Human IgA Monoclonal Antibodies That Neutralize Poliovirus, Produced by Hybridomas and Recombinant Expression

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Volunteer Blood Donors
2.2. B Cell Cultures and Hybridoma Creation
2.3. Immunocapture ELISAs
2.4. OCMS Analysis of mAbs Expressed by Human Hybridomas
2.5. Poliovirus Microneutralization Test
2.6. FPLC Gel Filtration Analysis and Immunoblotting
2.7. Expression Constructs for Recombinant IgA Expression
2.8. Recombinant mAb Expression and Purification
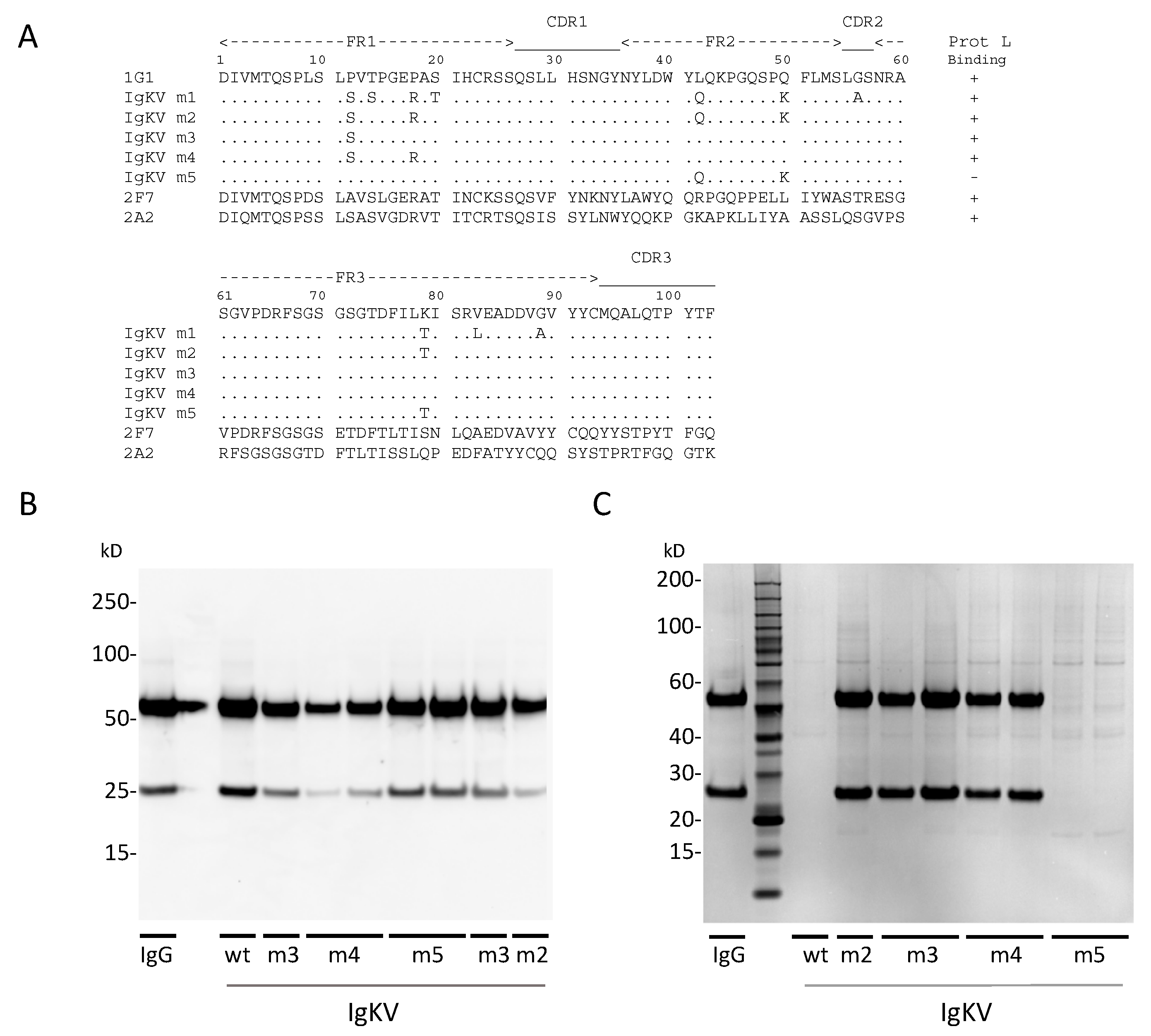
2.9. Generation and Screening of κ-Chain Mutants
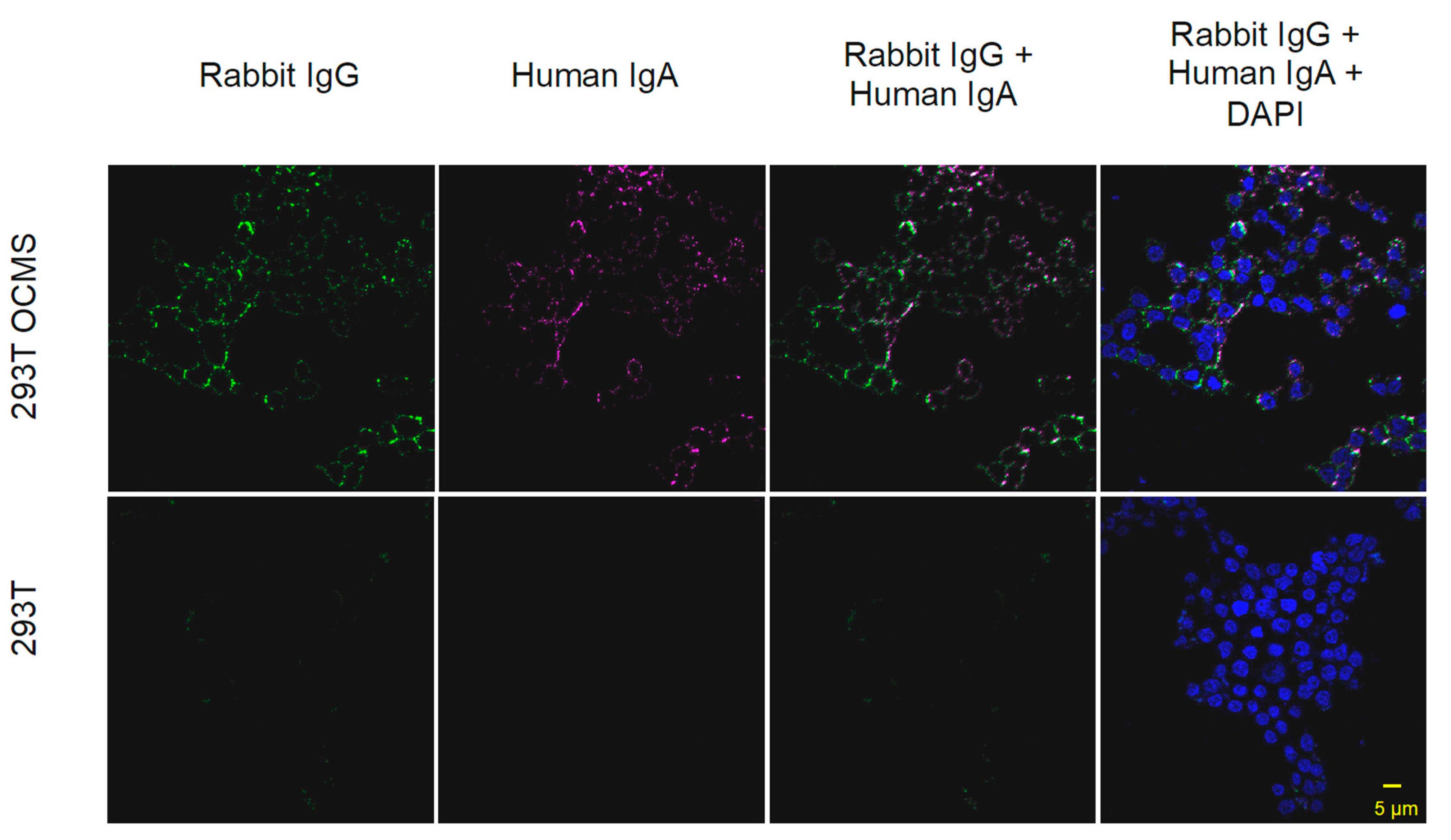
2.10. Binding of Recombinant IgA to 293T OCMS Cells
3. Results
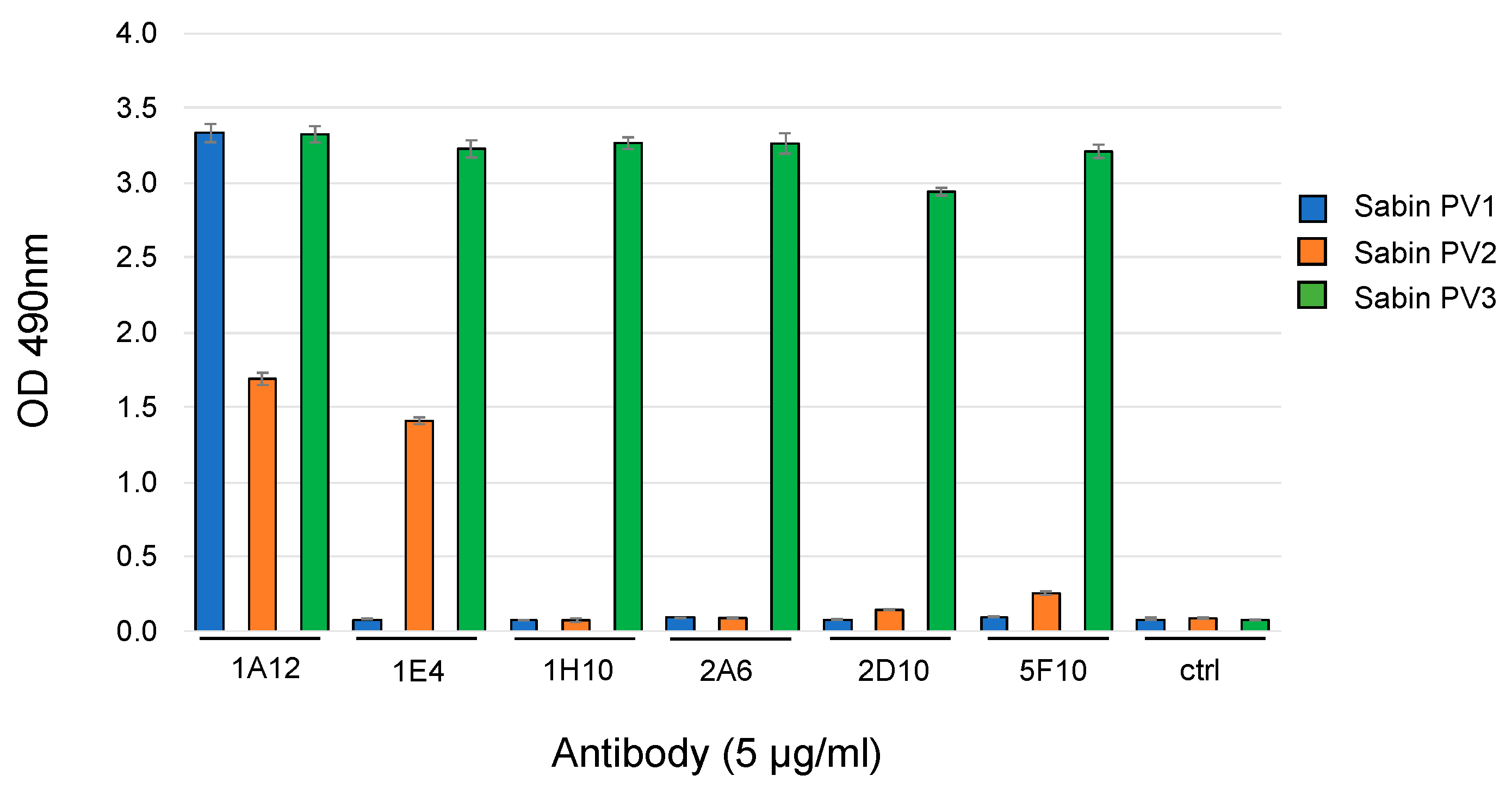
3.1. Human Monoclonal IgA Antibodies Specific for Poliovirus the Other Four IgAs Demonstrated
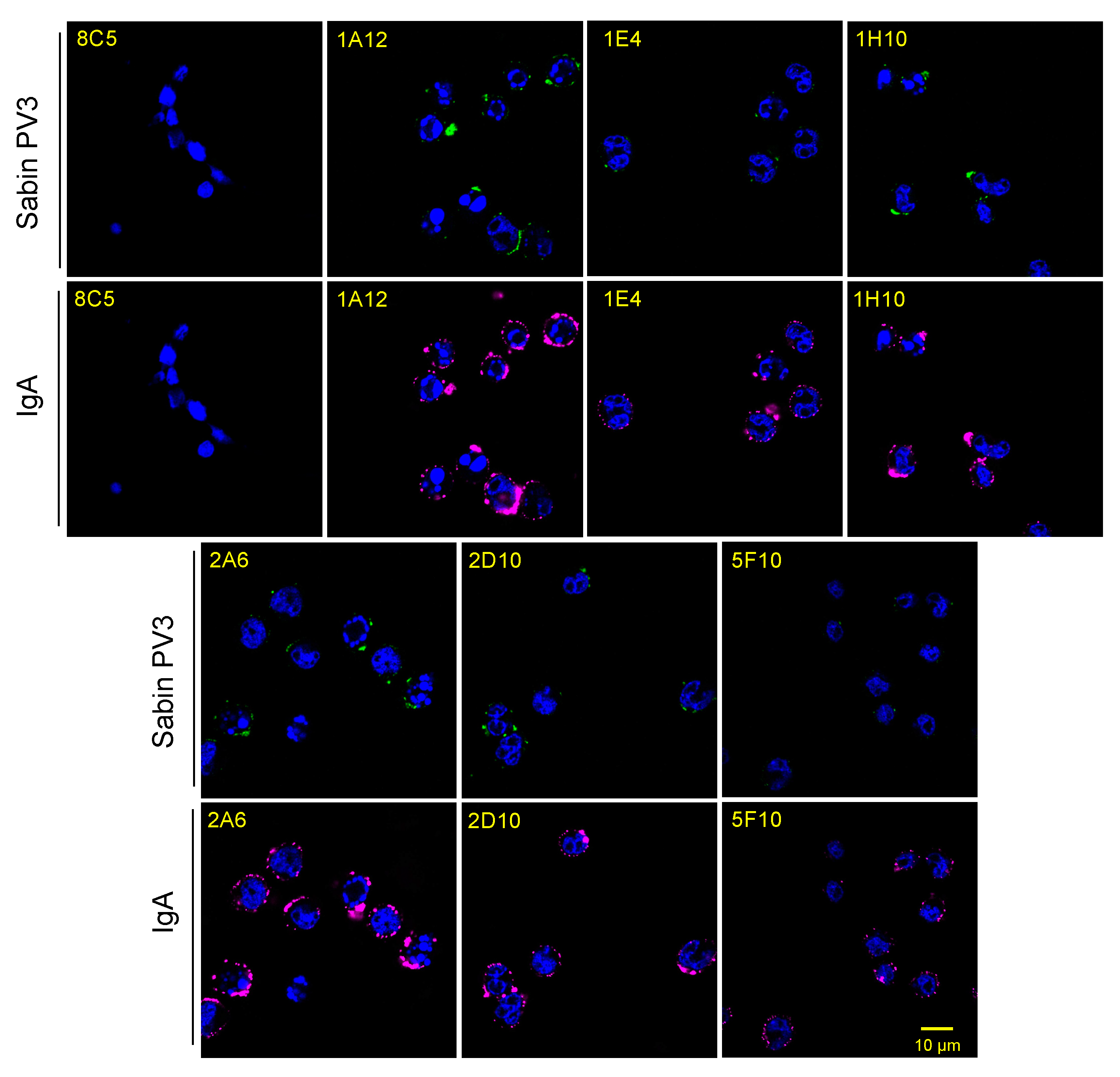
3.2. OCMS Testing of Hybridomas Secreting Human Anti-PV IgA mAbs
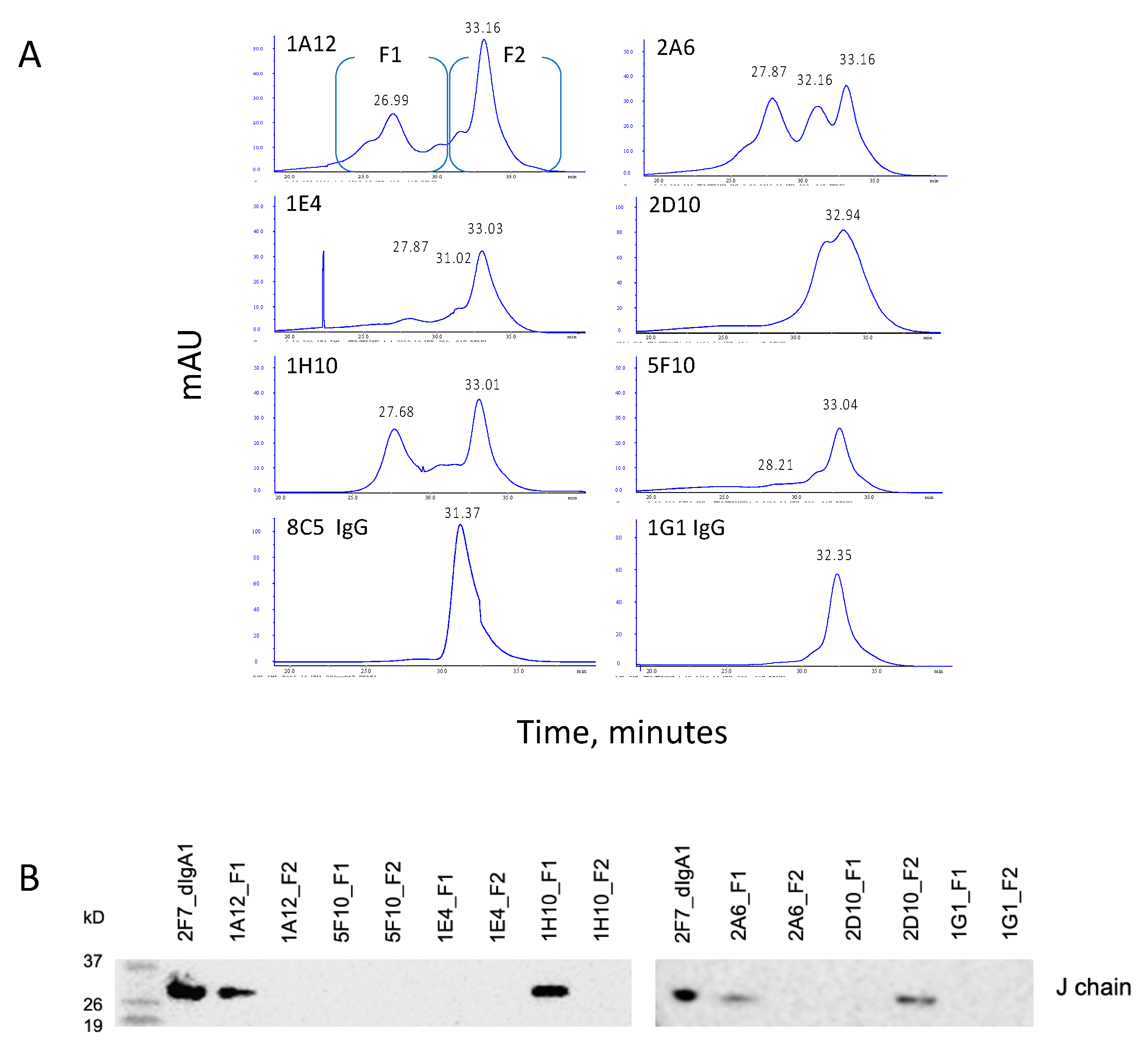
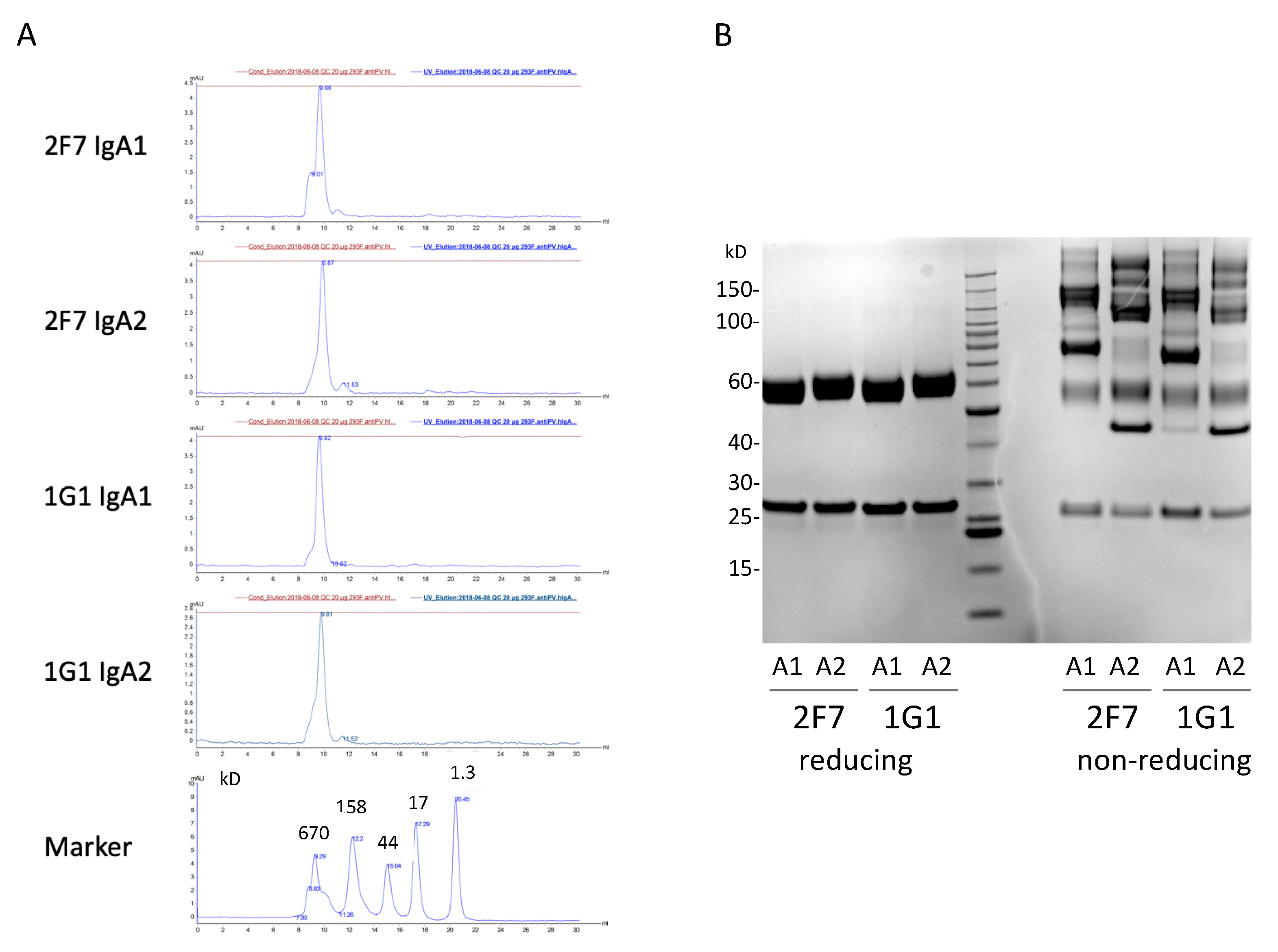
3.3. Gel Filtration Analysis of Hybridoma Expressed IgAs
3.4. Poliovirus Neutralizing Studies with Hybridoma-Expressed IgA mAbs
3.5. Expression of Recombinant Dimeric IgAs
3.6. Production and Purification and Testing of Dimeric IgAs
3.7. Neutralization of PV by Recombinant mAbs
3.8. Assessment of Recombinant IgA Expression in Transiently Transfected Cells Using OCMS
4. Discussion
5. Conclusions
- Human IgA monoclonal antibodies have been produced that have potent poliovirus neutralizing activity.
- IgA mAbs were cloned using by two methods: a hybridoma method with human memory B cells and by recombinant expression of isotype-switched immunoglobulin genes.
- On-Cell mAb Screening (OCMS) was used to characterize IgA expression by hybridomas and transiently transfected cells.
- These studies expand the toolkit for studying human IgA mAbs.
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hampton, L.M.; Farrell, M.; Ramirez-Gonzalez, A.; Menning, L.; Shendale, S.; Lewis, I.; Rubin, J.; Garon, J.; Harris, J.; Hyde, T.; et al. Cessation of Trivalent Oral Poliovirus Vaccine and Introduction of Inactivated Poliovirus Vaccine—Worldwide, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Hird, T.R.; Grassly, N.C. Systematic review of mucosal immunity induced by oral and inactivated poliovirus vaccines against virus shedding following oral poliovirus challenge. PLoS Pathog. 2012, 8, e1002599. [Google Scholar] [CrossRef] [PubMed]
