Antibody Structure and Function: The Basis for Engineering Therapeutics


Abstract
1. Introduction
1.1. Overall Features of the Immunoglobulin
1.2. Fab Region
1.2.1. Fab Overall Features
1.2.2. The Fab Antigen-Binding Site
1.2.3. Relationship between Binding and Affinity
1.2.4. Canonical Structures of the CDRs
1.2.5. CDR-H3
1.2.6. Antibody Modeling
1.3. Fc Region
1.3.1. The Fc CH2–CH3 Interface
1.3.2. The Fc CH2 Carbohydrate
1.4. Hinge
2. Structure-Based Antibody Engineering
2.1. Humanization
2.1.1. CDR Definitions
2.1.2. Human Germline Selection
2.1.3. VH–VL Pairing
2.1.4. Back Mutations
2.1.5. Deimmunization
2.1.6. Resurfacing
2.1.7. Super-Humanization
2.1.8. Humanness Optimization
2.2. Lambda to Kappa Chain Switching
2.3. Affinity Maturation
2.4. Specificity
2.5. Chemistry, Manufacturing, and Control (CMC) Considerations
2.5.1. Solubility
2.5.2. Stability
3. Engineering Antibody Activity
3.1. Binding Domain Engineering
3.2. Avidity Modulation
3.3. Antibody–Drug Conjugates
3.4. Fc Activity Engineering
3.4.1. Mutations that Modulate Effector Function
3.4.2. Mutations that Alter Pharmacokinetics
3.5. Bispecific Antibodies
3.5.1. Bispecific Fragments
Fusion of Antigen-Binding Fragments
Fusion of Single-Chain Variable Fragments
Fusion of Single-Domain Antibodies
3.5.2. Fc-Dependent Bispecific Antibodies
Heavy Chain Heterodimerization
Light Chain Control
3.5.3. Considerations for Selection
4. Evolving Applications
4.1. Multispecific Molecules
4.2. Intracellular Targeting
5. Conclusions
Funding
Conflicts of Interest
References
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. mAbs 2019, 11, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2018. mAbs 2018, 10, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, G.L.; Luo, J.; Vafa, O.; Almagro, J.C. Leveraging SBDD in protein therapeutic development: Antibody engineering. Methods Mol. Biol. 2012, 841, 321–349. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Padlan, E.A. Structural basis for the specificity of antibody-antigen reactions and structural mechanisms for the diversification of antigen-binding specificities. Q. Rev. Biophys. 1977, 10, 35–65. [Google Scholar] [CrossRef] [PubMed]
- Amzel, L.M.; Poljak, R.J. Three-dimensional structure of immunoglobulins. Annu. Rev. Biochem. 1979, 48, 961–997. [Google Scholar] [CrossRef]
- Davies, D.R.; Metzger, H. Structural basis of antibody function. Annu. Rev. Immunol. 1983, 1, 87–117. [Google Scholar] [CrossRef]
- Wilson, I.A.; Stanfield, R.L. Antibody-antigen interactions: New structures and new conformational changes. Curr. Opin. Struct. Biol. 1994, 4, 857–867. [Google Scholar] [CrossRef]
- Poljak, R.; Amzel, L.; Avey, H.; Chen, B.; Phizackerley, R.; Saul, F. Three-dimensional structure of the Fab’ fragment of a human immunoglobulin at 2,8-A resolution. Proc. Natl. Acad. Sci. USA 1973, 70, 3305–3310. [Google Scholar] [CrossRef]
- Poljak, R.; Amzel, L.; Chen, B.; Phizackerley, R.; Saul, F. The three-dimensional structure of the Fab’ fragment of a human myeloma immunoglobulin at 2.0-angstrom resolution. Proc. Natl. Acad. Sci. USA 1974, 71, 3440–3444. [Google Scholar] [CrossRef]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 1997, 36, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Luo, J.; Muzammil, S.; Sweet, R.; Almagro, J.C.; Gilliland, G.L. Structural diversity in a human antibody germline library. mAbs 2016, 8, 1045–1063. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, M.; Girling, R.L.; Ely, K.R.; Edmundson, A.B. Structure of a lambda-type Bence-Jones protein at 3.5-A resolution. Biochemistry 1973, 12, 4620–4631. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Zemla, A.; Wilson, I.A.; Rupp, B. Antibody elbow angles are influenced by their light chain class. J. Mol. Biol. 2006, 357, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Lesk, A.M.; Chothia, C. Elbow motion in the immunoglobulins involves a molecular ball-and-socket joint. Nature 1988, 335, 188–190. [Google Scholar] [CrossRef]
- Love, R.A.; Villafranca, J.E.; Aust, R.M.; Nakamura, K.K.; Jue, R.A.; Major, J.G., Jr.; Radhakrishnan, R.; Butler, W.F. How the anti-(metal chelate) antibody CHA255 is specific for the metal ion of its antigen: X-ray structures for two Fab’/hapten complexes with different metals in the chelate. Biochemistry 1993, 32, 10950–10959. [Google Scholar] [CrossRef]
- Wu, T.T.; Kabat, E.A. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J. Exp. Med. 1970, 132, 211–250. [Google Scholar] [CrossRef]
- Kabat, E.A.; Wu, T.T. Attempts to locate complementarity-determining residues in the variable positions of light and heavy chains. Ann. N. Y. Acad. Sci. 1971, 190, 382–393. [Google Scholar] [CrossRef]
- Almagro, J.C. Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: Implications for the rational design of antibody repertoires. J. Mol. Recognit. 2004, 17, 132–143. [Google Scholar] [CrossRef]
- Raghunathan, G.; Smart, J.; Williams, J.; Almagro, J.C. Antigen-binding site anatomy and somatic mutations in antibodies that recognize different types of antigens. J. Mol. Recognit. 2012, 25, 103–113. [Google Scholar] [CrossRef]
- Persson, H.; Lantto, J.; Ohlin, M. A focused antibody library for improved hapten recognition. J. Mol. Biol. 2006, 357, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Cobaugh, C.W.; Almagro, J.C.; Pogson, M.; Iverson, B.; Georgiou, G. Synthetic antibody libraries focused towards peptide ligands. J. Mol. Biol. 2008, 378, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Quintero-Hernandez, V.; Ortiz-Leon, M.; Velandia, A.; Smith, S.L.; Becerril, B. Design and validation of a synthetic VH repertoire with tailored diversity for protein recognition. J. Mol. Recognit. 2006, 19, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Cohen, G.H. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. USA 1996, 93, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Padlan, E.A. Twisting into shape. Curr. Biol. 1992, 2, 254–256. [Google Scholar] [CrossRef]
- Vogt, A.D.; Pozzi, N.; Chen, Z.; di Cera, E. Essential role of conformational selection in ligand binding. Biophys. Chem. 2014, 186, 13–21. [Google Scholar] [CrossRef]
- Foote, J.; Milstein, C. Conformational isomerism and the diversity of antibodies. Proc. Natl. Acad. Sci. USA 1994, 91, 10370–10374. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical. Relaxation Rates. PLoS Comput. Biol. 2016, 12, e1005067. [Google Scholar] [CrossRef]
- Ma, B.; Zhao, J.; Nussinov, R. Conformational selection in amyloid-based immunotherapy: Survey of crystal structures of antibody-amyloid complexes. Biochim. Biophys. Acta 2016, 1860, 2672–2681. [Google Scholar] [CrossRef]
- Kenakin, T.P.; Morgan, P.H. Theoretical effects of single and multiple transducer receptor coupling proteins on estimates of the relative potency of agonists. Mol. Pharmacol. 1989, 35, 214–222. [Google Scholar]
- Chothia, C.; Lesk, A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M.; Tramontano, A.; Levitt, M.; Smith-Gill, S.J.; Air, G.; Sheriff, S.; Padlan, E.A.; Davies, D.; Tulip, W.R.; et al. Conformations of immunoglobulin hypervariable regions. Nature 1989, 342, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M.; Gherardi, E.; Tomlinson, I.M.; Walter, G.; Marks, J.D.; Llewelyn, M.B.; Winter, G. Structural repertoire of the human VH segments. J. Mol. Biol. 1992, 227, 799–817. [Google Scholar] [CrossRef]
- Tomlinson, I.M.; Cox, J.P.; Gherardi, E.; Lesk, A.M.; Chothia, C. The structural repertoire of the human V kappa domain. EMBO J. 1995, 14, 4628–4638. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Madrazo, E.; Lara-Ochoa, F.; Almagro, J.C. Canonical structure repertoire of the antigen-binding site of immunoglobulins suggests strong geometrical restrictions associated to the mechanism of immune recognition. J. Mol. Biol. 1995, 254, 497–504. [Google Scholar] [CrossRef]
- Collis, A.V.; Brouwer, A.P.; Martin, A.C. Analysis of the antigen combining site: Correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J. Mol. Biol. 2003, 325, 337–354. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Lesk, A.M.; Chothia, C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997, 273, 927–948. [Google Scholar] [CrossRef]
- North, B.; Lehmann, A.; Dunbrack, R.L., Jr. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef]
- Mas, M.T.; Smith, K.C.; Yarmush, D.L.; Aisaka, K.; Fine, R.M. Modeling the anti-CEA antibody combining site by homology and conformational search. Proteins 1992, 14, 483–498. [Google Scholar] [CrossRef]
- Shirai, H.; Kidera, A.; Nakamura, H. Structural classification of CDR-H3 in antibodies. FEBS Lett. 1996, 399, 1–8. [Google Scholar] [CrossRef]
- Morea, V.; Tramontano, A.; Rustici, M.; Chothia, C.; Lesk, A.M. Conformations of the third hypervariable region in the VH domain of immunoglobulins. J. Mol. Biol. 1998, 275, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Dunbrack, R.L., Jr.; Gray, J.J. The origin of CDR H3 structural diversity. Structure 2015, 23, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.C.; Thornton, J.M. Structural families in loops of homologous proteins: Automatic classification, modelling and application to antibodies. J. Mol. Biol. 1996, 263, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Beavers, M.P.; Hernandez-Guzman, F.; Maier, J.; Shaulsky, J.; Butenhof, K.; Labute, P.; Thorsteinson, N.; Kelly, K.; Teplyakov, A.; et al. Antibody modeling assessment. Proteins 2011, 79, 3050–3066. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Teplyakov, A.; Luo, J.; Sweet, R.W.; Kodangattil, S.; Hernandez-Guzman, F.; Gilliland, G.L. Second antibody modeling assessment (AMA-II). Proteins 2014, 82, 1553–1562. [Google Scholar] [CrossRef]
