From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry

Abstract
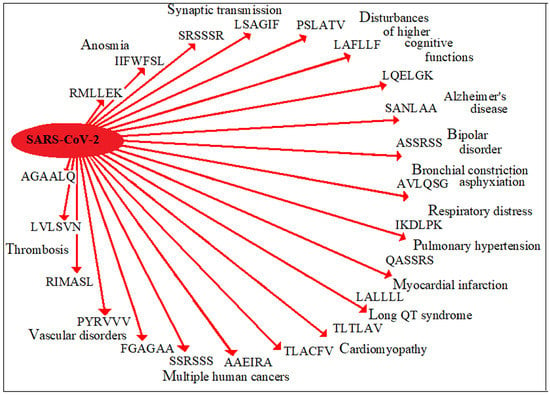
1. Introduction
- In 2008, it was reported [6] that prior immunization with SARS-CoV nucleocapsid protein N causes severe pneumonia in mice infected with SARS-CoV;
- In 2012, immunization with SARS coronavirus vaccines was found to lead to lung immunopathology on challenge with the SARS virus [7];
- In 2016, Agrawal et al. [8] demonstrated that immunization with inactivated Middle East Respiratory Syndrome (MERS) coronavirus vaccine leads to lung immunopathology on challenge with live virus. Moreover, the Authors documented that immunopathology with SARS-CoV vaccines occurred for whole-virus vaccines, subunit vaccines, different inactivation methods, different preparation substrates, and with recombinant surface spike protein. Finally, the Authors also underscored that, even if studies with vector vaccines point to the nucleoprotein N as responsible for the immunopathological effects and indicate that the S protein might be free of risk, nonetheless, also recombinant spike protein induced the immunopathology [6,7,8,9].
2. Materials and Methods
2.1. SARS-CoV-2 Epitopes
2.2. Analyses of the Peptide Sharing between SARS-CoV-2 Epitopes and the Human Proteome
2.3. Calculation of the Expected Value for Hexapeptide Sharing
3. Results
3.1. Numerical Description of the Peptide Sharing between SARS-CoV-2 Epitopes and the Human Proteome
3.2. Distribution of the Shared Hexapeptides through the SARS-CoV-2 Epitopes
3.3. Distribution of the Hexapeptide Sharing through the Human Proteins and the Potential Diseasome
- Pulmonary disorders
- Mothers against decapentaplegic homolog 9 protein (QASSRS), alterations of which may lead to pulmonary hypertension with proliferating endothelial cells in pulmonary arterioles, right ventricular failure, and death [21];
- Adenylate cyclase type 9 (KQLSSN) that is expressed in multiple cells of the lung with expression highest in airway smooth muscle [24];
- Acetylcholinesterase (AVLQSG) where imbalances in the neurotransmitter acetylcholine relate to neurological conditions, such as Alzheimer’s disease, Parkinson’s disease, and myastenia gravis; irreversible inhibition of acetylcholinesterase may lead to muscular paralysis, convulsions, bronchial constriction, and death by asphyxiation [25].
- Low-density lipoprotein receptor-related protein 8 (LALLLL), alterations of which can lead to myocardial infarction [37];
- Dol-P-Glc:Glc(2)Man(9)GlcNAc(2)-PP-Dol alpha-1,2-glucosyltransferase (TLTLAV) is implicated in susceptibility to the long QT syndrome [38];
- Presenilin-2 (TLACFV) relates to dilated cardiomyopathy and heart failure [39];
- Nuclear receptor coactivator 6 (PSLATV) can cause dilated cardiomyopathy [42];
- Latent-transforming growth factor beta-binding protein 3 (LALLLL) can associate with skin thickening, cardiac valvular thickening, tracheal stenosis, and respiratory insufficiency [43].
- Ephrin-B3 (LALLLL) is involved in blood pressure control and vascular smooth muscle cell contractility [44];
- Endoglin (LVLSVN) is required for normal structure and integrity of adult vasculature [45];
- Filamin-A (PYRVVV), alterations of filamin-A can cause disorders related to the vascular system and to a large phenotypic spectrum of disorders such as deafness, urogenital defects, malformations, intestinal obstruction, constipation, recurrent vomiting, and diarrhea [48];
- Glomulin (RIMASL) is crucial in vascular morphogenesis—especially in cutaneous veins [49].
