, T.P.; Cho, C.W.; Yun, Y.S. Environmental fate and toxicity of ionic liquids: A review. Water Res. 2010, 44, 352–372. [Google Scholar] [CrossRef]
- Mallakpour, S.; Dinari, M. Ionic liquids as green solvents: Progress and Prospects. In Green Solvents II: Properties and Applications of Ionic Liquids; Springer Science & Business Media: Berlin, Germany, 2012; pp. 1–32. ISBN 978-940-072-891-2. [Google Scholar]
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Ortolani, M.; Nucara, A.; Baldassarre, L.; Missori, M.; Fastampa, R.; Caminiti, R. Stabilization of the Tensile Strength of Aged Cellulose Paper by Cholinium-Amino Acid Ionic Liquid Treatment. J. Phys. Chem. C 2016, 120, 24088–24097. [Google Scholar] [CrossRef]
- Ke, M.; Zhou, A.; Song, Z.; Jiang, Q. Toxicity of ionic liquids. Prog. Chem. 2007, 19, 671–679. [Google Scholar]
- Visser, A.E.; Swatloski, R.P.; Reichert, W.M.; Mayton, R.; Sheff, S.; Wierzbicki, A.; Davis, J.H.; Rogers, R.D. Task-specific ionic liquids incorporating novel cations for the coordination and extraction of Hg 2+ and Cd 2+ : Synthesis, characterization, and extraction studies. Environ. Sci. Technol. 2002, 36, 2523–2529. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.D.; Seddon, K.R. Ionic Liquids - Solvents of the Future? Science 2003, 302, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Chiappe, C.; Pieraccini, D. Ionic liquids: Solvent properties and organic reactivity. J. Phys. Org. Chem. 2005, 18, 275–297. [Google Scholar] [CrossRef]
- Galiński, M.; Lewandowski, A.; Stępniak, I. Ionic liquids as electrolytes. Electrochim. Acta 2006, 51, 5567–5580. [Google Scholar] [CrossRef]
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Evolving Structure–Property Relationships and Expanding Applications. Chem. Rev. 2015, 115, 11379–11448. [Google Scholar] [CrossRef]
- Greaves, T.L.; Weerawardena, A.; Fong, C.; Krodkiewska, I.; Drummond, C.J. Protic Ionic Liquids: Solvents with Tunable Phase Behavior and Physicochemical Properties. J. Phys. Chem. B 2006, 110, 22479–22487. [Google Scholar] [CrossRef]
- Campetella, M.; Montagna, M.; Gontrani, L.; Scarpellini, E.; Bodo, E. Unexpected proton mobility in the bulk phase of cholinium-based ionic liquids: New insights from theoretical calculations. Phys. Chem. Chem. Phys. 2017, 19, 11869–11880. [Google Scholar] [CrossRef] [PubMed]
- Bodo, E.; Mangialardo, S.; Ramondo, F.; Ceccacci, F.; Postorino, P. Unravelling the Structure of Protic Ionic Liquids with Theoretical and Experimental Methods: Ethyl-, Propyl- and Butylammonium Nitrate Explored by Raman Spectroscopy and DFT Calculations. J. Phys. Chem. B 2012, 116, 13878–13888. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.L.; Kennedy, D.F.; Kirby, N.; Drummond, C.J. Nanostructure changes in protic ionic liquids (PILs) through adding solutes and mixing PILs. Phys. Chem. Chem. Phys. 2011, 13, 13501. [Google Scholar] [CrossRef] [PubMed]
- Russina, O.; Macchiagodena, M.; Kirchner, B.; Mariani, A.; Aoun, B.; Russina, M.; Caminiti, R.; Triolo, A. Association in ethylammonium nitrate–dimethyl sulfoxide mixtures: First structural and dynamical evidences. J. Non. Cryst. Solids 2015, 407, 333–338. [Google Scholar] [CrossRef]
- Marsh, K.; Boxall, J.; Lichtenthaler, R. Room temperature ionic liquids and their mixtures—A review. Fluid Phase Equilib. 2004, 219, 93–98. [Google Scholar] [CrossRef]
- Jiang, H.J.; FitzGerald, P.A.; Dolan, A.; Atkin, R.; Warr, G.G. Amphiphilic Self-Assembly of Alkanols in Protic Ionic Liquids. J. Phys. Chem. B 2014, 118, 9983–9990. [Google Scholar] [CrossRef]
- Mariani, A.; Dattani, R.; Caminiti, R.; Gontrani, L. Nanoscale Density Fluctuations in Ionic Liquid Binary Mixtures with Nonamphiphilic Compounds: First Experimental Evidence. J. Phys. Chem. B 2016, 120, 10540–10546. [Google Scholar] [CrossRef]