- Fasnacht, M.; Butenhof, K.; Goupil-Lamy, A.; Hernandez-Guzman, F.; Huang, H.; Yan, L. Automated antibody structure prediction using Accelrys tools: Results and best practices. Proteins 2014, 82, 1583–1598. [Google Scholar] [CrossRef]
- Maier, J.K.; Labute, P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014, 82, 1599–1610. [Google Scholar] [CrossRef]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014, 82, 1646–1655. [Google Scholar] [CrossRef]
- Weitzner, B.D.; Kuroda, D.; Marze, N.; Xu, J.; Gray, J.J. Blind prediction performance of Rosetta Antibody 3.0: Grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins 2014, 82, 1611–1623. [Google Scholar] [CrossRef]
- Berrondo, M.; Kaufmann, S.; Berrondo, M. Automated Aufbau of antibody structures from given sequences using Macromoltek’s SmrtMolAntibody. Proteins 2014, 82, 1636–1645. [Google Scholar] [CrossRef]
- Shirai, H.; Ikeda, K.; Yamashita, K.; Tsuchiya, Y.; Sarmiento, J.; Liang, S.; Morokata, T.; Mizuguchi, K.; Higo, J.; Standley, D.M.; et al. High-resolution modeling of antibody structures by a combination of bioinformatics, expert knowledge, and molecular simulations. Proteins 2014, 82, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Marcatili, P.; Rosi, A.; Tramontano, A. PIGS: Automatic prediction of antibody structures. Bioinformatics 2008, 24, 1953–1954. [Google Scholar] [CrossRef] [PubMed]
- Lepore, R.; Olimpieri, P.P.; Messih, M.A.; Tramontano, A. PIGSPro: Prediction of immunoGlobulin structures v2. Nucleic Acids Res. 2017, 45, W17–W23. [Google Scholar] [CrossRef] [PubMed]
- Teplyakov, A.; Luo, J.; Obmolova, G.; Malia, T.J.; Sweet, R.; Stanfield, R.L.; Kodangattil, S.; Almagro, J.C.; Gilliland, G.L. Antibody modeling assessment II. Structures and models. Proteins 2014, 82, 1563–1582. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.R. The formation of a specific inhibitor by hydrolysis of rabbit antiovalbumin. Biochem. J. 1950, 46, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.R. A chemical study of rabbit antiovalbumin. Biochem. J. 1950, 46, 473–478. [Google Scholar] [CrossRef]
- Porter, R.R. The hydrolysis of rabbit y-globulin and antibodies with crystalline papain. Biochem. J. 1959, 73, 119–126. [Google Scholar] [CrossRef]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef]
- Teplyakov, A.; Zhao, Y.; Malia, T.J.; Obmolova, G.; Gilliland, G.L. IgG2 Fc structure and the dynamic features of the IgG CH2-CH3 interface. Mol. Immunol. 2013, 56, 131–139. [Google Scholar] [CrossRef]
- Daeron, M. Fc receptor biology. Annu. Rev. Immunol. 1997, 15, 203–234. [Google Scholar] [CrossRef]
- Rouard, H.; Tamasdan, S.; Moncuit, J.; Moutel, S.; Michon, J.; Fridman, W.H.; Teillaud, J.L. Fc receptors as targets for immunotherapy. Int. Rev. Immunol. 1997, 16, 147–185. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Lindorfer, M.A. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: Historical precedence and recent developments. Blood 2015, 125, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, F.; Kuramochi, T.; Katada, H.; Igawa, T.; Hattori, K. Fc Engineering to Improve the Function of Therapeutic Antibodies. Curr. Pharm. Biotechnol. 2016, 17, 1298–1314. [Google Scholar] [CrossRef] [PubMed]
- Derebe, M.G.; Nanjunda, R.K.; Gilliland, G.L.; Lacy, E.R.; Chiu, M.L. Human IgG subclass cross-species reactivity to mouse and cynomolgus monkey Fcgamma receptors. Immunol. Lett. 2018, 197, 1–8. [Google Scholar] [CrossRef]
- Matsumiya, S.; Yamaguchi, Y.; Saito, J.; Nagano, M.; Sasakawa, H.; Otaki, S.; Satoh, M.; Shitara, K.; Kato, K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. [Google Scholar] [CrossRef]
- Saphire, E.O.; Stanfield, R.L.; Crispin, M.D.; Parren, P.W.; Rudd, P.M.; Dwek, R.A.; Burton, D.R.; Wilson, I.A. Contrasting IgG structures reveal extreme asymmetry and flexibility. J. Mol. Biol. 2002, 319, 9–18. [Google Scholar] [CrossRef]
- Borrok, M.J.; Jung, S.T.; Kang, T.H.; Monzingo, A.F.; Georgiou, G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem. Biol. 2012, 7, 1596–1602. [Google Scholar] [CrossRef]
- Landolfi, N.F.; Thakur, A.B.; Fu, H.; Vasquez, M.; Queen, C.; Tsurushita, N. The integrity of the ball-and-socket joint between V and C domains is essential for complete activity of a humanized antibody. J. Immunol. 2001, 166, 1748–1754. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation of natural and recombinant antibody molecules. Adv. Exp. Med. Biol. 2005, 564, 143–148. [Google Scholar]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 2015, 5, 8926. [Google Scholar] [CrossRef]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Frank, M.; Walker, R.C.; Lanzilotta, W.N.; Prestegard, J.H.; Barb, A.W. Immunoglobulin G1 Fc domain motions: Implications for Fc engineering. J. Mol. Biol. 2014, 426, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Sazinsky, S.L.; Ott, R.G.; Silver, N.W.; Tidor, B.; Ravetch, J.V.; Wittrup, K.D. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20167–20172. [Google Scholar] [CrossRef] [PubMed]
- Feige, M.J.; Groscurth, S.; Marcinowski, M.; Shimizu, Y.; Kessler, H.; Hendershot, L.M.; Buchner, J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 2009, 34, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Kronimus, Y.; Dodel, R.; Galuska, S.P.; Neumann, S. IgG Fc N-glycosylation: Alterations in neurologic diseases and potential therapeutic target? J. Autoimmun. 2019, 96, 14–23. [Google Scholar] [CrossRef]
- Hayashi, Y.; Miura, N.; Isobe, J.; Shinyashiki, N.; Yagihara, S. Molecular dynamics of hinge-bending motion of IgG vanishing with hydrolysis by papain. Biophys. J. 2000, 79, 1023–1029. [Google Scholar] [CrossRef][Green Version]
- Jay, J.W.; Bray, B.; Qi, Y.; Igbinigie, E.; Wu, H.; Li, J.; Ren, G. IgG Antibody 3D Structures and Dynamics. Antibodies 2018, 7, 18. [Google Scholar] [CrossRef]
- Van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martinez-Martinez, P.; Vermeulen, E.; den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007, 317, 1554–1557. [Google Scholar] [CrossRef]
- Turner, M.W.; Bennich, H. Subfragments from the Fc fragment of human immunoglobulin G. Isolation and physicochemical charaterization. Biochem. J. 1968, 107, 171–178. [Google Scholar]
- Ryan, M.H.; Petrone, D.; Nemeth, J.F.; Barnathan, E.; Bjorck, L.; Jordan, R.E. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol. Immunol. 2008, 45, 1837–1846. [Google Scholar] [CrossRef]
- Kinder, M.; Greenplate, A.R.; Grugan, K.D.; Soring, K.L.; Heeringa, K.A.; McCarthy, S.G.; Bannish, G.; Perpetua, M.; Lynch, F.; Jordan, R.E.; et al. Engineered protease-resistant antibodies with selectable cell-killing functions. J. Biol. Chem. 2013, 288, 30843–30854. [Google Scholar] [CrossRef] [PubMed]
- Brezski, R.J.; Oberholtzer, A.; Strake, B.; Jordan, R.E. The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. mAbs 2011, 3, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Senior, B.W.; Woof, J.M. The influences of hinge length and composition on the susceptibility of human IgA to cleavage by diverse bacterial IgA1 proteases. J. Immunol. 2005, 174, 7792–7799. [Google Scholar] [CrossRef] [PubMed]
- Senior, B.W.; Woof, J.M. Effect of mutations in the human immunoglobulin A1 (IgA1) hinge on its susceptibility to cleavage by diverse bacterial IgA1 proteases. Infect. Immun. 2005, 73, 1515–1522. [Google Scholar] [CrossRef][Green Version]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef]
- Fonseca, M.H.G.; Furtado, G.P.; Bezerra, M.R.L.; Pontes, L.Q.; Fernandes, C.F.C. Boosting half-life and effector functions of therapeutic antibodies by Fc-engineering: An interaction-function review. Int. J. Biol. Macromol. 2018, 119, 306–311. [Google Scholar] [CrossRef]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Kruse, T.; Schmidt, A.; Kampmann, M.; Strube, J. Integrated Clarification and Purification of Monoclonal Antibodies by Membrane Based Separation of Aqueous Two-Phase Systems. Antibodies 2019, 8, 40. [Google Scholar] [CrossRef]
- Lensink, M.F.; Wodak, S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013, 81, 2082–2095. [Google Scholar] [CrossRef]
- Klee, G.G. Human anti-mouse antibodies. Arch. Pathol. Lab. Med. 2000, 124, 921–923. [Google Scholar] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Verhoeyen, M.; Milstein, C.; Winter, G. Reshaping human antibodies: Grafting an antilysozyme activity. Science 1988, 239, 1534–1536. [Google Scholar] [CrossRef]
- Roguska, M.A.; Pedersen, J.T.; Keddy, C.A.; Henry, A.H.; Searle, S.J.; Lambert, J.M.; Goldmacher, V.S.; Blattler, W.A.; Rees, A.R.; Guild, B.C. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc. Natl. Acad. Sci. USA 1994, 91, 969–973. [Google Scholar] [CrossRef]
- Tan, P.; Mitchell, D.A.; Buss, T.N.; Holmes, M.A.; Anasetti, C.; Foote, J. “Superhumanized” antibodies: Reduction of immunogenic potential by complementarity-determining region grafting with human germline sequences: Application to an anti-CD28. J. Immunol. 2002, 169, 1119–1125. [Google Scholar] [CrossRef]
- Lazar, G.A.; Desjarlais, J.R.; Jacinto, J.; Karki, S.; Hammond, P.W. A molecular immunology approach to antibody humanization and functional optimization. Mol. Immunol. 2007, 44, 1986–1998. [Google Scholar] [CrossRef]
- Kabat, E.A.; Wu, T.T.; Bilofsky, H. Attempts to locate residues in complementarity-determining regions of antibody combining sites that make contact with antigen. Proc. Natl. Acad. Sci. USA 1976, 73, 617–619. [Google Scholar] [CrossRef]
- Martin, A.C. Protein Sequence and Structure Analysis of Antibody Variable Domains. In Antibody Engineering; Kontermann, R.E., Dübel, S., Eds.; Springer: Berlin, Germany, 2014; pp. 33–51. [Google Scholar]
- Adolf-Bryfogle, J.; Xu, Q.; North, B.; Lehmann, A.; Dunbrack, R.L., Jr. PyIgClassify: A database of antibody CDR structural classifications. Nucleic Acids Res. 2015, 43, D432–D438. [Google Scholar] [CrossRef]