- In addition, cross-reactive peptide sharing involving additional proteins can further affect the level of thrombomodulin. Indeed, cross-reactions with retinoic acid receptor RXR-alpha (LGFSTG) and prostaglandin G/H synthase 2 (TVLLKE) might completely eliminate thrombomodulin from blood circulation because (1) retinoic acid receptor RXR-alpha promotes the thrombomodulin gene transcription [55] and (2) prostaglandin G/H synthase 2 stimulates the expression of functionally active thrombomodulin in human smooth muscle cells [56].
- Adding up to such pro-thrombotic scenario, it is also worth of mention the potential cross-reactivity with tyrosine-protein kinase JAK2 (LLDDFV) that is involved in myelofibrosis, myeloid leukocytosis, and thrombocytosis with excessive platelet production resulting in increased numbers of circulating platelets, hemorrhages, and thrombotic episodes [57,58,59].
- Circadian locomotor output cycles protein kaput (ASSRSS) relates to bipolar disorder [62];
- Adenosine receptor A1 (VLPPLL), where sleep is significantly attenuated by the loss of adenosine A1 receptor expression [63];
- Calcium/calmodulin-dependent protein kinase kinase 2 (PSLATV) is linked to disturbances of higher cognitive functions, such as working memory and executive function. as well as schizophrenia [67];
- Endoplasmic reticulum mannosyl-oligosaccharide 1,2-alpha-mannosidase (LAFLLF) can cause mental retardation [68];
- Glutaminase kidney isoform, mitochondrial (LQELGK) can associate with epileptic encephalopathy, infantile cataract, skin abnormalities leukocytoclasia at the surface of the dermis, focal vacuolar alterations, hyperkeratosis, parakeratosis, glutamate excess, and impaired intellectual development, global developmental delay, progressive ataxia, and elevated glutamine [69,70,71];
- Mitochondrial glutamate carrier 1 (RLQSLQ) relates to neonatal myoclonic epilepsy [72].
4. Discussion
Supplementary Materials
Funding
Conflicts of Interest
References
- Cameron, M.J.; Bermejo-Martin, J.F.; Danesh, A.; Muller, M.P.; Kelvin, D.J. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 2008, 133, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.M.; Poon, L.L.; Lee, K.C.; Ng, W.F.; Lai, S.T.; Leung, C.Y.; Chu, C.M.; Hui, P.K.; Mak, K.L.; Lim, W.; et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003, 361, 1773–1778. [Google Scholar] [CrossRef]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.L.; Peiris, J.S. Pathogenesis of severe acute respiratory syndrome. Curr. Opin. Immunol. 2005, 17, 404–410. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- Yasui, F.; Kai, C.; Kitabatake, M.; Inoue, S.; Yoneda, M.; Yokochi, S.; Kase, R.; Sekiguchi, S.; Morita, K.; Hishima, T.; et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 2008, 181, 6337–6348. [Google Scholar] [CrossRef]
- Tseng, C.T.; Sbrana, E.; Iwata-Yoshikawa, N.; Newman, P.C.; Garron, T.; Atmar, R.L.; Peters, C.J.; Couch, R.B. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 2012, 7, e35421. [Google Scholar] [CrossRef]
- Agrawal, A.S.; Tao, X.; Algaissi, A.; Garron, T.; Narayanan, K.; Peng, B.H.; Couch, R.B.; Tseng, C.T. Immunization with inactivated Middle East Respiratory Syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum. Vaccin. Immunother. 2016, 12, 2351–2356. [Google Scholar] [CrossRef]
- Deming, D.; Sheahan, T.; Heise, M.; Yount, B.; Davis, N.; Sims, A.; Suthar, M.; Harkema, J.; Whitmore, A.; Pickles, R.; et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 2006, 3, 2359–2375. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. On the molecular determinants of the SARS-CoV-2 attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef]