- Troy, S.B.; Ferreyra-Reyes, L.; Huang, C.; Sarnquist, C.; Canizales-Quintero, S.; Nelson, C.; Baez-Saldana, R.; Holubar, M.; Ferreira-Guerrero, E.; Garcia-Garcia, L.; et al. Community circulation patterns of oral polio vaccine serotypes 1, 2, and 3 after Mexican national immunization weeks. J. Infect. Dis. 2014, 209, 1693–1699. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jorba, J.; Diop, O.M.; Iber, J.; Henderson, E.; Zhao, K.; Quddus, A.; Sutter, R.; Vertefeuille, J.F.; Wenger, J.; Wassilak, S.G.F.; et al. Update on Vaccine-Derived Poliovirus Outbreaks—Worldwide, January 2018-June 2019. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 1024–1028. [Google Scholar] [CrossRef]
- Van Damme, P.; De Coster, I.; Bandyopadhyay, A.S.; Revets, H.; Withanage, K.; De Smedt, P.; Suykens, L.; Oberste, M.S.; Weldon, W.C.; Costa-Clemens, S.A.; et al. The safety and immunogenicity of two novel live attenuated monovalent (serotype 2) oral poliovirus vaccines in healthy adults: A double-blind, single-centre phase 1 study. Lancet 2019, 394, 148–158. [Google Scholar] [CrossRef]
- Council, N.R. Exploring the Role of Antiviral Drugs in the Eradication of Polio: Workshop Report; The National Academies Press: Washington, DC, USA, 2006. [Google Scholar]
- Collett, M.S.; Hincks, J.R.; Benschop, K.; Duizer, E.; van der Avoort, H.; Rhoden, E.; Liu, H.; Oberste, M.S.; McKinlay, M.A.; Hartford, M. Antiviral Activity of Pocapavir in a Randomized, Blinded, Placebo-Controlled Human Oral Poliovirus Vaccine Challenge Model. J. Infect. Dis. 2017, 215, 335–343. [Google Scholar] [CrossRef]
- Puligedda, R.D.; Kouiavskaia, D.; Adekar, S.P.; Sharma, R.; Devi Kattala, C.; Rezapkin, G.; Bidzhieva, B.; Dessain, S.K.; Chumakov, K. Human monoclonal antibodies that neutralize vaccine and wild-type poliovirus strains. Antivir. Res. 2014, 108, 36–43. [Google Scholar] [CrossRef]