- Honegger, A.; Pluckthun, A. Yet another numbering scheme for immunoglobulin variable domains: An automatic modeling and analysis tool. J. Mol. Biol. 2001, 309, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, M.P. IMGT, the international ImMunoGeneTics information system: A standardized approach for immunogenetics and immunoinformatics. Immunome Res 2005, 1, 3. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lefranc, M.P.; Giudicelli, V.; Kaas, Q.; Duprat, E.; Jabado-Michaloud, J.; Scaviner, D.; Ginestoux, C.; Clement, O.; Chaume, D.; Lefranc, G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2005, 33, D593–D597. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, M.P.; Ehrenmann, F.; Kossida, S.; Giudicelli, V.; Duroux, P. Use of IMGT((R)) Databases and Tools for Antibody Engineering and Humanization. Methods Mol. Biol. 2018, 1827, 35–69. [Google Scholar]
- Kunik, V.; Ashkenazi, S.; Ofran, Y. Paratome: An online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Res. 2012, 40, W521–W524. [Google Scholar] [CrossRef] [PubMed]
- Kunik, V.; Peters, B.; Ofran, Y. Structural consensus among antibodies defines the antigen binding site. PLoS Comput. Biol. 2012, 8, e1002388. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping human antibodies for therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 10029–10033. [Google Scholar] [CrossRef]
- Graziano, R.F.; Tempest, P.R.; White, P.; Keler, T.; Deo, Y.; Ghebremariam, H.; Coleman, K.; Pfefferkorn, L.C.; Fanger, M.W.; Guyre, P.M. Construction and characterization of a humanized anti-gamma-Ig receptor type I (Fc gamma RI) monoclonal antibody. J. Immunol. 1995, 155, 4996–5002. [Google Scholar]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef]
- Presta, L.G.; Lahr, S.J.; Shields, R.L.; Porter, J.P.; Gorman, C.M.; Fendly, B.M.; Jardieu, P.M. Humanization of an antibody directed against IgE. J. Immunol. 1993, 151, 2623–2632. [Google Scholar] [PubMed]
- Neuberger, M.S.; Milstein, C. Somatic hypermutation. Curr. Opin. Immunol. 1995, 7, 248–254. [Google Scholar] [CrossRef]
- Gorman, S.D.; Clark, M.R. Humanisation of monoclonal antibodies for therapy. Semin. Immunol. 1990, 2, 457–466. [Google Scholar] [PubMed]
- Rother, R.P.; Wu, D. Hybrid Antibodies. U.S. Patent 8,282,924, 9 October 2012. [Google Scholar]
- Lebozec, K.; Jandrot-Perrus, M.; Avenard, G.; Favre-Bulle, O.; Billiald, P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. mAbs 2017, 9, 945–958. [Google Scholar] [CrossRef]
- Schlapschy, M.; Fogarasi, M.; Gruber, H.; Gresch, O.; Schafer, C.; Aguib, Y.; Skerra, A. Functional humanization of an anti-CD16 Fab fragment: Obstacles of switching from murine {lambda} to human {lambda} or {kappa} light chains. Protein Eng. Des. Sel. 2009, 22, 175–188. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tsumoto, K.; Yokota, A.; Kondo, H.; Kumagai, I. Critical contribution of VH-VL interaction to reshaping of an antibody: The case of humanization of anti-lysozyme antibody, HyHEL-10. Protein Sci. 2008, 17, 261–270. [Google Scholar] [CrossRef]
- Brezinschek, H.P.; Foster, S.J.; Dorner, T.; Brezinschek, R.I.; Lipsky, P.E. Pairing of variable heavy and variable kappa chains in individual naive and memory B cells. J. Immunol. 1998, 160, 4762–4767. [Google Scholar]
- De Wildt, R.M.; Hoet, R.M.; van Venrooij, W.J.; Tomlinson, I.M.; Winter, G. Analysis of heavy and light chain pairings indicates that receptor editing shapes the human antibody repertoire. J. Mol. Biol. 1999, 285, 895–901. [Google Scholar] [CrossRef]
- De Wildt, R.M.; van Venrooij, W.J.; Winter, G.; Hoet, R.M.; Tomlinson, I.M. Somatic insertions and deletions shape the human antibody repertoire. J. Mol. Biol. 1999, 294, 701–710. [Google Scholar] [CrossRef]
- Edwards, B.M.; Barash, S.C.; Main, S.H.; Choi, G.H.; Minter, R.; Ullrich, S.; Williams, E.; Fou, L.D.; Wilton, J.; Albert, V.R.; et al. The remarkable flexibility of the human antibody repertoire; isolation of over one thousand different antibodies to a single protein, BLyS. J. Mol. Biol. 2003, 334, 103–118. [Google Scholar] [CrossRef]
- Lloyd, C.; Lowe, D.; Edwards, B.; Welsh, F.; Dilks, T.; Hardman, C.; Vaughan, T. Modelling the human immune response: Performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 2009, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, N.; Bhowmick, P.; Martin, A.C. Germline VH/VL pairing in antibodies. Protein Eng. Des. Sel. 2012, 25, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Sellers, B.D.; Jacobson, M.P. Energy-based analysis and prediction of the orientation between light- and heavy-chain antibody variable domains. J. Mol. Biol. 2009, 388, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Sircar, A.; Kim, E.T.; Gray, J.J. RosettaAntibody: Antibody variable region homology modeling server. Nucleic Acids Res. 2009, 37, W474–W479. [Google Scholar] [CrossRef]
- Abhinandan, K.R.; Martin, A.C. Analysis and prediction of VH/VL packing in antibodies. Protein Eng. Des. Sel. 2010, 23, 689–697. [Google Scholar] [CrossRef]
- Dunbar, J.; Fuchs, A.; Shi, J.; Deane, C.M. ABangle: Characterising the VH-VL orientation in antibodies. Protein Eng. Des. Sel. 2013, 26, 611–620. [Google Scholar] [CrossRef]
- Foote, J.; Winter, G. Antibody framework residues affecting the conformation of the hypervariable loops. J. Mol. Biol. 1992, 224, 487–499. [Google Scholar] [CrossRef]
- Tramontano, A.; Chothia, C.; Lesk, A.M. Framework residue 71 is a major determinant of the position and conformation of the second hypervariable region in the VH domains of immunoglobulins. J. Mol. Biol. 1990, 215, 175–182. [Google Scholar] [CrossRef]
- Papanastasiou, D.; Mamalaki, A.; Eliopoulos, E.; Poulas, K.; Liolitsas, C.; Tzartos, S.J. Construction and characterization of a humanized single chain Fv antibody fragment against the main immunogenic region of the acetylcholine receptor. J. Neuroimmunol. 1999, 94, 182–195. [Google Scholar] [CrossRef]
- Xiang, J.; Sha, Y.; Jia, Z.; Prasad, L.; Delbaere, L.T. Framework residues 71 and 93 of the chimeric B72.3 antibody are major determinants of the conformation of heavy-chain hypervariable loops. J. Mol. Biol. 1995, 253, 385–390. [Google Scholar] [CrossRef]
- Tempest, P.R.; White, P.; Buttle, M.; Carr, F.J.; Harris, W.J. Identification of framework residues required to restore antigen binding during reshaping of a monoclonal antibody against the glycoprotein gB of human cytomegalovirus. Int. J. Biol. Macromol. 1995, 17, 37–42. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Raghunathan, G.; Martinez, C.; Fransson, J.; Edwards, W.; Connor, J.; Husovsky, M.; Beck, H.; et al. Structural insights into humanization of anti-tissue factor antibody 10H10. mAbs 2018, 10, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Shembekar, N.; Mallajosyula, V.V.; Chaudhary, P.; Upadhyay, V.; Varadarajan, R.; Gupta, S.K. Humanized antibody neutralizing 2009 pandemic H1N1 virus. Biotechnol. J. 2014, 9, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wang, W.; Xu, Z.; Wang, S.; Wang, T.; Wang, M.; Wu, M. A humanized anti-DLL4 antibody promotes dysfunctional angiogenesis and inhibits breast tumor growth. Sci. Rep. 2016, 6, 27985. [Google Scholar] [CrossRef] [PubMed]
- Villani, M.E.; Morea, V.; Consalvi, V.; Chiaraluce, R.; Desiderio, A.; Benvenuto, E.; Donini, M. Humanization of a highly stable single-chain antibody by structure-based antigen-binding site grafting. Mol. Immunol. 2008, 45, 2474–2485. [Google Scholar] [CrossRef]
- Schrade, A.; Bujotzek, A.; Spick, C.; Wagner, M.; Goerl, J.; Wezler, X.; Georges, G.; Kontermann, R.E.; Brinkmann, U. Back-to-Germline (B2G) Procedure for Antibody Devolution. Antibodies 2019, 8, 45. [Google Scholar] [CrossRef]
- Bernett, M.J.; Karki, S.; Moore, G.L.; Leung, I.W.; Chen, H.; Pong, E.; Nguyen, D.H.; Jacinto, J.; Zalevsky, J.; Muchhal, U.S.; et al. Engineering fully human monoclonal antibodies from murine variable regions. J. Mol. Biol. 2010, 396, 1474–1490. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Hamilton, A.; King, S.; Liu, H.; Moy, P.; Bander, N.; Carr, F. A novel humanized antibody against prostate-specific membrane antigen also reacts with tumor vascular endothelium [abstract]. Proc. Am. Assoc. Cancer Res. 1998, 39, 440. [Google Scholar]
- Maynard, J.A.; Maassen, C.B.; Leppla, S.H.; Brasky, K.; Patterson, J.L.; Iverson, B.L.; Georgiou, G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat. Biotechnol. 2002, 20, 597–601. [Google Scholar] [CrossRef]
- Wirth, M.; Heidenreich, A.; Gschwend, J.E.; Gil, T.; Zastrow, S.; Laniado, M.; Gerloff, J.; Zuhlsdorf, M.; Mordenti, G.; Uhl, W.; et al. A multicenter phase 1 study of EMD 525797 (DI17E6), a novel humanized monoclonal antibody targeting alphav integrins, in progressive castration-resistant prostate cancer with bone metastases after chemotherapy. Eur. Urol. 2014, 65, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.S.; Choi, Y.; Griswold, K.E.; Bailey-Kellogg, C. Structure-guided deimmunization of therapeutic proteins. J. Comput. Biol. 2013, 20, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.D.; Crompton, L.J.; Carr, F.J.; Baker, M.P. Deimmunization of monoclonal antibodies. Methods Mol. Biol. 2009, 525, 405–423. [Google Scholar] [PubMed]
- Padlan, E.A. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol. Immunol. 1991, 28, 489–498. [Google Scholar] [CrossRef]
- Roguska, M.A.; Pedersen, J.T.; Henry, A.H.; Searle, S.M.; Roja, C.M.; Avery, B.; Hoffee, M.; Cook, S.; Lambert, J.M.; Blattler, W.A.; et al. A comparison of two murine monoclonal antibodies humanized by CDR-grafting and variable domain resurfacing. Protein Eng. 1996, 9, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A.; Wu, T.T.; Reid-Miller, M.; Gottesman, K. Sequences of Proteins of Immunological Interest; DHHS: Washington, DC, USA, 1991.