- Pieczenik, G. Are the universes of antibodies and antigens symmetrical? Reprod. Biomed. Online 2003, 6, 154–156. [Google Scholar] [CrossRef]
- Kanduc, D. Pentapeptides as minimal functional units in cell biology and immunology. Curr. Protein Pept. Sci. 2013, 14, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Rothbard, J.B.; Koszinowski, U.H. A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature 1989, 337, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. Available online: www.iedb.org (accessed on 7 July 2020). [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef]
- Chen, C.; Li, Z.; Huang, H.; Suzek, B.E.; Wu, C.H.; UniProt Consortium. A fast Peptide Match service for UniProt knowledgebase. Bioinformatics 2013, 29, 2808–2809. Available online: https://proteininformationresource.org/pirwww/dbinfo/ (accessed on 7 July 2020). [CrossRef]
- Morris, B.; Adams, D.J.; van der Weyden, L. Renin gene expression: The switch and the fingers. Clin. Exp. Pharmacol. Physiol. 2001, 28, 1044–1047. [Google Scholar] [CrossRef]
- Kanduc, D. The comparative biochemistry of viruses and humans: An evolutionary path towards autoimmunity. Biol. Chem. 2019, 400, 629–638. [Google Scholar] [CrossRef]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef]
- Huang, X.; Dai, Z.; Cai, L.; Sun, K.; Cho, J.; Albertine, K.H.; Malik, A.B.; Schraufnagel, D.E.; Zhao, Y.Y. Endothelial p110γPI3K mediates endothelial regeneration and vascular repair after inflammatory vascular injury. Circulation 2016, 133, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.S.; Tao, J.Q.; Feinstein, S.I.; Zhang, L.; Fisher, A.B.; Bates, S.R. Role of the PI3-kinase signaling pathway in trafficking of the surfactant protein A receptor P63 (CKAP4) on type II pneumocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L794–L807. [Google Scholar] [CrossRef][Green Version]
- Teixeira, H.M.; Alcantara-Neves, N.M.; Barreto, M.; Figueiredo, C.A.; Costa, R.S. Adenylyl cyclase type 9 gene polymorphisms are associated with asthma and allergy in Brazilian children. Mol. Immunol. 2017, 82, 137–1145. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, T.P. Actions of drugs on the autonomic nervous system. In Pharmacology for Chemists: Drug Discovery in Context; Hill, R., Kenakin, T., Blackburn, T.P., Eds.; The Royal Society of Chemistry: London, UK, 2018; pp. 73–129. ISBN 978-1-78262-142-3. [Google Scholar]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Dai, S.D.; Wang, E.H. Down-regulation of NKD1 increases the invasive potential of non-small-cell lung cancer and correlates with a poor prognosis. BMC Cancer 2011, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.D.; Zhang, L.; Liu, X.P.; Jin, L.Y.; Dong, Q.; Li, F.N.; Wang, H.B.; Kong, B. NKD1 down-regulation is associated with poor prognosis in breast invasive ductal carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 4015–4021. [Google Scholar]
