A Critical Note on Symmetry Contact Artifacts and the Evaluation of the Quality of Homology Models

Abstract
:1. Introduction
2. Methods
- Conserved versus Mutated in structure superposition bases sequence alignment of the template and the model.
- Symmetry Contact (SMC) versus No Symmetry Contact (NoSMC). Obviously, residues in homology models do not make symmetry contacts. Therefore, a residue is called SMC if either the residue in the template structure, or the equivalent residue in the real structure, or both of them make a symmetry contact. A residue is called making a symmetry contact if any atom in the residue’s side chain makes a tight contact with any symmetry related atom other than water. A contact is called tight when there is maximally 0.25 Å space between their Van der Waals’ surfaces (or 1.4 Å, which is half of the diameter of a water molecule, for Table 1).
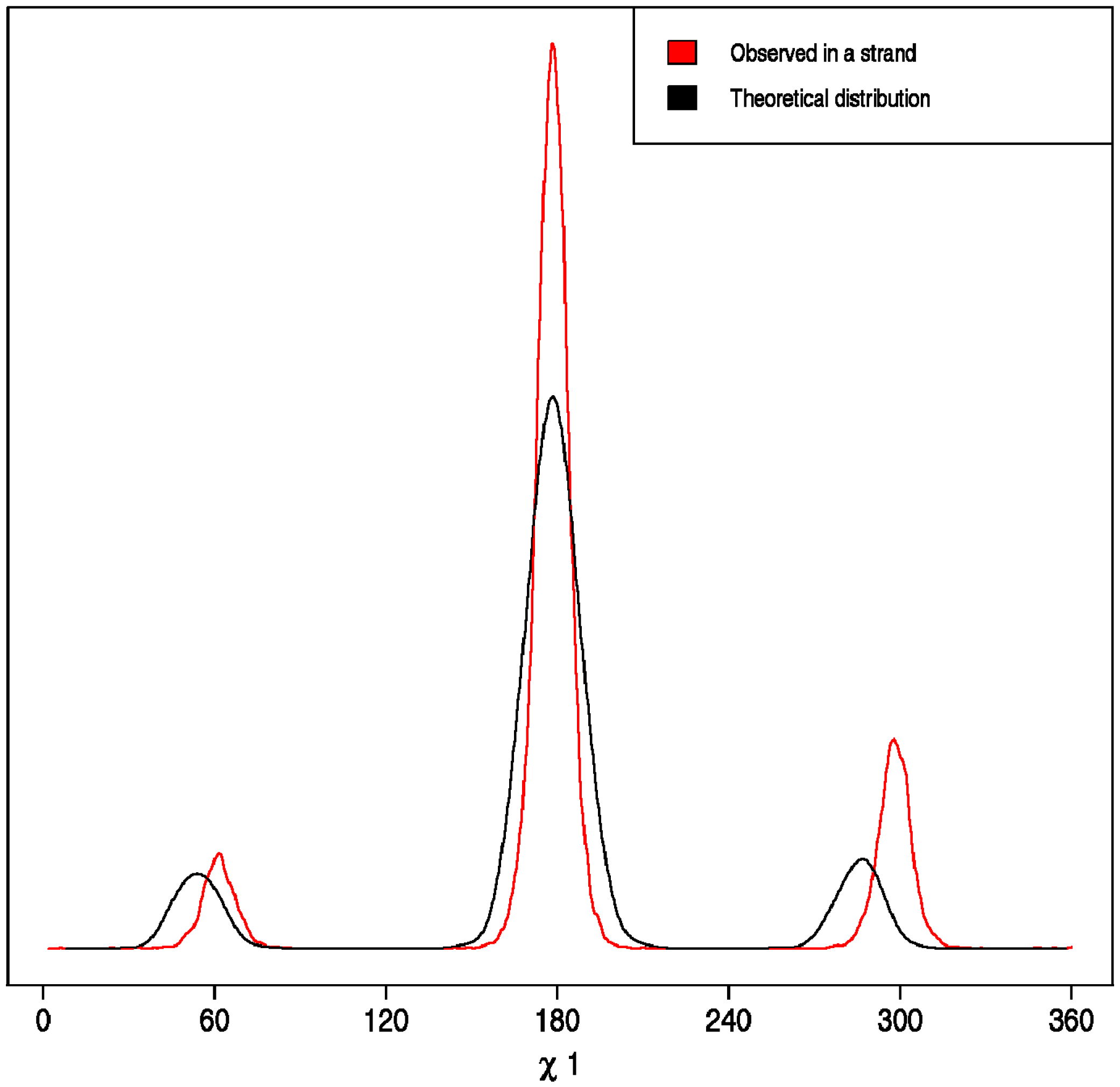
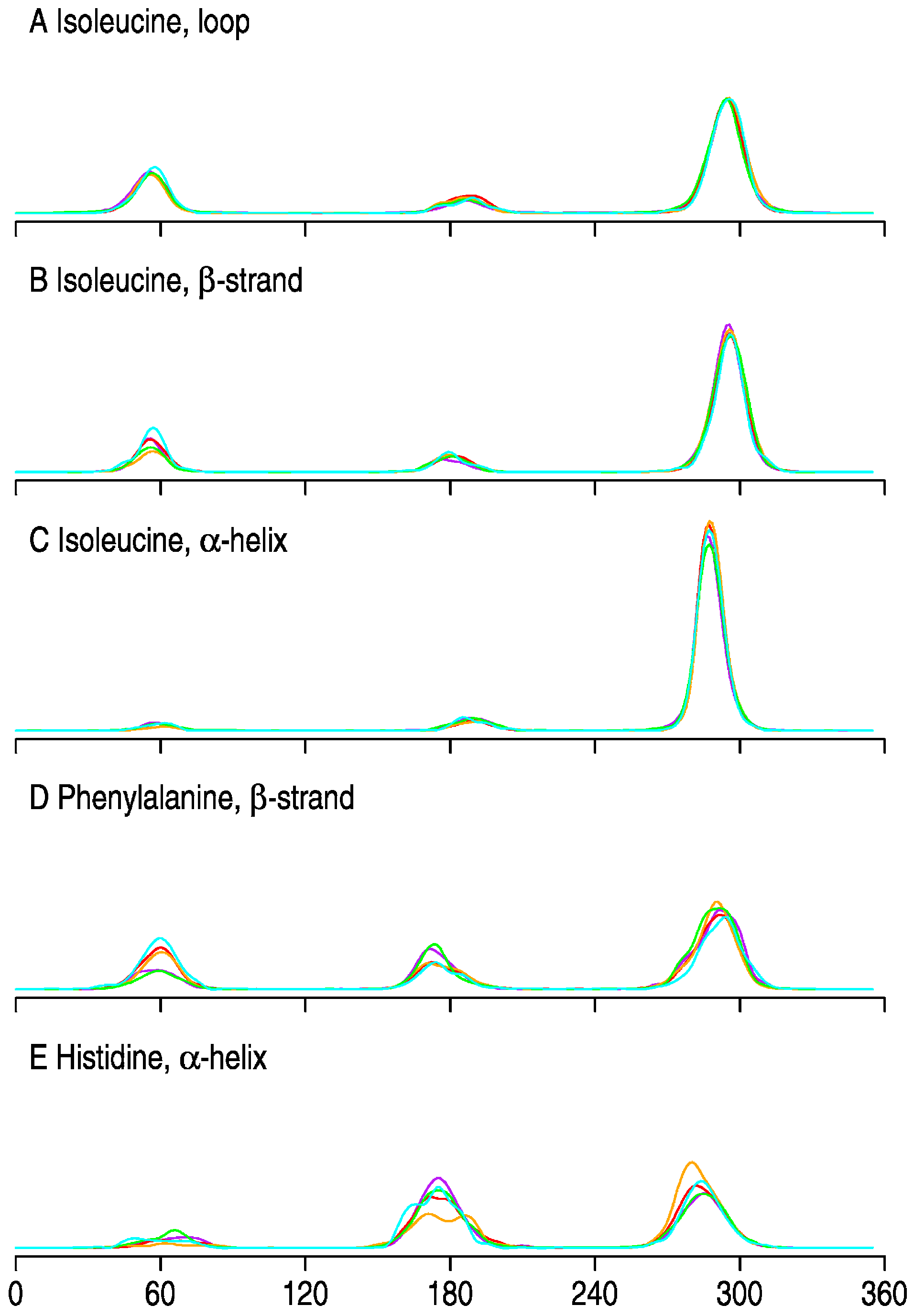
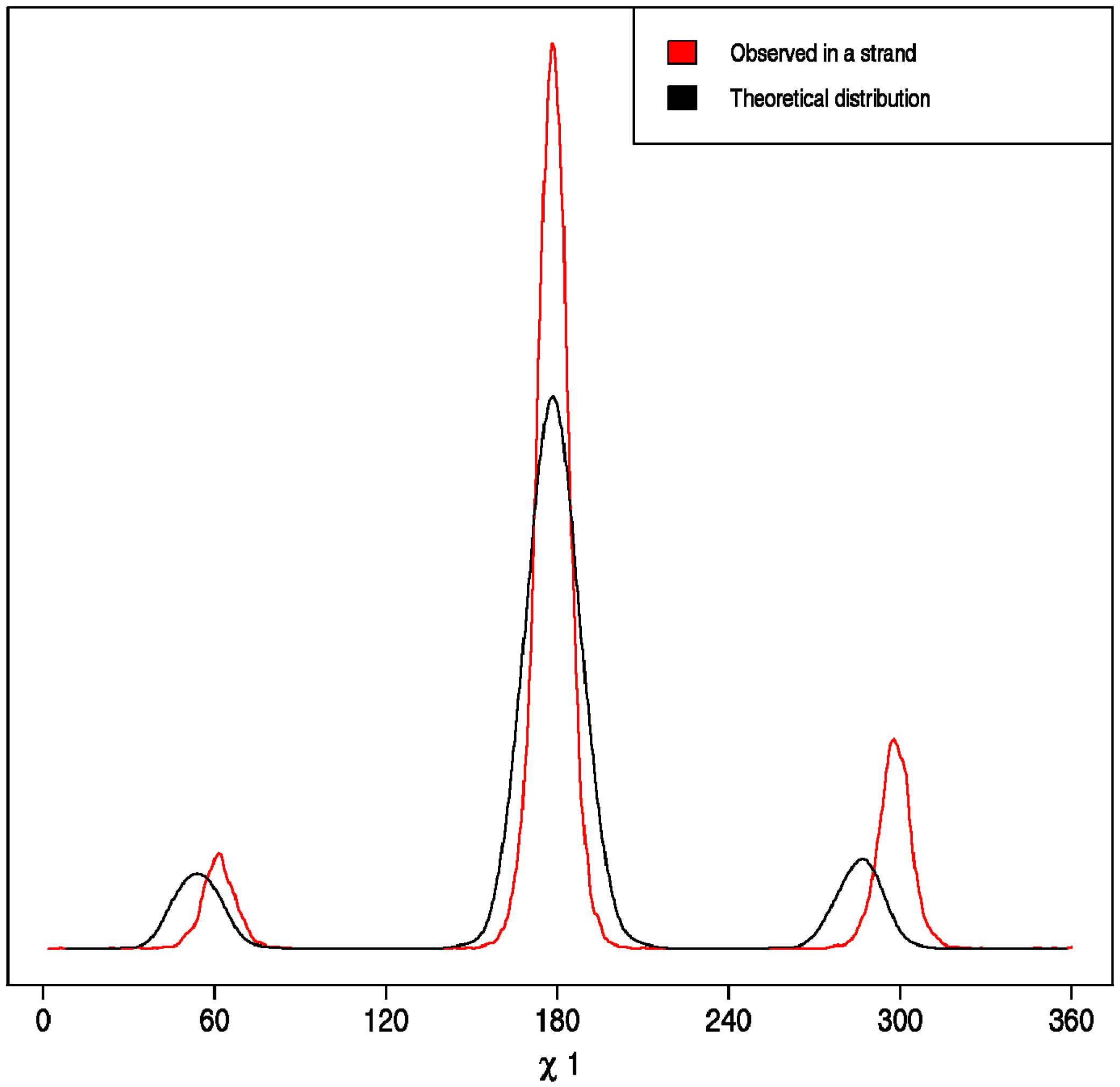
- Correct versus Wrong. A side chain is said to have the correct rotamer if the χ1 torsion angles of the model and the real structure differ less than 45°.