- Fan, C.Y.; Huang, S.Y.; Chou, M.Y.; Lyu, P.C. De novo protein sequencing, humanization and in vitro effects of an antihuman CD34 mouse monoclonal antibody. Biochem. Biophys. Rep. 2017, 9, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Skrlj, N.; Vranac, T.; Popovic, M.; Serbec, V.C.; Dolinar, M. Specific binding of the pathogenic prion isoform: Development and characterization of a humanized single-chain variable antibody fragment. PLoS ONE 2011, 6, e15783. [Google Scholar] [CrossRef]
- Bugelski, P.J.; Achuthanandam, R.; Capocasale, R.J.; Treacy, G.; Bouman-Thio, E. Monoclonal antibody-induced cytokine-release syndrome. Expert Rev. Clin. Immunol. 2009, 5, 499–521. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Almagro, J.C.; Buss, T.N.; Tan, P.; Foote, J. Use of human germline genes in a CDR homology-based approach to antibody humanization. Methods 2005, 36, 35–42. [Google Scholar] [CrossRef]
- Hu, W.G.; Chau, D.; Wu, J.; Jager, S.; Nagata, L.P. Humanization and mammalian expression of a murine monoclonal antibody against Venezuelan equine encephalitis virus. Vaccine 2007, 25, 3210–3214. [Google Scholar] [CrossRef]
- Pelat, T.; Bedouelle, H.; Rees, A.R.; Crennell, S.J.; Lefranc, M.P.; Thullier, P. Germline humanization of a non-human primate antibody that neutralizes the anthrax toxin, by in vitro and in silico engineering. J. Mol. Biol. 2008, 384, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Hua, C.; Sentman, C.L.; Ackerman, M.E.; Bailey-Kellogg, C. Antibody humanization by structure-based computational protein design. mAbs 2015, 7, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Abhinandan, K.R.; Martin, A.C. Analyzing the “degree of humanness” of antibody sequences. J. Mol. Biol. 2007, 369, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.H.; Huang, K.; Tu, H.; Adler, A.S. Monoclonal antibody humanness score and its applications. BMC Biotechnol. 2013, 13, 55. [Google Scholar] [CrossRef]
- Seeliger, D. Development of scoring functions for antibody sequence assessment and optimization. PLoS ONE 2013, 8, e1002388. [Google Scholar] [CrossRef]
- Clavero-Alvarez, A.; di Mambro, T.; Perez-Gaviro, S.; Magnani, M.; Bruscolini, P. Humanization of Antibodies using a Statistical Inference Approach. Sci. Rep. 2018, 8, 14820. [Google Scholar] [CrossRef]
- Worn, A.; der Maur, A.A.; Escher, D.; Honegger, A.; Barberis, A.; Pluckthun, A. Correlation between in vitro stability and in vivo performance of anti-GCN4 intrabodies as cytoplasmic inhibitors. J. Biol. Chem. 2000, 275, 2795–2803. [Google Scholar] [CrossRef]
- Lehmann, A.; Wixted, J.H.; Shapovalov, M.V.; Roder, H.; Dunbrack, R.L., Jr.; Robinson, M.K. Stability engineering of anti-EGFR scFv antibodies by rational design of a lambda-to-kappa swap of the VL framework using a structure-guided approach. mAbs 2015, 7, 1058–1071. [Google Scholar] [CrossRef]
- Tiller, T.; Schuster, I.; Deppe, D.; Siegers, K.; Strohner, R.; Herrmann, T.; Berenguer, M.; Poujol, D.; Stehle, J.; Stark, Y.; et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs 2013, 5, 445–470. [Google Scholar] [CrossRef]
- Foote, J.; Eisen, H.N. Kinetic and affinity limits on antibodies produced during immune responses. Proc. Natl. Acad. Sci. USA 1995, 92, 1254–1256. [Google Scholar] [CrossRef]
- Batista, F.D.; Neuberger, M.S. Affinity dependence of the B cell response to antigen: A threshold, a ceiling, and the importance of off-rate. Immunity 1998, 8, 751–759. [Google Scholar] [CrossRef]
- Yanaka, S.; Moriwaki, Y.; Tsumoto, K.; Sugase, K. Elucidation of potential sites for antibody engineering by fluctuation editing. Sci. Rep. 2019, 7, 9597. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Boriack-Sjodin, P.A.; Eldredge, J.; Fitch, C.; Friedman, B.; Hanf, K.J.; Jarpe, M.; Liparoto, S.F.; Li, Y.; Lugovskoy, A.; et al. Affinity enhancement of an in vivo matured therapeutic antibody using structure-based computational design. Protein Sci. 2006, 15, 949–960. [Google Scholar] [CrossRef]
- Lippow, S.M.; Tidor, B. Progress in computational protein design. Curr. Opin. Biotechnol. 2007, 18, 305–311. [Google Scholar] [CrossRef]
- Lippow, S.M.; Wittrup, K.D.; Tidor, B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat. Biotechnol. 2007, 25, 1171–1176. [Google Scholar] [CrossRef]
- Li, B.; Zhao, L.; Wang, C.; Guo, H.; Wu, L.; Zhang, X.; Qian, W.; Wang, H.; Guo, Y. The protein-protein interface evolution acts in a similar way to antibody affinity maturation. J. Biol. Chem. 2010, 285, 3865–3871. [Google Scholar] [CrossRef]
- Berek, C.; Milstein, C. Mutation drift and repertoire shift in the maturation of the immune response. Immunol. Rev. 1987, 96, 23–41. [Google Scholar] [CrossRef]
- Rogozin, I.; Kondrashov, F.; Glazko, G. Use of mutation spectra analysis software. Hum. Mutat. 2001, 17, 83–102. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Pavlov, Y.I.; Bebenek, K.; Matsuda, T.; Kunkel, T.A. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat. Immunol. 2001, 2, 530–536. [Google Scholar] [CrossRef]
- Kiyoshi, M.; Caaveiro, J.M.; Miura, E.; Nagatoishi, S.; Nakakido, M.; Soga, S.; Shirai, H.; Kawabata, S.; Tsumoto, K. Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS ONE 2014, 9, e87099. [Google Scholar] [CrossRef]
- Babor, M.; Mandell, D.J.; Kortemme, T. Assessment of flexible backbone protein design methods for sequence library prediction in the therapeutic antibody Herceptin-HER2 interface. Protein Sci. 2011, 20, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177–2198. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Suganami, A.; Ishida, I.; Tamura, Y.; Maeda, Y. Affinity maturation of a CDR3-grafted VHH using in silico analysis and surface plasmon resonance. J. Biochem. 2013, 154, 325–332. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Epa, V.C.; Colman, P.M. Electrostatic complementarity at protein/protein interfaces. J. Mol. Biol. 1997, 268, 570–584. [Google Scholar] [CrossRef]
- Pedotti, M.; Simonelli, L.; Livoti, E.; Varani, L. Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin. Int. J. Mol. Sci. 2011, 12, 226–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Liang, W.; Zheng, J.; Li, S.; Hu, C.; Chen, A. A Highly Sensitive Detection System based on Proximity-dependent Hybridization with Computer-aided Affinity Maturation of a scFv Antibody. Sci. Rep. 2018, 8, 3837. [Google Scholar] [CrossRef] [PubMed]
- Barderas, R.; Desmet, J.; Timmerman, P.; Meloen, R.; Casal, J.I. Affinity maturation of antibodies assisted by in silico modeling. Proc. Natl. Acad. Sci. USA 2008, 105, 9029–9034. [Google Scholar] [CrossRef] [PubMed]
- Barderas, R.; Shochat, S.; Timmerman, P.; Hollestelle, M.J.; Martinez-Torrecuadrada, J.L.; Hoppener, J.W.; Altschuh, D.; Meloen, R.; Casal, J.I. Designing antibodies for the inhibition of gastrin activity in tumoral cell lines. Int. J. Cancer 2008, 122, 2351–2359. [Google Scholar] [CrossRef]
- Yang, W.P.; Green, K.; Pinz-Sweeney, S.; Briones, A.T.; Burton, D.R.; Barbas, C.F., III. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol. 1995, 254, 392–403. [Google Scholar] [CrossRef]
- Lamdan, H.; Gavilondo, J.V.; Munoz, Y.; Pupo, A.; Huerta, V.; Musacchio, A.; Perez, L.; Ayala, M.; Rojas, G.; Balint, R.F.; et al. Affinity maturation and fine functional mapping of an antibody fragment against a novel neutralizing epitope on human vascular endothelial growth factor. Mol. Biosyst. 2013, 9, 2097–2106. [Google Scholar] [CrossRef]
- Colley, C.S.; Popovic, B.; Sridharan, S.; Debreczeni, J.E.; Hargeaves, D.; Fung, M.; An, L.L.; Edwards, B.; Arnold, J.; England, E.; et al. Structure and characterization of a high affinity C5a monoclonal antibody that blocks binding to C5aR1 and C5aR2 receptors. mAbs 2018, 10, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Laffly, E.; Pelat, T.; Cedrone, F.; Blesa, S.; Bedouelle, H.; Thullier, P. Improvement of an antibody neutralizing the anthrax toxin by simultaneous mutagenesis of its six hypervariable loops. J. Mol. Biol. 2008, 378, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Lord, D.M.; Bird, J.J.; Honey, D.M.; Best, A.; Park, A.; Wei, R.R.; Qiu, H. Structure-based engineering to restore high affinity binding of an isoform-selective anti-TGFbeta1 antibody. mAbs 2018, 10, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Fuh, G.; Wu, P.; Liang, W.C.; Ultsch, M.; Lee, C.V.; Moffat, B.; Wiesmann, C. Structure-function studies of two synthetic anti-vascular endothelial growth factor Fabs and comparison with the Avastin Fab. J. Biol. Chem. 2006, 281, 6625–6631. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.V.; Hymowitz, S.G.; Wallweber, H.J.; Gordon, N.C.; Billeci, K.L.; Tsai, S.P.; Compaan, D.M.; Yin, J.; Gong, Q.; Kelley, R.F.; et al. Synthetic anti-BR3 antibodies that mimic BAFF binding and target both human and murine B cells. Blood 2006, 108, 3103–3111. [Google Scholar] [CrossRef]
- Sanders, B.M.; Martin, L.S.; Nakagawa, P.A.; Hunter, D.A.; Miller, S.; Ullrich, S.J. Specific cross-reactivity of antibodies raised against two major stress proteins, stress 70 and chaperonin 60, in diverse species. Environ. Toxicol. Chem. 1994, 13, 1241–1249. [Google Scholar] [CrossRef]
- Hamdani, N.; van der Velden, J. Lack of specificity of antibodies directed against human beta-adrenergic receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 403–407. [Google Scholar] [CrossRef]
- Liang, W.C.; Wu, X.; Peale, F.V.; Lee, C.V.; Meng, Y.G.; Gutierrez, J.; Fu, L.; Malik, A.K.; Gerber, H.P.; Ferrara, N.; et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J. Biol. Chem. 2006, 281, 951–961. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Levy, R.; Arndt, J.W.; Forsyth, C.M.; Razai, A.; Lou, J.; Geren, I.; Stevens, R.C.; Marks, J.D. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat. Biotechnol. 2007, 25, 107–116. [Google Scholar] [CrossRef]