- Mariani, A.; Caminiti, R.; Ramondo, F.; Salvitti, G.; Mocci, F.; Gontrani, L. Inhomogeneity in Ethylammonium Nitrate—Acetonitrile Binary Mixtures: The Highest “Low q Excess” Reported to Date. J. Phys. Chem. Lett. 2017, 8, 3512–3522. [Google Scholar] [CrossRef]
- Campetella, M.; Mariani, A.; Sadun, C.; Wu, B.; Castner, E.W.; Gontrani, L. Structure and dynamics of propylammonium nitrate-acetonitrile mixtures: An intricate multi-scale system probed with experimental and theoretical techniques. J. Chem. Phys. 2018, 148, 134507. [Google Scholar] [CrossRef]
- Mariani, A.; Bonomo, M.; Wu, B.; Centrella, B.; Dini, D.; Castner, E.W.; Gontrani, L. Intriguing transport dynamics of ethylammonium nitrate–Acetonitrile binary mixtures arising from nano-inhomogeneity. Phys. Chem. Chem. Phys. 2017, 19, 27212–27220. [Google Scholar] [CrossRef]
- Van Vaerenbergh, P.; Léonardon, J.; Sztucki, M.; Boesecke, P.; Gorini, J.; Claustre, L.; Sever, F.; Morse, J.; Narayanan, T. An upgrade beamline for combined wide, small and ultra small-angle x-ray scattering at the ESRF. In Proceedings of the 12th International Conference on Synchrtron Radiation Instrumentation, AIP Conference Proceedings, New York, NY, USA, 6–10 July 2015; Volume 1741, p. 030034. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, K.G.; Jaeger, V.W.; Pfaendtner, J. The General AMBER Force Field (GAFF) Can Accurately Predict Thermodynamic and Transport Properties of Many Ionic Liquids. J. Phys. Chem. B 2015, 119, 5882–5895. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, version D.01; Gaussian; Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Mariani, A.; Caminiti, R.; Campetella, M.; Gontrani, L. Pressure-induced mesoscopic disorder in protic ionic liquids: First computational study. Phys. Chem. Chem. Phys. 2016, 18, 2297–2302. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. TRAVIS—A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef]
- Bodo, E.; Mangialardo, S.; Capitani, F.; Gontrani, L.; Leonelli, F.; Postorino, P. Interaction of a long alkyl chain protic ionic liquid and water. J. Chem. Phys. 2014, 140, 204503. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Model | EAN | DAB | EtNH2 | ABAN | Side [Å] |
|---|---|---|---|---|---|---|
| EAN+DAB | No Transfer | 5280 | 47524 | N/A | N/A | 208.41 |
| EtNH2+ABAN+DAB | Total Transfer | N/A | 42244 | 4837 | 5280 | 208.68 |
| EAN+EtNH2+ABAN+DAB | Partial Transfer | 267 | 42511 | 5013 | 5013 | 208.66 |
| ABAN+DAB | No EtNH2 | N/A | 42244 | N/A | 5280 | 203.30 |
| Experimental [g/mL] | Computed [g/mL] | |||||
|---|---|---|---|---|---|---|
| System | 1 Day Old | 1 Year Old | No Transfer | Total Transfer | Partial Transfer | No EtNH2 |
| Density | 0.86885 | 0.86144 | 0.8716 | 0.8683 | 0.8685 | 0.8920 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mariani, A.; Bonomo, M.; Passerini, S. Statistic-Driven Proton Transfer Affecting Nanoscopic Organization in an Ethylammonium Nitrate Ionic Liquid and 1,4-Diaminobutane Binary Mixture: A Steamy Pizza Model. Symmetry 2019, 11, 1425. https://doi.org/10.3390/sym11111425
Mariani A, Bonomo M, Passerini S. Statistic-Driven Proton Transfer Affecting Nanoscopic Organization in an Ethylammonium Nitrate Ionic Liquid and 1,4-Diaminobutane Binary Mixture: A Steamy Pizza Model. Symmetry. 2019; 11(11):1425. https://doi.org/10.3390/sym11111425
Chicago/Turabian StyleMariani, Alessandro, Matteo Bonomo, and Stefano Passerini. 2019. "Statistic-Driven Proton Transfer Affecting Nanoscopic Organization in an Ethylammonium Nitrate Ionic Liquid and 1,4-Diaminobutane Binary Mixture: A Steamy Pizza Model" Symmetry 11, no. 11: 1425. https://doi.org/10.3390/sym11111425
APA StyleMariani, A., Bonomo, M., & Passerini, S. (2019). Statistic-Driven Proton Transfer Affecting Nanoscopic Organization in an Ethylammonium Nitrate Ionic Liquid and 1,4-Diaminobutane Binary Mixture: A Steamy Pizza Model. Symmetry, 11(11), 1425. https://doi.org/10.3390/sym11111425