3. Results
4. Discussion
Author Contributions
Conflicts of Interest
List of Abbreviations
| PDB | Protein Data Bank |
| CASP | Critical Assessment of protein Structure Prediction |
| 3D | Three-Dimensional |
| SCOP | Structural Classification of Proteins |
| SMC | Symmetry Contact |
| NoSMC | No Symmetry Contact |
References
- Moult, J.; Pedersen, J.T.; Judson, R.; Fidelis, K. A large-scale experiment to assess protein structure prediction methods. Proteins 1995, 23, ii–v. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Hubbard, T.; Bryant, S.H.; Fidelis, K.; Pedersen, J.T. Critical assessment of methods of protein structure prediction (CASP): Round II. Proteins 1997, 37, 2–6. [Google Scholar] [CrossRef]
- Moult, J.; Hubbard, T.; Fidelis, K.; Pedersen, J.T. Critical assessment of methods of protein structure prediction (CASP): Round III. Proteins 1999, 3, 2–6. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Zemla, A.; Hubbard, T. Critical assessment of methods of protein structure prediction (CASP): Round IV. Proteins 2001, 5, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Zemla, A.; Hubbard, T. Critical assessment of methods of protein structure prediction (CASP): Round V. Proteins 2003, 53, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Rost, B.; Hubbard, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP): Round VI. Proteins 2005, 61, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Rost, B.; Hubbard, T.; Tramontano, A. Critical assessment of methods of protein structure prediction—Round VII. Proteins 2007, 69, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Rost, B.; Tramontano, A. Critical assessment of methods of protein structure prediction—Round VIII. Proteins 2009, 77, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Tramontano, A. Critical assessment of methods of protein structure prediction—Round IX. Proteins 2011, 79, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A.; Venclovas, C.; Moult, J.; Fidelis, K. Processing and analysis of CASP3 protein structure predictions. Proteins 1999, 3, 22–29. [Google Scholar] [CrossRef]
- Venclovas, C.; Zemla, A.; Fidelis, K.; Moult, J. Criteria for evaluating protein structures derived from comparative modeling. Proteins 1997, 1, 7–13. [Google Scholar] [CrossRef]
- Mosimann, S.; Meleshko, R.; James, M.N. A critical assessment of comparative molecular modeling of tertiary structures of proteins. Proteins 1995, 23, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Siew, N.; Elofsson, A.; Rychlewski, L.; Fischer, D. MaxSub: An automated measure for the assessment of protein structure prediction quality. Bioinformatics 2000, 16, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.R.; Strauss, C.E.; Olmea, O. MAMMOTH (matching molecular models obtained from theory): An automated method for model comparison. Protein Sci. 2002, 11, 2606–2621. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Milostan, M.; Szajkowski, L.; Daniluk, P.; Fidelis, K. CASP6 data processing and automatic evaluation at the protein structure prediction center. Proteins 2005, 61, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Cozzetto, D.; Kryshtafovych, A.; Fidelis, K.; Moult, J.; Rost, B.; Tramontano, A. Evaluation of template-based models in CASP8 with standard measures. Proteins 2009, 77, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Olechnovic, K.; Kulberkyte, E.; Venclovas, C. CAD-score: A new contact area difference-based function for evaluation of protein structural models. Proteins 2013, 81, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A.; Venclovas, C.; Moult, J.; Fidelis, K. Processing and evaluations of predictions in CASP4. Proteins 2001, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Boltzmann Distribution. Available online: https://en.wikipedia.org/wiki/Boltzmann_distribution (accessed on 1 November 2017).