- Wright, P.F.; Wieland-Alter, W.; Ilyushina, N.A.; Hoen, A.G.; Arita, M.; Boesch, A.W.; Ackerman, M.E.; van der Avoort, H.; Oberste, M.S.; Pallansch, M.A.; et al. Intestinal immunity is a determinant of clearance of poliovirus after oral vaccination. J. Infect. Dis. 2014, 209, 1628–1634. [Google Scholar] [CrossRef]
- MacLennan, C.; Dunn, G.; Huissoon, A.P.; Kumararatne, D.S.; Martin, J.; O’Leary, P.; Thompson, R.A.; Osman, H.; Wood, P.; Minor, P.; et al. Failure to clear persistent vaccine-derived neurovirulent poliovirus infection in an immunodeficient man. Lancet 2004, 363, 1509–1513. [Google Scholar] [CrossRef]
- Odineal, D.D.; Gershwin, M.E. The Epidemiology and Clinical Manifestations of Autoimmunity in Selective IgA Deficiency. Clin. Rev. Allergy Immunol. 2020, 58, 107–133. [Google Scholar] [CrossRef]
- Dzidic, M.; Abrahamsson, T.R.; Artacho, A.; Bjorksten, B.; Collado, M.C.; Mira, A.; Jenmalm, M.C. Aberrant IgA responses to the gut microbiota during infancy precede asthma and allergy development. J. Allergy Clin. Immunol. 2017, 139, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Wills, S.; Hwang, K.K.; Liu, P.; Dennison, S.M.; Tay, M.Z.; Shen, X.; Pollara, J.; Lucas, J.T.; Parks, R.; Rerks-Ngarm, S.; et al. HIV-1-Specific IgA Monoclonal Antibodies from an HIV-1 Vaccinee Mediate Galactosylceramide Blocking and Phagocytosis. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Fadlallah, J.; El Kafsi, H.; Sterlin, D.; Juste, C.; Parizot, C.; Dorgham, K.; Autaa, G.; Gouas, D.; Almeida, M.; Lepage, P.; et al. Microbial ecology perturbation in human IgA deficiency. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.J.; Erickson, S.A.; Flynn, T.M.; Henry, C.; Koval, J.C.; Meisel, M.; Jabri, B.; Antonopoulos, D.A.; Wilson, P.C.; Bendelac, A. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 2017, 358. [Google Scholar] [CrossRef]
- Okai, S.; Usui, F.; Yokota, S.; Hori, I.Y.; Hasegawa, M.; Nakamura, T.; Kurosawa, M.; Okada, S.; Yamamoto, K.; Nishiyama, E.; et al. High-affinity monoclonal IgA regulates gut microbiota and prevents colitis in mice. Nat. Microbiol. 2016, 1, 16103. [Google Scholar] [CrossRef]
- Pabst, O. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 2012, 12, 821–832. [Google Scholar] [CrossRef]