- Harada, H.; Nagai, H.; Tsuneizumi, M.; Mikami, I.; Sugano, S.; Emi, M. Identification of DMC1, a novel gene in the TOC region on 17q25.1 that shows loss of expression in multiple human cancers. J. Hum. Genet. 2001, 46, 90–95. [Google Scholar] [CrossRef]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ouyang, P.; Sugrue, S.P. Characterization of the gene encoding pinin/DRS/memA and evidence for its potential tumor suppressor function. Oncogene 2000, 19, 289–297. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lalonde, J.P.; Lim, R.; Ingley, E.; Tilbrook, P.A.; Thompson, M.J.; McCulloch, R.; Beaumont, J.G.; Wicking, C.; Eyre, H.J.; Sutherland, G.R.; et al. HLS5, a novel RBCC (ring finger, B box, coiled-coil) family member isolated from a hemopoietic lineage switch, is a candidate tumor suppressor. J. Biol. Chem. 2004, 279, 8181–8189. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Yi, F.; Wen, P.; Liu, J.; Chen, X.; Ren, J.; Li, X.; Shang, Y.; Nie, Y.; Wu, K.; et al. Myo9b is a key player in SLIT/ROBO-mediated lung tumor suppression. J. Clin. Invest. 2015, 125, 4407–4420. [Google Scholar] [CrossRef]
- Tschan, M.P.; Gullberg, U.; Shan, D.; Torbett, B.E.; Fey, M.F.; Tobler, A. The hDMP1 tumor suppressor is a new WT1 target in myeloid leukemias. Leukemia 2008, 22, 1087–1090. [Google Scholar] [CrossRef][Green Version]
- Shen, G.Q.; Li, L.; Girelli, D.; Seidelmann, S.B.; Rao, S.; Fan, C.; Park, J.E.; Xi, Q.; Li, J.; Hu, Y.; et al. An LRP8 variant is associated with familial and premature coronary artery disease and myocardial infarction. Am. J. Hum. Genet. 2007, 81, 780–791. [Google Scholar] [CrossRef]
- Hayashi, K.; Fujino, N.; Ino, H.; Uchiyama, K.; Sakata, K.; Konno, T.; Masuta, E.; Funada, A.; Sakamoto, Y.; Tsubokawa, T.; et al. A KCR1 variant implicated in susceptibility to the long QT syndrome. J. Mol. Cell. Cardiol. 2011, 50, 50–57. [Google Scholar] [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef]
- Szabadosova, V.; Boronova, I.; Ferenc, P.; Tothova, I.; Bernasovska, J.; Zigova, M.; Kmec, J.; Bernasovsky, I. Analysis of selected genes associated with cardiomyopathy by next-generation sequencing. J. Clin. Lab. Anal. 2018, 32. [Google Scholar] [CrossRef]
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z.; et al. Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum. Mol. Genet. 2017, 26, 2258–2276. [Google Scholar] [CrossRef]
- Roh, J.I.; Cheong, C.; Sung, Y.H.; Lee, J.; Oh, J.; Lee, B.S.; Lee, J.E.; Gho, Y.S.; Kim, D.K.; Park, C.B.; et al. Perturbation of NCOA6 leads to dilated cardiomyopathy. Cell Rep. 2014, 8, 991–998. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McInerney-Leo, A.M.; Le Goff, C.; Leo, P.J.; Kenna, T.J.; Keith, P.; Harris, J.E.; Steer, R.; Bole-Feysot, C.; Nitschke, P.; Kielty, C.; et al. Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. J. Med. Genet. 2016, 53, 457–464. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.; Luo, H.; Peng, J.; Raelson, J.; Ehret, G.B.; Munroe, P.B.; Stoyanova, E.; Qin, Z.; Cloutier, G.; et al. The role of GRIP1 and ephrin B3 in blood pressure control and vascular smooth muscle cell contractility. Sci. Rep. 2016, 6, 38976. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.C.; He, L.; Giro, M.; Yong, S.L.; Tiller, G.E.; Davidson, J.M. Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN). J. Biol. Chem. 1999, 274, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Urbán, Z.; Michels, V.V.; Thibodeau, S.N.; Davis, E.C.; Bonnefont, J.P.; Munnich, A.; Eyskens, B.; Gewillig, M.; Devriendt, K.; Boyd, C.D. Isolated supravalvular aortic stenosis: Functional haploinsufficiency of the elastin gene as a result of nonsense-mediated decay. Hum. Genet. 2000, 106, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, E.; Frentz, S.; Morgan, T.; García-Miñaúr, S.; Leventer, R.J.; McGillivray, G.; Pariani, M.; van der Steen, A.; Pope, M.; Holder-Espinasse, M.; et al. Vascular and connective tissue anomalies associated with X-linked periventricular heterotopia due to mutations in Filamin A. Eur. J. Hum. Genet. 2013, 21, 494–502. [Google Scholar] [CrossRef]