- McGregor, M.J.; Islam, S.A.; Sternberg, M.J. Analysis of the relationship between side chain conformation and secondary structure in globular proteins. J. Mol. Biol. 1987, 198, 295–310. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Pal, D. Main-chain conformation features at different conformations of the side-chains in proteins. Protein Eng. 1998, 11, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Wodak, S.; Levitt, M.; Maigret, B. Conformation of amino acid side chains in proteins. J. Mol. Biol. 1978, 125, 357–386. [Google Scholar] [CrossRef]
- Benedetti, E.; Morelli, G.; Nemethy, G.; Scheraga, H.A. Statistical and energetic analysis of side-chain conformations in oligopeptides. Int. J. Peptide Protein Res. 1983, 22, 1–15. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Dunbrack, R.L., Jr.; Karplus, M. Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains. Nat. Struct. Mol. Biol. 1994, 1, 334–340. [Google Scholar] [CrossRef]
- Jones, T.A.; Thirup, S. Using known substructures in protein model building and crystallography. EMBO J. 1986, 5, 819–822. [Google Scholar] [PubMed]
- De Filippis, V.; Sander, C.; Vriend, G. Predicting local structural changes that result from point mutations. Protein Eng. 1994, 7, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Chinea, G.; Padron, G.; Hooft, R.W.W.; Sander, C.; Vriend, G. The use of position-specific rotamers in model building by homology. Proteins 1995, 23, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Dunbrack, R.L., Jr.; Karplus, M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 1993, 230, 543–574. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, M.; Böhm, G.; Jaenicke, R. Structural relationships of homologous proteins as a fundamental principle in homology modeling. Proteins Struct. Funct. Bioinform. 1993, 17, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO 1986, 5, 823–826. [Google Scholar]
- Rodriguez, R.; Chinea, G.; Lopez, N.; Pons, T.; Vriend, G. Homology modeling, model and software evaluation: Three related resources. Bioinformatics 1998, 14, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modelling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Vásquez, M. Modeling side-chain conformation. Curr. Opin. Struct. Biol. 1996, 6, 217–221. [Google Scholar] [CrossRef]
- Wilson, C.; Gregoret, L.M.; Agard, D.A. Modeling Side-chain Conformation for Homologous Proteins Using an Energy-based Rotamer Search. J. Mol. Biol. 1993, 229, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.Y.; Subbiah, S. How similar must a template protein be for homology modeling by side-chain packing methods? Pac. Symp. Biocomput. 1996, 126–141. [Google Scholar]
- Dunbrack, R.L., Jr. Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 2002, 12, 431–440. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Joosten, R.P.; Chinea, G.; Kleywegt, G.J.; Vriend, G. Validation of protein structure models. In Comprehensive Medicinal Chemistry II; Taylor, J., Triggle, D., Eds.; Elsevier: Oxford, UK, 2007; Volume 3, pp. 507–530. [Google Scholar]
- Berntsen, K.R.M.; Vriend, G. Anomalies in the refinement of isoleucine. Acta Crystallogr. D 2014, 70, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Bower, M.J.; Cohen, F.; Dunbrack, R.L. Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: A new homology modeling tool. J. Mol. Biol. 1997, 267, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Conte, L.L.; Ailey, B.; Hubbard, T.; Brenner, S.E.; Murzin, A.G.; Chothia, C. SCOP: A structural classification of proteins database. Nucleic Acids Res. 2000, 28, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G.; Sander, C. Detection of common three-dimentional substructures in protein. Proteins 1991, 11, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. WHAT IF: A molecular modelling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentilas for the Amber ff99SB protein force field. Proteins 2012, 78, 1950–1958. [Google Scholar]