- Woof, J.M.; Russell, M.W. Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590–597. [Google Scholar] [CrossRef]
- Fernandes, J.R.; Snider, D.P. Polymeric IgA-secreting and mucosal homing pre-plasma cells in normal human peripheral blood. Int. Immunol. 2010, 22, 527–540. [Google Scholar] [CrossRef]
- Mei, H.E.; Yoshida, T.; Sime, W.; Hiepe, F.; Thiele, K.; Manz, R.A.; Radbruch, A.; Dorner, T. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood 2009, 113, 2461–2469. [Google Scholar] [CrossRef]
- Iversen, R.; Snir, O.; Stensland, M.; Kroll, J.E.; Steinsbo, O.; Korponay-Szabo, I.R.; Lundin, K.E.A.; de Souza, G.A.; Sollid, L.M. Strong Clonal Relatedness between Serum and Gut IgA despite Different Plasma Cell Origins. Cell Rep. 2017, 20, 2357–2367. [Google Scholar] [CrossRef]
- Sapparapu, G.; Czako, R.; Alvarado, G.; Shanker, S.; Prasad, B.V.; Atmar, R.L.; Estes, M.K.; Crowe, J.E., Jr. Frequent Use of the IgA Isotype in Human B Cells Encoding Potent Norovirus-Specific Monoclonal Antibodies That Block HBGA Binding. PLoS Pathog. 2016, 12, e1005719. [Google Scholar] [CrossRef] [PubMed]
- El Bannoudi, H.; Anquetil, C.; Braunstein, M.J.; Pond, S.L.K.; Silverman, G.J. Unbiased RACE-Based Massive Parallel Surveys of Human IgA Antibody Repertoires. Methods Mol. Biol. 2017, 1643, 45–73. [Google Scholar] [CrossRef] [PubMed]
- Holtmeier, W.; Hennemann, A.; Caspary, W.F. IgA and IgM V(H) repertoires in human colon: Evidence for clonally expanded B cells that are widely disseminated. Gastroenterology 2000, 119, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Lombana, T.N.; Rajan, S.; Zorn, J.A.; Mandikian, D.; Chen, E.C.; Estevez, A.; Yip, V.; Bravo, D.D.; Phung, W.; Farahi, F.; et al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. mAbs 2019, 11, 1122–1138. [Google Scholar] [CrossRef]
- Westerhof, L.B.; Wilbers, R.H.; van Raaij, D.R.; van Wijk, C.Z.; Goverse, A.; Bakker, J.; Schots, A. Transient Expression of Secretory IgA In Planta is Optimal Using a Multi-Gene Vector and may be Further Enhanced by Improving Joining Chain Incorporation. Front. Plant Sci. 2015, 6, 1200. [Google Scholar] [CrossRef]