- Joachimiak, L.A.; Kortemme, T.; Stoddard, B.L.; Baker, D. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J. Mol. Biol. 2006, 361, 195–208. [Google Scholar] [CrossRef]
- Farady, C.J.; Sellers, B.D.; Jacobson, M.P.; Craik, C.S. Improving the species cross-reactivity of an antibody using computational design. Bioorg. Med. Chem. Lett. 2009, 19, 3744–3747. [Google Scholar] [CrossRef] [PubMed]
- Grossman, I.; Ilani, T.; Fleishman, S.J.; Fass, D. Overcoming a species-specificity barrier in development of an inhibitory antibody targeting a modulator of tumor stroma. Protein Eng. Des. Sel. 2016, 29, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.; Adams, J.; Kuglstatter, A.; Li, Z.; Harris, S.F.; Liu, Y.; Bohini, S.; Ma, H.; Klumpp, K.; Gao, J.; et al. Structure-Guided Combinatorial Engineering Facilitates Affinity and Specificity Optimization of Anti-CD81 Antibodies. J. Mol. Biol. 2018, 430, 2139–2152. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, O.; Bossus, M.; Graille, M.; Bilous, M.; Savatier, A.; Jolivet, M.; Menez, A.; Stura, E.; Ducancel, F. Fine tuning of the specificity of an anti-progesterone antibody by first and second sphere residue engineering. J. Biol. Chem. 2005, 280, 24880–24887. [Google Scholar] [CrossRef]
- Koenig, P.; Sanowar, S.; Lee, C.V.; Fuh, G. Tuning the specificity of a Two-in-One Fab against three angiogenic antigens by fully utilizing the information of deep mutational scanning. mAbs 2017, 9, 959–967. [Google Scholar] [CrossRef]
- James, L.C.; Roversi, P.; Tawfik, D.S. Antibody multispecificity mediated by conformational diversity. Science 2003, 299, 1362–1367. [Google Scholar] [CrossRef]
- James, L.C.; Tawfik, D.S. The specificity of cross-reactivity: Promiscuous antibody binding involves specific hydrogen bonds rather than nonspecific hydrophobic stickiness. Protein Sci. 2003, 12, 2183–2193. [Google Scholar] [CrossRef]
- Bostrom, J.; Lee, C.V.; Haber, L.; Fuh, G. Improving antibody binding affinity and specificity for therapeutic development. Methods Mol. Biol. 2009, 525, 353–376. [Google Scholar]
- Bostrom, J.; Yu, S.F.; Kan, D.; Appleton, B.A.; Lee, C.V.; Billeci, K.; Man, W.; Peale, F.; Ross, S.; Wiesmann, C.; et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 2009, 323, 1610–1614. [Google Scholar] [CrossRef]
- Schaefer, G.; Haber, L.; Crocker, L.M.; Shia, S.; Shao, L.; Dowbenko, D.; Totpal, K.; Wong, A.; Lee, C.V.; Stawicki, S.; et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011, 20, 472–486. [Google Scholar] [CrossRef]
- Jenkins, N.; Murphy, L.; Tyther, R. Post-translational modifications of recombinant proteins: Significance for biopharmaceuticals. Mol. Biotechnol. 2008, 39, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Jain, T.; Lynaugh, H.; Nobrega, R.P.; Lu, X.; Boland, T.; Burnina, I.; Sun, T.; Caffry, I.; Brown, M.; et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. mAbs 2017, 9, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nobrega, R.P.; Lynaugh, H.; Jain, T.; Barlow, K.; Boland, T.; Sivasubramanian, A.; Vasquez, M.; Xu, Y. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. mAbs 2019, 11, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Vlasak, J.; Ionescu, R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr. Pharm. Biotechnol. 2008, 9, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Tesar, D.B.; Mukhyala, K.; Theil, F.P.; Fielder, P.J.; Khawli, L.A. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug. Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, D.; Boswell, C.A.; Fielder, P.J.; Khawli, L.A. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J. 2012, 14, 554–558. [Google Scholar] [CrossRef]
- Yang, N.; Tang, Q.; Hu, P.; Lewis, M.J. Use of In Vitro Systems to Model In Vivo Degradation of Therapeutic Monoclonal Antibodies. Anal. Chem. 2018, 90, 7896–7902. [Google Scholar] [CrossRef]
- Jimenez del Val, I.; Nagy, J.M.; Kontoravdi, C. A dynamic mathematical model for monoclonal antibody N-linked glycosylation and nucleotide sugar donor transport within a maturing Golgi apparatus. Biotechnol. Prog. 2011, 27, 1730–1743. [Google Scholar] [CrossRef]
- Loebrich, S.; Clark, E.; Ladd, K.; Takahashi, S.; Brousseau, A.; Kitchener, S.; Herbst, R.; Ryll, T. Comprehensive manipulation of glycosylation profiles across development scales. mAbs 2019, 11, 335–349. [Google Scholar] [CrossRef]
- Wang, Q.; Chung, C.Y.; Chough, S.; Betenbaugh, M.J. Antibody glycoengineering strategies in mammalian cells. Biotechnol. Bioeng. 2018, 115, 1378–1393. [Google Scholar] [CrossRef]
- Liu, L.; Stadheim, A.; Hamuro, L.; Pittman, T.; Wang, W.; Zha, D.; Hochman, J.; Prueksaritanont, T. Pharmacokinetics of IgG1 monoclonal antibodies produced in humanized Pichia pastoris with specific glycoforms: A comparative study with CHO produced materials. Biologicals 2011, 39, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, S.; Yogo, R.; Inoue, R.; Sugiyama, M.; Itoh, S.G.; Okumura, H.; Miyanoiri, H.; Yagi, H.; Satoh, T.; Yamaguchi, T.; et al. Dynamic Views of the Fc Region of Immunoglobulin G Provided by Experimental and Computational Observations. Antibodies 2019, 8, 39. [Google Scholar] [CrossRef]
- Liu, Y.D.; van Enk, J.Z.; Flynn, G.C. Human antibody Fc deamidation in vivo. Biologicals 2009, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Pastuskovas, C.V.; Khawli, L.A.; Stults, J.T. Characterization of therapeutic monoclonal antibodies reveals differences between in vitro and in vivo time-course studies. Pharm. Res. 2013, 30, 167–178. [Google Scholar] [CrossRef]
- Chung, S.; Tian, J.; Tan, Z.; Chen, J.; Lee, J.; Borys, M.; Li, Z.J. Industrial bioprocessing perspectives on managing therapeutic protein charge variant profiles. Biotechnol. Bioeng. 2018, 115, 1646–1665. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Tian, J.; Tan, Z.; Chen, J.; Zhang, N.; Huang, Y.; Vandermark, E.; Lee, J.; Borys, M.; Li, Z.J. Modulating cell culture oxidative stress reduces protein glycation and acidic charge variant formation. mAbs 2019, 11, 205–2016. [Google Scholar] [CrossRef]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar]
- Aswad, D.W. Deamidation and Isoaspartate Formation in Peptides and Proteins; Aswad, D.W., Ed.; CRC Series in Analytical Biotechnology; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Phillips, J.J.; Buchanan, A.; Andrews, J.; Chodorge, M.; Sridharan, S.; Mitchell, L.; Burmeister, N.; Kippen, A.D.; Vaughan, T.J.; Higazi, D.R.; et al. Rate of Asparagine Deamidation in a Monoclonal Antibody Correlating with Hydrogen Exchange Rate at Adjacent Downstream Residues. Anal. Chem. 2017, 89, 2361–2368. [Google Scholar] [CrossRef]
- Ni, W.; Dai, S.; Karger, B.L.; Zhou, Z.S. Analysis of isoaspartic Acid by selective proteolysis with Asp-N and electron transfer dissociation mass spectrometry. Anal. Chem. 2010, 82, 7485–7491. [Google Scholar] [CrossRef]
- Alam, M.E.; Barnett, G.V.; Slaney, T.R.; Starr, C.G.; Das, T.K.; Tessier, P.M. Deamidation Can Compromise Antibody Colloidal Stability and Enhance Aggregation in a pH-Dependent Manner. Mol. Pharm. 2019, 16, 1939–1949. [Google Scholar] [CrossRef]
- Rehder, D.S.; Chelius, D.; McAuley, A.; Dillon, T.M.; Xiao, G.; Crouse-Zeineddini, J.; Vardanyan, L.; Perico, N.; Mukku, V.; Brems, D.N.; et al. Isomerization of a single aspartyl residue of anti-epidermal growth factor receptor immunoglobulin gamma2 antibody highlights the role avidity plays in antibody activity. Biochemistry 2008, 47, 2518–2530. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Wei, R.; Jaworski, J.; Boudanova, E.; Hughes, H.; VanPatten, S.; Lund, A.; Day, J.; Zhou, Y.; McSherry, T.; et al. Engineering an anti-CD52 antibody for enhanced deamidation stability. mAbs 2019, 11, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gaza-Bulseco, G.; Chumsae, C. Glutamine deamidation of a recombinant monoclonal antibody. Rapid Commun. Mass Spectrom. 2008, 22, 4081–4088. [Google Scholar] [CrossRef]
- Schechter, Y. Selective oxidation and reduction of methionine residues in peptides and proteins by oxygen exchange between sulfoxide and sulfide. J. Biol. Chem. 1986, 261, 66–70. [Google Scholar]
- Schechter, Y.; Burstein, Y.; Patchornik, A. Proceedings: Selective oxidation of methionine residues in proteins. Isr. J. Med. Sci. 1975, 11, 1171. [Google Scholar] [CrossRef]
- Brot, N.; Weissbach, H. Biochemistry and physiological role of methionine sulfoxide residues in proteins. Arch. Biochem. Biophys. 1983, 223, 271–281. [Google Scholar] [CrossRef]
- Pan, H.; Chen, K.; Chu, L.; Kinderman, F.; Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [Google Scholar] [CrossRef]
- Cymer, F.; Thomann, M.; Wegele, H.; Avenal, C.; Schlothauer, T.; Gygax, D.; Beck, H. Oxidation of M252 but not M428 in hu-IgG1 is responsible for decreased binding to and activation of hu-FcgammaRIIa (His131). Biologicals 2017, 50, 125–128. [Google Scholar] [CrossRef]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky, P.; Fang, Y.; McKelvey, T.; Li, Y.; Li, Y.; Drummond, J.; Prueksaritanont, T.; et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol. Immunol. 2009, 46, 1878–1882. [Google Scholar] [CrossRef]
- Wang, W.; Vlasak, J.; Li, Y.; Pristatsky, P.; Fang, Y.; Pittman, T.; Roman, J.; Wang, Y.; Prueksaritanont, T.; Ionescu, R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol. Immunol. 2011, 48, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Stracke, J.; Emrich, T.; Rueger, P.; Schlothauer, T.; Kling, L.; Knaupp, A.; Hertenberger, H.; Wolfert, A.; Spick, C.; Lau, W.; et al. A novel approach to investigate the effect of methionine oxidation on pharmacokinetic properties of therapeutic antibodies. mAbs 2014, 6, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Creed, D. The photophysics and photochemistry of the near-uv absorbing amino acids–i. Tryptophan and its simple derivatives. Photochem. Photobiol. 1984, 39, 537–562. [Google Scholar] [CrossRef]