- Brouillard, P.; Boon, L.M.; Mulliken, J.B.; Enjolras, O.; Ghassibé, M.; Warman, M.L.; Tan, O.T.; Olsen, B.R.; Vikkula, M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am. J. Hum. Genet. 2002, 70, 866–874. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Aganna, E.; Hammond, L.; Hawkins, P.N.; Aldea, A.; McKee, S.A.; van Amstel, H.K.; Mischung, C.; Kusuhara, K.; Saulsbury, F.T.; Lachmann, H.J.; et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 2003, 48, 2632–2644. [Google Scholar] [CrossRef]
- Esmon, C.T.; Owen, W.G. The discovery of thrombomodulin. J. Thromb. Haemost. 2004, 2, 209–213. [Google Scholar] [CrossRef]
- Ito, T.; Thachil, J.; Asakura, H.; Levy, J.H.; Iba, T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions-a multi-faceted anticoagulant protein with therapeutic potential. Crit. Care 2019, 23, 280. [Google Scholar] [CrossRef] [PubMed]
- Isermann, B.; Sood, R.; Pawlinski, R.; Zogg, M.; Kalloway, S.; Degen, J.L.; Mackman, N.; Weiler, H. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat. Med. 2003, 9, 331–337. [Google Scholar] [CrossRef]
- Horie, S.; Ishii, H.; Matsumoto, F.; Kusano, M.; Kizaki, K.; Matsuda, J.; Kazama, M. Acceleration of thrombomodulin gene transcription by retinoic acid: Retinoic acid receptors and Sp1 regulate the promoter activity through interactions with two different sequences in the 5’-flanking region of human gene. J. Biol. Chem. 2001, 276, 2440–2450. [Google Scholar] [CrossRef] [PubMed]
- Rabausch, K.; Bretschneider, E.; Sarbia, M.; Meyer-Kirchrath, J.; Censarek, P.; Pape, R.; Fischer, J.W.; Schrör, K.; Weber, A.A. Regulation of thrombomodulin expression in human vascular smooth muscle cells by COX-2-derived prostaglandins. Circ. Res 2005, 96, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Rugless, M.J.; Jacobsen, S.E.; Schuh, A. Germline JAK2 mutation in a family with hereditary thrombocytosis. N. Engl. J. Med. 2012, 366, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Nicholas, B.; Rudrasingham, V.; Nash, S.; Kirov, G.; Owen, M.J.; Wimpory, D.C. Association of Per1 and Npas2 with autistic disorder: Support for the clock genes/social timing hypothesis. Mol. Psychiatry. 2007, 12, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Matsumoto, A.; Nakayama, K.; Jimbo, E.F.; Kojima, K.; Nagata, K.; Iwamoto, S.; Yamagata, T. Circadian-relevant genes are highly polymorphic in autism spectrum disorder patients. Brain Dev. 2016, 38, 91–99. [Google Scholar] [CrossRef]
- McClung, C.A. Role for the Clock gene in bipolar disorder. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Bjorness, T.E.; Kelly, C.L.; Gao, T.; Poffenberger, V.; Greene, R.W. Control and function of the homeostatic sleep response by adenosine A1 receptors. J. Neurosci. 2009, 29, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Ribases, M.; Gratacos, M.; Badia, A.; Jimenez, L.; Solano, R.; Vallejo, J.; Fernandez-Aranda, F.; Estivill, X. Contribution of NTRK2 to the genetic susceptibility to anorexia nervosa, harm avoidance and minimum body mass index. Mol. Psychiatry 2005, 10, 851–860. [Google Scholar] [CrossRef]
- Chen, Z.; Simmons, M.S.; Perry, R.T.; Wiener, H.W.; Harrell, L.E.; Go, R.C. Genetic association of neurotrophic tyrosine kinase receptor type 2 (NTRK2) with Alzheimer’s disease. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008, 147, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.M.; Siebert, M.; Bock, H.; Mota, S.M.; Krammer, B.R.; Duarte, J.Á.; Bragatti, J.A.; Castan, J.U.; de Castro, L.A.; Saraiva-Pereira, M.L.; et al. NTRK2 (TrkB gene)variants and temporal lobe epilepsy: A genetic association study. Epilepsy Res. 2017, 137, 1–8. [Google Scholar] [CrossRef]