- Shoji, K.; Takahashi, T.; Kurohane, K.; Iwata, K.; Matsuoka, T.; Tsuruta, S.; Sugino, T.; Miyake, M.; Suzuki, T.; Imai, Y. Recombinant immunoglobulin A specific for influenza A virus hemagglutinin: Production, functional analysis, and formation of secretory immunoglobulin A. Viral Immunol. 2015, 28, 170–178. [Google Scholar] [CrossRef]
- Lorin, V.; Mouquet, H. Efficient generation of human IgA monoclonal antibodies. J. Immunol. Methods 2015, 422, 102–110. [Google Scholar] [CrossRef]
- Paul, M.; Reljic, R.; Klein, K.; Drake, P.M.; van Dolleweerd, C.; Pabst, M.; Windwarder, M.; Arcalis, E.; Stoger, E.; Altmann, F.; et al. Characterization of a plant-produced recombinant human secretory IgA with broad neutralizing activity against HIV. mAbs 2014, 6, 1585–1597. [Google Scholar] [CrossRef]
- Moldt, B.; Saye-Francisco, K.; Schultz, N.; Burton, D.R.; Hessell, A.J. Simplifying the synthesis of SIgA: Combination of dIgA and rhSC using affinity chromatography. Methods 2014, 65, 127–132. [Google Scholar] [CrossRef]
- Adekar, S.P.; Jones, R.M.; Elias, M.D.; Al-Saleem, F.H.; Root, M.J.; Simpson, L.L.; Dessain, S.K. Hybridoma populations enriched for affinity-matured human IgGs yield high-affinity antibodies specific for botulinum neurotoxins. J. Immunol. Methods 2008, 333, 156–166. [Google Scholar] [CrossRef]
- Puligedda, R.D.; Kouiavskaia, D.; Al-Saleem, F.H.; Kattala, C.D.; Nabi, U.; Yaqoob, H.; Bhagavathula, V.S.; Sharma, R.; Chumakov, K.; Dessain, S.K. Characterization of human monoclonal antibodies that neutralize multiple poliovirus serotypes. Vaccine 2017, 35, 5455–5462. [Google Scholar] [CrossRef]
- Puligedda, R.D.; Sharma, R.; Al-Saleem, F.H.; Kouiavskaia, D.; Velu, A.B.; Kattala, C.D.; Prendergast, G.C.; Lynch, D.R.; Chumakov, K.; Dessain, S.K. Capture and display of antibodies secreted by hybridoma cells enables fluorescent on-cell screening. mAbs 2019, 11, 546–558. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- World Health Organization. Manual for the Virological Investigation of Polio; World Health Organization: Geneva, Switzerland, 1997. [Google Scholar]
- Carbonetti, S.; Oliver, B.G.; Vigdorovich, V.; Dambrauskas, N.; Sack, B.; Bergl, E.; Kappe, S.H.I.; Sather, D.N. A method for the isolation and characterization of functional murine monoclonal antibodies by single B cell cloning. J. Immunol. Methods 2017, 448, 66–73. [Google Scholar] [CrossRef]