- Scouras, A.D.; Daggett, V. The Dynameomics rotamer library: Amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci. 2011, 20, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Fasman, G.D. Prediction of the protein conformation. Biochemistry 1974, 13, 222–245. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Fasman, G.D. Conformational parameters for amino acids in helical, beta-sheet and random coil regions calculated from proteins. Biochemistry 1974, 13, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Desmet, J.; de Maeyer, M.; Hazes, B.; Lasters, I. The dead-end elimination theorem and its use in protein side-chain positioning. Nature 1992, 356, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Hekkelman, M.L.; Beek, T.A.T.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics Web service collection. Nucleic Acids Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | X-Contact % |
|---|---|
| Arg | 27.1 |
| Lys | 24.3 |
| Gln | 24.1 |
| Glu | 22.9 |
| Asn | 20.8 |
| Pro | 20.2 |
| His | 20.0 |
| Asp | 19.0 |
| Thr | 17.3 |
| Ser | 16.7 |
| Tyr | 16.6 |
| Trp | 16.0 |
| Met | 14.3 |
| Gly | 13.6 |
| Phe | 12.4 |
| Ala | 12.2 |
| Leu | 11.5 |
| Val | 10.1 |
| Ile | 9.8 |
| Cys | 6.4 |
| Total = 26,312 | |||
|---|---|---|---|
| α | β | α/β | α + β |
| 5503 | 8071 | 7537 | 5201 |
| Category | Number | Percentage |
|---|---|---|
| Conserved | 15,399 | 58.5 |
| Mutated | 10,913 | 41.4 |
| SMC | 6973 | 26.5 |
| NoSMC | 19,339 | 73.4 |
| Correct | 20,665 | 78.5 |
| Wrong | 5647 | 21.4 |
| A | (Sub-)Category | Observed | Predicted | Preference | |
| Conserved | SMC | Correct | 2933 | 3205 | −0.09 |
| Wrong | 536 | 876 | −0.49 | ||
| NoSMC | Correct | 11,108 | 8889 | +0.22 | |
| Wrong | 822 | 2429 | −1.08 | ||
| Mutated | SMC | Correct | 2056 | 2271 | −0.10 |
| Wrong | 1448 | 621 | +0.85 | ||
| NoSMC | Correct | 4568 | 6299 | −0.32 | |
| Wrong | 2841 | 1721 | +0.50 | ||
| B | Without Modeling | Observed | Predicted | Preference | |
| Conserved | SMC | 3469 | 4081 | −0.16 | |
| NoSMC | 11,930 | 11,318 | +0.05 | ||
| Mutated | SMC | 3504 | 2892 | +0.19 | |
| NoSMC | 7409 | 8021 | −0.08 | ||
| C | Without Symmetry | Observed | Predicted | Preference | |
| Conserved | Correct | 14,041 | 12,094 | +0.15 | |
| Wrong | 1358 | 3305 | −0.89 | ||
| Mutated | Correct | 6624 | 8571 | −0.26 | |
| Wrong | 4289 | 2342 | +0.60 | ||
| D | Without Conservation | Observed | Predicted | Preference | |
| SMC | Correct | 4989 | 5476 | −0.09 | |
| Wrong | 1984 | 1497 | +0.28 | ||
| NoSMC | Correct | 15,676 | 15,189 | +0.03 | |
| Wrong | 3663 | 4150 | −0.12 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, D.; Berntsen, K.R.M.; Baakman, C.; Vriend, G.; Lahiri, T. A Critical Note on Symmetry Contact Artifacts and the Evaluation of the Quality of Homology Models. Symmetry 2018, 10, 25. https://doi.org/10.3390/sym10010025
Singh D, Berntsen KRM, Baakman C, Vriend G, Lahiri T. A Critical Note on Symmetry Contact Artifacts and the Evaluation of the Quality of Homology Models. Symmetry. 2018; 10(1):25. https://doi.org/10.3390/sym10010025
Chicago/Turabian StyleSingh, Dipali, Karen R. M. Berntsen, Coos Baakman, Gert Vriend, and Tapobrata Lahiri. 2018. "A Critical Note on Symmetry Contact Artifacts and the Evaluation of the Quality of Homology Models" Symmetry 10, no. 1: 25. https://doi.org/10.3390/sym10010025
APA StyleSingh, D., Berntsen, K. R. M., Baakman, C., Vriend, G., & Lahiri, T. (2018). A Critical Note on Symmetry Contact Artifacts and the Evaluation of the Quality of Homology Models. Symmetry, 10(1), 25. https://doi.org/10.3390/sym10010025