- Yu, P.; Chen, X.; Zhao, W.; Zhang, Z.; Zhang, Q.; Han, B.; Zhai, J.; Chen, M.; Du, B.; Deng, X.; et al. Effect of rs1063843 in the CAMKK2 gene on the dorsolateral prefrontal cortex. Hum. Brain Mapp. 2016, 37, 2398–2406. [Google Scholar] [CrossRef]
- Rafiq, M.A.; Kuss, A.W.; Puettmann, L.; Noor, A.; Ramiah, A.; Ali, G.; Hu, H.; Kerio, N.A.; Xiang, Y.; Garshasbi, M.; et al. Mutations in the alpha 1,2-mannosidase gene, MAN1B1, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2011, 89, 176–182. [Google Scholar] [CrossRef]
- Rumping, L.; Büttner, B.; Maier, O.; Rehmann, H.; Lequin, M.; Schlump, J.U.; Schmitt, B.; Schiebergen-Bronkhorst, B.; Prinsen, H.C.M.T.; Losa, M.; et al. Identification of a loss-of-function mutation in the context of glutaminase deficiency and neonatal epileptic encephalopathy. JAMA Neurol. 2019, 76, 342–350. [Google Scholar] [CrossRef]
- Rumping, L.; Tessadori, F.; Pouwels, P.J.W.; Vringer, E.; Wijnen, J.P.; Bhogal, A.A.; Savelberg, S.M.C.; Duran, K.J.; Bakkers, M.J.G.; Ramos, R.J.J.; et al. GLS hyperactivity causes glutamate excess, infantile cataract and profound developmental delay. Hum. Mol. Genet. 2019, 28, 96–104. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.P.; Tarailo-Graovac, M.; Richmond, P.A.; Drögemöller, B.I.; Pouladi, M.A.; Leen, R.; Brand-Arzamendi, K.; Dobritzsch, D.; Dolzhenko, E.; Eberle, M.A.; et al. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N. Engl. J. Med. 2019, 380, 1433–1441. [Google Scholar] [CrossRef]
- Molinari, F.; Raas-Rothschild, A.; Rio, M.; Fiermonte, G.; Encha-Razavi, F.; Palmieri, L.; Palmieri, F.; Ben-Neriah, Z.; Kadhom, N.; Vekemans, M.; et al. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 2005, 76, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.S.; Deng, T.C.; Garcia, T.; Sellers, Z.M.; Best, P.M. Calcium channel gamma subunits: A functionally diverse protein family. Cell. Biochem. Biophys. 2007, 47, 178–186. [Google Scholar] [CrossRef]
- Payne, H.L. The role of transmembrane AMPA receptor regulatory proteins (TARPs) in neurotransmission and receptor trafficking. Mol. Membr. Biol. 2008, 25, 353–362. [Google Scholar] [CrossRef]
- Malnic, B.; Godfrey, P.A.; Buck, L.B. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. USA 2004, 101, 2584–2589. [Google Scholar] [CrossRef]
- Di Gennaro, F.; Pizzol, D.; Marotta, C.; Antunes, M.; Racalbuto, V.; Veronese, N.; Smith, L. Coronavirus Diseases (COVID-19) current status and future perspectives: A narrative review. Int. J. Environ. Res. Public. Health 2020, 17, 2690. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti–spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158. [Google Scholar] [CrossRef]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-mediated inflammatory re-sponses: From mechanisms to potential therapeutic tools. Virol. Sin. 2020, 1–6. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, F.; Yu, W.; He, T.; Yu, J.; Yi, C.E.; Ba, L.; Li, W.; Farzan, M.; Chen, Z.; et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J. Med. Virol. 2006, 78, 1–8. [Google Scholar] [CrossRef]