- Belnoue, E.; Pihlgren, M.; McGaha, T.L.; Tougne, C.; Rochat, A.F.; Bossen, C.; Schneider, P.; Huard, B.; Lambert, P.H.; Siegrist, C.A. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 2008, 111, 2755–2764. [Google Scholar] [CrossRef]
- Bossen, C.; Cachero, T.G.; Tardivel, A.; Ingold, K.; Willen, L.; Dobles, M.; Scott, M.L.; Maquelin, A.; Belnoue, E.; Siegrist, C.A.; et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 2008, 111, 1004–1012. [Google Scholar] [CrossRef]
- Castigli, E.; Wilson, S.A.; Scott, S.; Dedeoglu, F.; Xu, S.; Lam, K.P.; Bram, R.J.; Jabara, H.; Geha, R.S. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 2005, 201, 35–39. [Google Scholar] [CrossRef]
- Litinskiy, M.B.; Nardelli, B.; Hilbert, D.M.; He, B.; Schaffer, A.; Casali, P.; Cerutti, A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 2002, 3, 822–829. [Google Scholar] [CrossRef]
- Bonner, A.; Furtado, P.B.; Almogren, A.; Kerr, M.A.; Perkins, S.J. Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. J. Immunol. 2008, 180, 1008–1018. [Google Scholar] [CrossRef]
- Nilson, B.H.; Solomon, A.; Bjorck, L.; Akerstrom, B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J. Biol. Chem. 1992, 267, 2234–2239. [Google Scholar]
- Graille, M.; Stura, E.A.; Housden, N.G.; Beckingham, J.A.; Bottomley, S.P.; Beale, D.; Taussig, M.J.; Sutton, B.J.; Gore, M.G.; Charbonnier, J.B. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure 2001, 9, 679–687. [Google Scholar] [CrossRef]
- Muzard, J.; Adi-Bessalem, S.; Juste, M.; Laraba-Djebari, F.; Aubrey, N.; Billiald, P. Grafting of protein L-binding activity onto recombinant antibody fragments. Anal. Biochem. 2009, 388, 331–338. [Google Scholar] [CrossRef]
- de Sousa-Pereira, P.; Woof, J.M. IgA: Structure, Function, and Developability. Antibodies 2019, 8, 57. [Google Scholar] [CrossRef]
- Virdi, V.; Juarez, P.; Boudolf, V.; Depicker, A. Recombinant IgA production for mucosal passive immunization, advancing beyond the hurdles. Cell. Mol. Life Sci. 2016, 73, 535–545. [Google Scholar] [CrossRef]