- Li, Y.; Polozova, A.; Gruia, F.; Feng, J. Characterization of the degradation products of a color-changed monoclonal antibody: Tryptophan-derived chromophores. Anal. Chem. 2014, 86, 6850–6857. [Google Scholar] [CrossRef]
- Lam, X.M.; Lai, W.G.; Chan, E.K.; Ling, V.; Hsu, C.C. Site-specific tryptophan oxidation induced by autocatalytic reaction of polysorbate 20 in protein formulation. Pharm. Res. 2011, 28, 2543–2555. [Google Scholar] [CrossRef]
- Barnett, G.V.; Balakrishnan, G.; Chennamsetty, N.; Hoffman, L.; Bongers, J.; Tao, L.; Huang, Y.; Slaney, T.; Das, T.K.; Leone, A.; et al. Probing the Tryptophan Environment in Therapeutic Proteins: Implications for Higher Order Structure on Tryptophan Oxidation. J. Pharm. Sci. 2019, 108, 1944–1952. [Google Scholar] [CrossRef]
- Wei, Z.; Feng, J.; Lin, H.Y.; Mullapudi, S.; Bishop, E.; Tous, G.I.; Casas-Finet, J.; Hakki, F.; Strouse, R.; Schenerman, M.A. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal. Chem. 2007, 79, 2797–2805. [Google Scholar] [CrossRef]
- Pavon, J.A.; Xiao, L.; Li, X.; Zhao, J.; Aldredge, D.; Dank, E.; Fridman, A.; Liu, Y.H. Selective Tryptophan Oxidation of Monoclonal Antibodies: Oxidative Stress and Modeling Prediction. Anal. Chem. 2019, 91, 2192–2200. [Google Scholar] [CrossRef]
- Glover, Z.K.; Basa, L.; Moore, B.; Laurence, J.S.; Sreedhara, A. Metal ion interactions with mAbs: Part 1. mAbs 2015, 7, 901–911. [Google Scholar] [CrossRef]
- Zhu, F.; Glover, M.S.; Shi, H.; Trinidad, J.C.; Clemmer, D.E. Populations of metal-glycan structures influence MS fragmentation patterns. J. Am. Soc. Mass Spectrom. 2015, 26, 25–35. [Google Scholar] [CrossRef]
- Moritz, B.; Stracke, J.O. Assessment of disulfide and hinge modifications in monoclonal antibodies. Electrophoresis 2017, 38, 769–785. [Google Scholar] [CrossRef] [PubMed]
- McSherry, T.; McSherry, J.; Ozaeta, P.; Longenecker, K.; Ramsay, C.; Fishpaugh, J.; Allen, S. Cysteinylation of a monoclonal antibody leads to its inactivation. mAbs 2016, 8, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Wust, C.J. Interference with antibody neutralization by coenzyme and reducing agents. Ann. N. Y. Acad. Sci. 1963, 103, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Chen, X.; Enk, J.Z.; Plant, M.; Dillon, T.M.; Flynn, G.C. Human IgG2 antibody disulfide rearrangement in vivo. J. Biol. Chem. 2008, 283, 29266–29272. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Chen, Y.; Yi, L.; Brantley, T.; Stanley, B.; Sosic, Z.; Zang, L. Discovery and Characterization of Histidine Oxidation Initiated Cross-links in an IgG1 Monoclonal Antibody. Anal. Chem. 2017, 89, 7915–7923. [Google Scholar] [CrossRef]
- Amano, M.; Kobayashi, N.; Yabuta, M.; Uchiyama, S.; Fukui, K. Detection of histidine oxidation in a monoclonal immunoglobulin gamma (IgG) 1 antibody. Anal. Chem. 2014, 86, 7536–7543. [Google Scholar] [CrossRef]
- Bane, J.; Mozziconacci, O.; Yi, L.; Wang, Y.J.; Sreedhara, A.; Schoneich, C. Photo-oxidation of IgG1 and Model Peptides: Detection and Analysis of Triply Oxidized His and Trp Side Chain Cleavage Products. Pharm. Res. 2017, 34, 229–242. [Google Scholar] [CrossRef]
- Inglis, A.S. Cleavage at aspartic acid. Methods Enzymol. 1983, 91, 324–332. [Google Scholar]
- Oliyai, C.; Borchardt, R.T. Chemical pathways of peptide degradation. IV. Pathways, kinetics, and mechanism of degradation of an aspartyl residue in a model hexapeptide. Pharm. Res. 1993, 10, 95–102. [Google Scholar] [CrossRef]
- Kameoka, D.; Ueda, T.; Imoto, T. Effect of the conformational stability of the CH2 domain on the aggregation and peptide cleavage of a humanized IgG. Appl. Biochem. Biotechnol. 2011, 164, 642–654. [Google Scholar] [CrossRef]
- Dick, L.W., Jr.; Kim, C.; Qiu, D.; Cheng, K.C. Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodies using model peptides. Biotechnol. Bioeng. 2007, 97, 544–553. [Google Scholar] [CrossRef]
- Chelius, D.; Jing, K.; Lueras, A.; Rehder, D.S.; Dillon, T.M.; Vizel, A.; Rajan, R.S.; Li, T.; Treuheit, M.J.; Bondarenko, P.V. Formation of pyroglutamic acid from N-terminal glutamic acid in immunoglobulin gamma antibodies. Anal. Chem. 2006, 78, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Van den Bremer, E.T.; Beurskens, F.J.; Voorhorst, M.; Engelberts, P.J.; de Jong, R.N.; van der Boom, B.G.; Cook, E.M.; Lindorfer, M.A.; Taylor, R.P.; van Berkel, P.H.; et al. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. mAbs 2015, 7, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Yuk, I.H.; Zhang, B.; Yang, Y.; Dutina, G.; Leach, K.D.; Vijayasankaran, N.; Shen, A.Y.; Andersen, D.C.; Snedecor, B.R.; Joly, J.C. Controlling glycation of recombinant antibody in fed-batch cell cultures. Biotechnol. Bioeng. 2011, 108, 2600–2610. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009, 8, 226–234. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation of antibody therapeutics: Optimisation for purpose. Methods Mol. Biol. 2009, 483, 223–238. [Google Scholar]
- Jefferis, R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Prog. 2005, 21, 11–16. [Google Scholar] [CrossRef]
- Jefferis, R. The glycosylation of antibody molecules: Functional significance. Glycoconj. J. 1993, 10, 358–361. [Google Scholar]
- Shibata-Koyama, M.; Iida, S.; Okazaki, A.; Mori, K.; Kitajima-Miyama, K.; Saitou, S.; Kakita, S.; Kanda, Y.; Shitara, K.; Kato, K.; et al. The N-linked oligosaccharide at Fc gamma RIIIa Asn-45: An inhibitory element for high Fc gamma RIIIa binding affinity to IgG glycoforms lacking core fucosylation. Glycobiology 2009, 19, 126–134. [Google Scholar] [CrossRef]
- Zheng, K.; Yarmarkovich, M.; Bantog, C.; Bayer, R.; Patapoff, T.W. Influence of glycosylation pattern on the molecular properties of monoclonal antibodies. mAbs 2014, 6, 649–658. [Google Scholar] [CrossRef]
- Zhong, X.; Ma, W.; Meade, C.L.; Tam, A.S.; Llewellyn, E.; Cornell, R.; Cote, K.; Scarcelli, J.J.; Marshall, J.K.; Tzvetkova, B.; et al. Transient CHO expression platform for robust antibody production and its enhanced N-glycan sialylation on therapeutic glycoproteins. Biotechnol. Prog. 2019, 35, e2724. [Google Scholar] [CrossRef]
- Yamane-Ohnuki, N.; Satoh, M. Production of therapeutic antibodies with controlled fucosylation. mAbs 2009, 1, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Recombinant antibody therapeutics: The impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Huhn, C.; Selman, M.H.; Ruhaak, L.R.; Deelder, A.M.; Wuhrer, M. IgG glycosylation analysis. Proteomics 2009, 9, 882–913. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Flynn, G.C. Effect of high mannose glycan pairing on IgG antibody clearance. Biologicals 2016, 44, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Nichols, P.; Li, L.; Kumar, S.; Buck, P.M.; Singh, S.K.; Goswami, S.; Balthazor, B.; Conley, T.R.; Sek, D.; Allen, M.J. Rational design of viscosity reducing mutants of a monoclonal antibody: Hydrophobic versus electrostatic inter-molecular interactions. mAbs 2015, 7, 212–230. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Tessier, P.M. Engineering aggregation-resistant antibodies. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 263–286. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Ladiwala, A.R.; Bhattacharya, M.; Tessier, P.M. Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng. Des. Sel. 2012, 25, 591–601. [Google Scholar] [CrossRef]
- Perchiacca, J.M.; Ladiwala, A.R.; Bhattacharya, M.; Tessier, P.M. Structure-based design of conformation- and sequence-specific antibodies against amyloid beta. Proc. Natl. Acad. Sci. USA 2012, 109, 84–89. [Google Scholar] [CrossRef]
- Wu, S.J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef]
- Douillard, P.; Freissmuth, M.; Antoine, G.; Thiele, M.; Fleischander, D.; Matthiessen, P.; Voelkel, D.; Kerschbaumer, R.J.; Scheiflinger, F.; Sabarth, N. Optimization of an Antibody Light Chain Framework Enhances Expression, Biophysical Properties and Pharmacokinetics. Antibodies 2019, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Jetha, A.; Thorsteinson, N.; Jmeian, Y.; Jeganathan, A.; Giblin, P.; Fransson, J. Homology modeling and structure-based design improve hydrophobic interaction chromatography behavior of integrin binding antibodies. mAbs 2018, 10, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Sankar, K.; Krystek, S.R., Jr.; Carl, S.M.; Day, T.; Maier, J.K.X. AggScore: Prediction of aggregation-prone regions in proteins based on the distribution of surface patches. Proteins 2018, 86, 1147–1156. [Google Scholar] [CrossRef]
- Worn, A.; Pluckthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.F.T.; Young, T.M.; Roberts, C.J. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J. Pharm. Sci. 2009, 98, 1246–1277. [Google Scholar] [CrossRef] [PubMed]
- He, X. Thermostability of biological systems: Fundamentals, challenges, and quantification. Open Biomed. Eng. J. 2011, 5, 47–73. [Google Scholar] [CrossRef]
- He, F.; Woods, C.E.; Trilisky, E.; Bower, K.M.; Litowski, J.R.; Kerwin, B.A.; Becker, G.W.; Narhi, L.O.; Razinkov, V.I. Screening of monoclonal antibody formulations based on high-throughput thermostability and viscosity measurements: Design of experiment and statistical analysis. J. Pharm. Sci. 2011, 100, 1330–1340. [Google Scholar] [CrossRef]
- Thiagarajan, G.; Semple, A.; James, J.K.; Cheung, J.K.; Shameem, M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. mAbs 2016, 8, 1088–1097. [Google Scholar] [CrossRef]
- Schermeyer, M.T.; Woll, A.K.; Kokke, B.; Eppink, M.; Hubbuch, J. Characterization of highly concentrated antibody solution—A toolbox for the description of protein long-term solution stability. mAbs 2017, 9, 1169–1185. [Google Scholar] [CrossRef]
- Remmele, R.L.; Gombotz, W.R. Differential scanning calorimetry: A practical tool for elucidating stability of liquid biopharmaceuticals. Biopharm 2000, 13, 36–46. [Google Scholar]