- Kanduc, D. Homology, similarity, and identity in peptide epitope immunodefinition. J. Pept. Sci. 2012, 18, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D. Peptide cross-reactivity: The original sin of vaccines. Front. Biosci. 2012, 4, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D. “Self-nonself” peptides in the design of vaccines. Curr. Pharm. Des. 2009, 15, 3283–3289. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D. Immunogenicity, immunopathogenicity, and immunotolerance in one graph. Anticancer Agents Med. Chem. 2015, 15, 1264–1268. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. From HBV to HPV: Designing vaccines for extensive and intensive vaccination campaigns worldwide. Autoimmun. Rev. 2016, 15, 1054–1061. [Google Scholar] [CrossRef]

{kind=link}
| Hexapeptides composing the 233 epitopes | 733 |
| Hexapeptides shared with the human proteome | 230 |
| Hexapeptides shared with the human proteome (including multiple occurrences) | 505 |
| Human proteins involved in the sharing | 460 |
| A 1 | B 2 | C 3 | A 1 | B 2 | C 3 |
|---|---|---|---|---|---|
| 956 | N | AEGSRGGSQA | 37,515 | N | lLLLDRLNql |
| 999 | S | aeiRASANLA | 37,544 | S | lLLQYGSfc |
| 1220 | S | aevqidrli | 37,583 | orf1b | Llmpiltlt |
| 1221 | S | aevqidrlit | 37,724 | S | LLQYGSfct |
| 1349 | ORF7b | AFLLFLVLI | 37,766 | orf1a | LLSAGIFGA |
| 1350 | ORF7b | AFLLFLVLIMLIIFw | 38,043 | orf1b | Lmierfvsl |
| 1946 | orf1a | AIILASFSA | 38,353 | S | lntLVKQLSSNFGAi |
| 2027 | orf1b | aimtrclav | 38,831 | S | LQDVVNQNAQALNTL |
| 2431 | N | ALALLLLDr | 38,855 | S | Lqipfamqm |
| 2682 | orf1b | ALLADKfpv | 38,874 | orf1b | LQLGFSTGv |
| 2801 | S | alntlvkql | 38,881 | N | LQLPQGttl |
| 2802 | N | ALNTPKdhi | 38,990 | S | lqslqtyvtQQLIRA |
| 2855 | orf1a | aLRANSAvk | 39,003 | S | lqtyvtQQLIRAAEI |
| 2998 | orf1a | alweiqqvv | 39,576 | N | Lsprwyfyy |
| 3589 | S | aphgvvflhv | 40,459 | orf1b | LVLSVNpyv |
| 3810 | N | APSASAFFgm | 40,677 | M | Lwllwpvtl |
| 3939 | S | aQALNTLvk | 40,685 | M | lWPVTLAcf |
| 3956 | N | AQFAPSASA | 41,962 | ORF7b | mLIIFWFSL |
| 3982 | S | aqkfnGLTVLPPLLT | 42,093 | orf1b | Mlwckdghv |
| 4307 | N | asaffgmsr | 42,128 | orf1b | Mmisagfsl |
| 4321 | S | ASANLAATk | 42,260 | orf1a | Mpaswvmri |
| 4936 | N | ATEGALNTPK | 42,648 | N | msriGMEVTPSGTWl |
| 5149 | M | aTSRTLSYY | 42,873 | S | mTSCCSCLk |
| 5150 | M | aTSRTLSYYK | 42,972 | orf1b | mvMCGGSLyv |
| 5209 | orf1b | ATVVIGtsk | 43,024 | M | mwSFNPETni |
| 5447 | orf1a | AVLQSGFRK | 44,501 | N | nkhidayktFPPTEP |
| 5908 | S | ayrfngiGVTQNVly | 44,814 | S | nlnESLIDL |
| 6184 | ORF7a | celyhyqecv | 44,913 | M | nlVIGFLFL |
| 6668 | S | cmTSCCSCLk | 60,380 | N | sQASSRSSSR |
| Shared Hexapeptide(s) | TARP |
|---|---|
| LSAGIF, SRSSSR | gamma-2 subunit |
| LSAGIF | gamma-3 subunit |
| SRSSSR | gamma-4 subunit |
| LGAGCF | gamma-6 subunit |
| SRSSSR | gamma-8 subunit |
| Shared Hexapeptide | Olfactory Receptor |
|---|---|
| AIILAS | 2B11 |
| AIILAS | 2W1 |
| SVLLSM | 51G1 |
| SVLLSM | 51G2 |
| RMLLEK | 51J1 |
| IFWFSL | 52N5 |
| IIFWFSL | 7D4 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanduc, D. From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry. Antibodies 2020, 9, 33. https://doi.org/10.3390/antib9030033
Kanduc D. From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry. Antibodies. 2020; 9(3):33. https://doi.org/10.3390/antib9030033
Chicago/Turabian StyleKanduc, Darja. 2020. "From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry" Antibodies 9, no. 3: 33. https://doi.org/10.3390/antib9030033
APA StyleKanduc, D. (2020). From Anti-SARS-CoV-2 Immune Responses to COVID-19 via Molecular Mimicry. Antibodies, 9(3), 33. https://doi.org/10.3390/antib9030033