- Juarez, P.; Huet-Trujillo, E.; Sarrion-Perdigones, A.; Falconi, E.E.; Granell, A.; Orzaez, D. Combinatorial Analysis of Secretory Immunoglobulin A (sIgA) Expression in Plants. Int. J. Mol. Sci. 2013, 14, 6205–6222. [Google Scholar] [CrossRef]
- Boes, A.; Spiegel, H.; Delbruck, H.; Fischer, R.; Schillberg, S.; Sack, M. Affinity purification of a framework 1 engineered mouse/human chimeric IgA2 antibody from tobacco. Biotechnol. Bioeng. 2011, 108, 2804–2814. [Google Scholar] [CrossRef]
- Reinhart, D.; Weik, R.; Kunert, R. Recombinant IgA production: Single step affinity purification using camelid ligands and product characterization. J. Immunol. Methods 2012, 378, 95–101. [Google Scholar] [CrossRef]
- Kouiavskaia, D.; Puligedda, R.D.; Dessain, S.K.; Chumakov, K. Universal ELISA for quantification of D-antigen in inactivated poliovirus vaccines. J. Virol. Methods 2019, 276, 113785. [Google Scholar] [CrossRef]
- He, B.; Xu, W.; Santini, P.A.; Polydorides, A.D.; Chiu, A.; Estrella, J.; Shan, M.; Chadburn, A.; Villanacci, V.; Plebani, A.; et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 2007, 26, 812–826. [Google Scholar] [CrossRef]
- Blanco, E.; Perez-Andres, M.; Sanoja-Flores, L.; Wentink, M.; Pelak, O.; Martin-Ayuso, M.; Grigore, G.; Torres-Canizales, J.; Lopez-Granados, E.; Kalina, T.; et al. Selection and validation of antibody clones against IgG and IgA subclasses in switched memory B-cells and plasma cells. J. Immunol. Methods 2019, 475, 112372. [Google Scholar] [CrossRef]
- Kutteh, W.H.; Prince, S.J.; Mestecky, J. Tissue origins of human polymeric and monomeric IgA. J. Immunol. 1982, 128, 990–995. [Google Scholar]
- Moldoveanu, Z.; Egan, M.L.; Mestecky, J. Cellular origins of human polymeric and monomeric IgA: Intracellular and secreted forms of IgA. J. Immunol. 1984, 133, 3156–3162. [Google Scholar]
- Phalipon, A.; Cardona, A.; Kraehenbuhl, J.P.; Edelman, L.; Sansonetti, P.J.; Corthesy, B. Secretory component: A new role in secretory IgA-mediated immune exclusion in vivo. Immunity 2002, 17, 107–115. [Google Scholar] [CrossRef]
- Lakhrif, Z.; Pugniere, M.; Henriquet, C.; di Tommaso, A.; Dimier-Poisson, I.; Billiald, P.; Juste, M.O.; Aubrey, N. A method to confer Protein L binding ability to any antibody fragment. mAbs 2016, 8, 379–388. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | FPLC | WT 1 | Sabin 3 | WT 3 |