- Maa, Y.F.; Hsu, C.C. Aggregation of recombinant human growth hormone induced by phenolic compounds. Int. J. Pharm. 1996, 140, 155–168. [Google Scholar] [CrossRef]
- Remmele, R.L.; Nightlinger, N.S.; Srinivasan, S.; Gombotz, W.R. Interleukin-1 receptor (IL-1R) liquid formulation development using differential scanning calorimetry. Pharm. Res. 1997, 15, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaisheva, E. Development of a multidose formulation for a humanized monoclonal antibody using experimental design techniques. AAPS PharmSci 2003, 5, E8. [Google Scholar] [CrossRef]
- Bedu-Addo, F.K.; Johnson, C.; Jeyarajah, S.; Henderson, I.; Advant, S.J. Use of biophysical characterization in preformulation development of a heavy-chain fragment of botulinum serotype B: Evaluation of suitable purification process conditions. Pharm. Res. 2004, 21, 1353–1361. [Google Scholar] [CrossRef]
- Henry, K.A.; Kim, D.Y.; Kandalaft, H.; Lowden, M.J.; Yang, Q.; Schrag, J.D.; Hussack, G.; MacKenzie, C.R.; Tanha, J. Stability-Diversity Tradeoffs Impose Fundamental Constraints on Selection of Synthetic Human VH/VL Single-Domain Antibodies from In Vitro Display Libraries. Front. Immunol. 2017, 8, 1759. [Google Scholar] [CrossRef]
- Ramaraj, T.; Angel, T.; Dratz, E.A.; Jesaitis, A.J.; Mumey, B. Antigen-antibody interface properties: Composition, residue interactions, and features of 53 non-redundant structures. Biochim. Biophys. Acta 2012, 1824, 520–532. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef]
- Abskharon, R.N.; Soror, S.H.; Pardon, E.; El Hassan, H.; Legname, G.; Steyaert, J.; Wohlkonig, A. Combining in-situ proteolysis and microseed matrix screening to promote crystallization of PrPc-nanobody complexes. Protein Eng. Des. Sel. 2011, 24, 737–741. [Google Scholar] [CrossRef]
- Domanska, K.; Vanderhaegen, S.; Srinivasan, V.; Pardon, E.; Dupeux, F.; Marquez, J.A.; Giorgetti, S.; Stoppini, M.; Wyns, L.; Bellotti, V.; et al. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic beta2-microglobulin variant. Proc. Natl. Acad. Sci. USA 2011, 108, 1314–1319. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta (2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Steyaert, J.; Kobilka, B.K. Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 2011, 21, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Pardon, E.; Wu, M.; Steyaert, J.; Hol, W.G. Crystal structure of a heterodimer of editosome interaction proteins in complex with two copies of a cross-reacting nanobody. Nucleic Acids Res. 2012, 40, 1828–1840. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh-Ghassabeh, G.; Devoogdt, N.; de Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Sheridan, C. Ablynx’s nanobody fragments go places antibodies cannot. Nat. Biotechnol. 2017, 35, 1115–1117. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, C.; Muyldermans, S. Nanobody-Based Delivery Systems for Diagnosis and Targeted Tumor Therapy. Front. Immunol. 2017, 8, 1442. [Google Scholar] [CrossRef]
- Arezumand, R.; Alibakhshi, A.; Ranjbari, J.; Ramazani, A.; Muyldermans, S. Nanobodies As Novel Agents for Targeting Angiogenesis in Solid Cancers. Front. Immunol. 2017, 8, 1746. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Hovinga, J.A.K.; Cataland, S.; Knobl, P.; Wu, H.; Artoni, A.; Westwood, J.P.; Taleghani, M.M.; Jilma, B.; et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef]
- Klarenbeek, A.; El Mazouari, K.; Desmyter, A.; Blanchetot, C.; Hultberg, A.; de Jonge, N.; Roovers, R.C.; Cambillau, C.; Spinelli, S.; Del-Favero, J.; et al. Camelid Ig V genes reveal significant human homology not seen in therapeutic target genes, providing for a powerful therapeutic antibody platform. mAbs 2015, 7, 693–706. [Google Scholar] [CrossRef]
- Konning, D.; Zielonka, S.; Grzeschik, J.; Empting, M.; Valldorf, B.; Krah, S.; Schroter, C.; Sellmann, C.; Hock, B.; Kolmar, H. Camelid and shark single domain antibodies: Structural features and therapeutic potential. Curr. Opin. Struct. Biol. 2017, 45, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, S.A.; Dooley, H.; Schoepp, R.J.; Flajnik, M.; Lonsdale, S.G. Isolation and characterisation of Ebolavirus-specific recombinant antibody fragments from murine and shark immune libraries. Mol. Immunol. 2011, 48, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Nuttall, S.; Revill, P.; Colledge, D.; Cabuang, L.; Soppe, S.; Dolezal, O.; Griffiths, K.; Bartholomeusz, A.; Locarnini, S. Targeting the hepatitis B virus precore antigen with a novel IgNAR single variable domain intrabody. Virology 2011, 411, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, S.D. Overview and discovery of IgNARs and generation of VNARs. Methods Mol. Biol. 2012, 911, 27–36. [Google Scholar]
- Kovalenko, O.V.; Olland, A.; Piche-Nicholas, N.; Godbole, A.; King, D.; Svenson, K.; Calabro, V.; Muller, M.R.; Barelle, C.J.; Somers, W.; et al. Atypical antigen recognition mode of a shark immunoglobulin new antigen receptor (IgNAR) variable domain characterized by humanization and structural analysis. J. Biol. Chem. 2013, 288, 17408–17419. [Google Scholar] [CrossRef]
- Kovaleva, M.; Ferguson, L.; Steven, J.; Porter, A.; Barelle, C. Shark variable new antigen receptor biologics—A novel technology platform for therapeutic drug development. Expert Opin. Biol. Ther. 2014, 14, 1527–1539. [Google Scholar] [CrossRef]
- Zielonka, S.; Empting, M.; Grzeschik, J.; Konning, D.; Barelle, C.J.; Kolmar, H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. mAbs 2015, 7, 15–25. [Google Scholar] [CrossRef]
- Grzeschik, J.; Yanakieva, D.; Roth, L.; Krah, S.; Hinz, S.C.; Elter, A.; Zollmann, T.; Schwall, G.; Zielonka, S.; Kolmar, H. Yeast Surface Display in Combination with Fluorescence-activated Cell Sorting Enables the Rapid Isolation of Antibody Fragments Derived from Immunized Chickens. Biotechnol. J. 2019, 14, e1800466. [Google Scholar] [CrossRef]
- Grzeschik, J.; Konning, D.; Hinz, S.C.; Krah, S.; Schroter, C.; Empting, M.; Kolmar, H.; Zielonka, S. Generation of Semi-Synthetic Shark IgNAR Single-Domain Antibody Libraries. Methods Mol. Biol. 2018, 1701, 147–167. [Google Scholar]
- Streltsov, V.A.; Varghese, J.N.; Carmichael, J.A.; Irving, R.A.; Hudson, P.J.; Nuttall, S.D. Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 12444–12449. [Google Scholar] [CrossRef]
- Griffiths, K.; Dolezal, O.; Cao, B.; Nilsson, S.K.; See, H.B.; Pfleger, K.D.; Roche, M.; Gorry, P.R.; Pow, A.; Viduka, K.; et al. I-bodies, Human Single Domain Antibodies That Antagonize Chemokine Receptor CXCR4. J. Biol. Chem. 2016, 291, 12641–12657. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Bian, H.; Wu, X.; Fu, T.; Fu, Y.; Hong, J.; Fleming, B.D.; Flajnik, M.F.; Ho, M. Construction and next-generation sequencing analysis of a large phage-displayed VNAR single-domain antibody library from six naive nurse sharks. Antib. Ther. 2019, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science, Taylor and Science Group: New York, NY, USA, 2017. [Google Scholar]
- Kitov, P.I.; Bundle, D.R. On the nature of the multivalency effect: A thermodynamic model. J. Am. Chem. Soc. 2003, 125, 16271–16284. [Google Scholar] [CrossRef]
- Vorup-Jensen, T. On the roles of polyvalent binding in immune recognition: Perspectives in the nanoscience of immunology and the immune response to nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1759–1781. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.S.; Gnanapragasam, P.N.; Galimidi, R.P.; Foglesong, C.P.; West, A.P., Jr.; Bjorkman, P.J. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc. Natl. Acad. Sci. USA 2009, 106, 7385–7390. [Google Scholar] [CrossRef]
- Vauquelin, G.; Charlton, S.J. Exploring avidity: Understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol. 2013, 168, 1771–1785. [Google Scholar] [CrossRef]
- Nesspor, T.C.; Raju, T.S.; Chin, C.N.; Vafa, O.; Brezski, R.J. Avidity confers FcgammaR binding and immune effector function to aglycosylated immunoglobulin G1. J. Mol. Recognit. 2012, 25, 147–154. [Google Scholar] [CrossRef]
- Loyau, J.; Malinge, P.; Daubeuf, B.; Shang, L.; Elson, G.; Kosco-Vilbois, M.; Fischer, N.; Rousseau, F. Maximizing the potency of an anti-TLR4 monoclonal antibody by exploiting proximity to Fcgamma receptors. mAbs 2014, 6, 1621–1630. [Google Scholar] [CrossRef][Green Version]
- Read, T.; Olkhov, R.V.; Williamson, E.D.; Shaw, A.M. Label-free Fab and Fc affinity/avidity profiling of the antibody complex half-life for polyclonal and monoclonal efficacy screening. Anal. Bioanal. Chem. 2015, 407, 7349–7357. [Google Scholar] [CrossRef]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Jain, A.; Olsen, H.S.; Vyzasatya, R.; Burch, E.; Sakoda, Y.; Merigeon, E.Y.; Cai, L.; Lu, C.; Tan, M.; Tamada, K.; et al. Fully recombinant IgG2a Fc multimers (stradomers) effectively treat collagen-induced arthritis and prevent idiopathic thrombocytopenic purpura in mice. Arthritis Res. Ther. 2012, 14, R192. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.F.; Lansing, J.C.; Rutitzky, L.; Kurtagic, E.; Prod’homme, T.; Choudhury, A.; Washburn, N.; Bhatnagar, N.; Beneduce, C.; Holte, K.; et al. Elucidating the interplay between IgG-Fc valency and FcgammaR activation for the design of immune complex inhibitors. Sci. Transl. Med. 2016, 8, 365ra158. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Rowley, T.F.; Junker, F.; Peters, S.J.; Crilly, S.; Compson, J.; Eddleston, A.; Bjorkelund, H.; Greenslade, K.; Parkinson, M.; et al. Multivalent Fcgamma-receptor engagement by a hexameric Fc-fusion protein triggers Fcgamma-receptor internalisation and modulation of Fcgamma-receptor functions. Sci. Rep. 2017, 7, 17049. [Google Scholar] [CrossRef] [PubMed]
- Spirig, R.; Campbell, I.K.; Koernig, S.; Chen, C.G.; Lewis, B.J.B.; Butcher, R.; Muir, I.; Taylor, S.; Chia, J.; Leong, D.; et al. rIgG1 Fc Hexamer Inhibits Antibody-Mediated Autoimmune Disease via Effects on Complement and FcgammaRs. J. Immunol. 2018, 200, 2542–2553. [Google Scholar] [CrossRef] [PubMed]