|---|---|---|---|---|
| 1A12 | two peaks | x | ND | 600 |
| 1E4 | monomer | x | 800 | 7600 |
| 1H10 | two peaks | x | 102,400 | 204,800 |
| 2A6 | three peaks | x | 688,900 | 409,600 |
| 2D10 | superimposed peaks | x | 86,100 | 102,400 |
| 5F10 | monomer | x | 400 | 1300 |
| Antibody | Source | Sabin 1 | WT 1 | Sabin 3 | WT 3 |
|---|---|---|---|---|---|
| 2F7_IgG | H | 409,600 | 579,262 | x | x |
| 2F7_rIgG | R | 237,449 | 949,797 | x | x |
| 2F7_dIgA1 | R | 402,265 | 1,137,778 | x | x |
| 2F7_dIgA2 | R | 402,265 | 100,566 | x | x |
| 1G1_IgG | H | x | x | 204,800 | 102,400 |
| 1G1_rIgG | R | x | x | 72,408 | 144,815 |
| 1G1_dIgA1 | R | x | x | 289,631 | 102,400 |
| 1G1_dIgA2 | R | x | x | 204,800 | 144,815 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puligedda, R.D.; Vigdorovich, V.; Kouiavskaia, D.; Kattala, C.D.; Zhao, J.-y.; Al-Saleem, F.H.; Chumakov, K.; Sather, D.N.; Dessain, S.K. Human IgA Monoclonal Antibodies That Neutralize Poliovirus, Produced by Hybridomas and Recombinant Expression. Antibodies 2020, 9, 5. https://doi.org/10.3390/antib9010005
Puligedda RD, Vigdorovich V, Kouiavskaia D, Kattala CD, Zhao J-y, Al-Saleem FH, Chumakov K, Sather DN, Dessain SK. Human IgA Monoclonal Antibodies That Neutralize Poliovirus, Produced by Hybridomas and Recombinant Expression. Antibodies. 2020; 9(1):5. https://doi.org/10.3390/antib9010005
Chicago/Turabian StylePuligedda, Rama Devudu, Vladimir Vigdorovich, Diana Kouiavskaia, Chandana Devi Kattala, Jiang-yang Zhao, Fetweh H. Al-Saleem, Konstantin Chumakov, D. Noah Sather, and Scott K. Dessain. 2020. "Human IgA Monoclonal Antibodies That Neutralize Poliovirus, Produced by Hybridomas and Recombinant Expression" Antibodies 9, no. 1: 5. https://doi.org/10.3390/antib9010005
APA StylePuligedda, R. D., Vigdorovich, V., Kouiavskaia, D., Kattala, C. D., Zhao, J.-y., Al-Saleem, F. H., Chumakov, K., Sather, D. N., & Dessain, S. K. (2020). Human IgA Monoclonal Antibodies That Neutralize Poliovirus, Produced by Hybridomas and Recombinant Expression. Antibodies, 9(1), 5. https://doi.org/10.3390/antib9010005