- Warncke, M.; Calzascia, T.; Coulot, M.; Balke, N.; Touil, R.; Kolbinger, F.; Heusser, C. Different adaptations of IgG effector function in human and nonhuman primates and implications for therapeutic antibody treatment. J. Immunol. 2012, 188, 4405–4411. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Schaer, D.A.; Hirschhorn-Cymerman, D.; Wolchok, J.D. Targeting tumor-necrosis factor receptor pathways for tumor immunotherapy. J. Immunother. Cancer 2014, 2, 7. [Google Scholar] [CrossRef]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Pollok, K.E.; Kim, Y.J.; Zhou, Z.; Hurtado, J.; Kim, K.K.; Pickard, R.T.; Kwon, B.S. Inducible T cell antigen 4-1BB. Analysis of expression and function. J. Immunol. 1993, 150, 771–781. [Google Scholar] [PubMed]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar] [PubMed]
- Kanamaru, F.; Youngnak, P.; Hashiguchi, M.; Nishioka, T.; Takahashi, T.; Sakaguchi, S.; Ishikawa, I.; Azuma, M. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J. Immunol. 2004, 172, 7306–7314. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, V.; Sundarapandiyan, K.; Zhao, B.; Bylesjo, M.; Marsh, H.C.; Keler, T. Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J. Immunother. Cancer 2015, 3, 37. [Google Scholar] [CrossRef]
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011, 19, 101–113. [Google Scholar] [CrossRef]
- He, L.Z.; Prostak, N.; Thomas, L.J.; Vitale, L.; Weidlick, J.; Crocker, A.; Pilsmaker, C.D.; Round, S.M.; Tutt, A.; Glennie, M.J.; et al. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J. Immunol. 2013, 191, 4174–4183. [Google Scholar] [CrossRef]
- Mangsbo, S.M.; Broos, S.; Fletcher, E.; Veitonmaki, N.; Furebring, C.; Dahlen, E.; Norlen, P.; Lindstedt, M.; Totterman, T.H.; Ellmark, P. The human agonistic CD40 antibody ADC-1013 eradicates bladder tumors and generates T-cell-dependent tumor immunity. Clin. Cancer Res. 2015, 21, 1115–1126. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910. [Google Scholar] [CrossRef]
- Morris, N.P.; Peters, C.; Montler, R.; Hu, H.M.; Curti, B.D.; Urba, W.J.; Weinberg, A.D. Development and characterization of recombinant human Fc:OX40L fusion protein linked via a coiled-coil trimerization domain. Mol. Immunol. 2007, 44, 3112–3121. [Google Scholar] [CrossRef]
- Stavenhagen, J.B.; Gorlatov, S.; Tuaillon, N.; Rankin, C.T.; Li, H.; Burke, S.; Huang, L.; Johnson, S.; Koenig, S.; Bonvini, E. Enhancing the potency of therapeutic monoclonal antibodies via Fc optimization. Adv. Enzym. Regul. 2008, 48, 152–164. [Google Scholar] [CrossRef]
- Kim, J.M.; Ashkenazi, A. Fcgamma receptors enable anticancer action of proapoptotic and immune-modulatory antibodies. J. Exp. Med. 2013, 210, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Furness, A.J.; Vargas, F.A.; Peggs, K.S.; Quezada, S.A. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014, 35, 290–298. [Google Scholar] [CrossRef] [PubMed]
- White, A.L.; Chan, H.T.; Roghanian, A.; French, R.R.; Mockridge, C.I.; Tutt, A.L.; Dixon, S.V.; Ajona, D.; Verbeek, J.S.; Al-Shamkhani, A.; et al. Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J. Immunol. 2011, 187, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ravetch, J.V. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proc. Natl. Acad. Sci. USA 2012, 109, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Szalai, A.J.; Zhou, T.; Zinn, K.R.; Chaudhuri, T.R.; Li, X.; Koopman, W.J.; Kimberly, R.P. Fc gamma Rs modulate cytotoxicity of anti-Fas antibodies: Implications for agonistic antibody-based therapeutics. J. Immunol. 2003, 171, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Jolicoeur, R.; Zhang, J.; Dranoff, G.; Wilson, N.S.; Brogdon, J.L. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol. Cell Biol. 2014, 92, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcgamma receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef]
- Chu, S.Y.; Vostiar, I.; Karki, S.; Moore, G.L.; Lazar, G.A.; Pong, E.; Joyce, P.F.; Szymkowski, D.E.; Desjarlais, J.R. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol. Immunol. 2008, 45, 3926–3933. [Google Scholar] [CrossRef]
- Mimoto, F.; Katada, H.; Kadono, S.; Igawa, T.; Kuramochi, T.; Muraoka, M.; Wada, Y.; Haraya, K.; Miyazaki, T.; Hattori, K. Engineered antibody Fc variant with selectively enhanced FcgammaRIIb binding over both FcgammaRIIa(R131) and FcgammaRIIa(H131). Protein Eng. Des. Sel. 2013, 26, 589–598. [Google Scholar] [CrossRef]
- Mimoto, F.; Igawa, T.; Kuramochi, T.; Katada, H.; Kadono, S.; Kamikawa, T.; Shida-Kawazoe, M.; Hattori, K. Novel asymmetrically engineered antibody Fc variant with superior FcgammaR binding affinity and specificity compared with afucosylated Fc variant. mAbs 2013, 5, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Whitaker, B.; Derebe, M.G.; Chiu, M.L. FcgammaRII-binding Centyrins mediate agonism and antibody-dependent cellular phagocytosis when fused to an anti-OX40 antibody. mAbs 2018, 10, 463–475. [Google Scholar] [CrossRef] [PubMed]
- White, A.L.; Chan, H.T.; French, R.R.; Willoughby, J.; Mockridge, C.I.; Roghanian, A.; Penfold, C.A.; Booth, S.G.; Dodhy, A.; Polak, M.E.; et al. Conformation of the human immunoglobulin g2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 2015, 27, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- De Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Goldberg, M.V.; Chiu, M.L. Fc Engineering Approaches to Enhance the Agonism and Effector Functions of an Anti-OX40 Antibody. J. Biol. Chem. 2016, 291, 27134–27146. [Google Scholar] [CrossRef]
- Zhang, D.; Armstrong, A.A.; Tam, S.H.; McCarthy, S.G.; Luo, J.; Gilliland, G.L.; Chiu, M.L. Functional optimization of agonistic antibodies to OX40 receptor with novel Fc mutations to promote antibody multimerization. mAbs 2017, 9, 1129–1142. [Google Scholar] [CrossRef]
- Rowley, T.F.; Peters, S.J.; Aylott, M.; Griffin, R.; Davies, N.L.; Healy, L.J.; Cutler, R.M.; Eddleston, A.; Pither, T.L.; Sopp, J.M.; et al. Engineered hexavalent Fc proteins with enhanced Fc-gamma receptor avidity provide insights into immune-complex interactions. Commun. Biol. 2018, 1, 146. [Google Scholar] [CrossRef]
- Piao, X.; Ozawa, T.; Hamana, H.; Shitaoka, K.; Jin, A.; Kishi, H.; Muraguchi, A. TRAIL-receptor 1 IgM antibodies strongly induce apoptosis in human cancer cells in vitro and in vivo. Oncoimmunology 2016, 5, e1131380. [Google Scholar] [CrossRef]
- Dubuisson, A.; Micheau, O. Antibodies and Derivatives Targeting DR4 and DR5 for Cancer Therapy. Antibodies 2017, 6, 16. [Google Scholar] [CrossRef]
- Roux, K.H.; Strelets, L.; Brekke, O.H.; Sandlie, I.; Michaelsen, T.E. Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes: A role for flexibility and geometry. J. Immunol. 1998, 161, 4083–4090. [Google Scholar] [PubMed]
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar] [PubMed]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Egan, P.C.; Reagan, J.L. The return of gemtuzumab ozogamicin: A humanized anti-CD33 monoclonal antibody-drug conjugate for the treatment of newly diagnosed acute myeloid leukemia. Onco Targets Ther. 2018, 11, 8265–8272. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Goad, M.E.; Dougher, M.M.; Boghaert, E.R.; Kunz, A.; Hamann, P.R.; Damle, N.K. Potent and specific antitumor efficacy of CMC-544, a CD22-targeted immunoconjugate of calicheamicin, against systemically disseminated B-cell lymphoma. Clin. Cancer Res. 2004, 10, 8620–8629. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Scott, L.J. Brentuximab Vedotin: A Review in CD30-Positive Hodgkin Lymphoma. Drugs 2017, 77, 435–445. [Google Scholar] [CrossRef]
- Chari, R.V.; Martell, B.A.; Gross, J.L.; Cook, S.B.; Shah, S.A.; Blattler, W.A.; McKenzie, S.J.; Goldmacher, V.S. Immunoconjugates containing novel maytansinoids: Promising anticancer drugs. Cancer Res. 1992, 52, 127–131. [Google Scholar] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Arrojo, R.; Liu, D.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. Bioconjug. Chem. 2015, 26, 919–931. [Google Scholar] [CrossRef]
- Mansfield, E.; Chiron, M.F.; Amlot, P.; Pastan, I.; FitzGerald, D.J. Recombinant RFB4 single-chain immunotoxin that is cytotoxic towards CD22-positive cells. Biochem. Soc. Trans. 1997, 25, 709–714. [Google Scholar] [CrossRef]
- Mansfield, E.; Amlot, P.; Pastan, I.; FitzGerald, D.J. Recombinant RFB4 immunotoxins exhibit potent cytotoxic activity for CD22-bearing cells and tumors. Blood 1997, 90, 2020–2026. [Google Scholar] [CrossRef]
- Bang, S.; Nagata, S.; Onda, M.; Kreitman, R.J.; Pastan, I. HA22 (R490A) is a recombinant immunotoxin with increased antitumor activity without an increase in animal toxicity. Clin. Cancer Res. 2005, 11, 1545–1550. [Google Scholar] [CrossRef]
- Knox, S.J.; Goris, M.L.; Trisler, K.; Negrin, R.; Davis, T.; Liles, T.M.; Grillo-Lopez, A.; Chinn, P.; Varns, C.; Ning, S.C.; et al. Yttrium-90-labeled anti-CD20 monoclonal antibody therapy of recurrent B-cell lymphoma. Clin. Cancer Res. 1996, 2, 457–470. [Google Scholar]
- Kaminski, M.S.; Zasadny, K.R.; Francis, I.R.; Milik, A.W.; Ross, C.W.; Moon, S.D.; Crawford, S.M.; Burgess, J.M.; Petry, N.A.; Butchko, G.M.; et al. Radioimmunotherapy of B-cell lymphoma with [131I]anti-B1(anti-CD20) antibody. N. Engl. J. Med. 1993, 329, 459–465. [Google Scholar] [CrossRef]
- Prasad, V. The withdrawal of drugs for commercial reasons: The incomplete story of tositumomab. JAMA Intern. Med. 2014, 174, 1887–1888. [Google Scholar] [CrossRef]
- Teeling, J.L.; Mackus, W.J.; Wiegman, L.J.; van den Brakel, J.H.; Beers, S.A.; French, R.R.; van Meerten, T.; Ebeling, S.; Vink, T.; Slootstra, J.W.; et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J. Immunol. 2006, 177, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Leusen, J.H. Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar] [PubMed]
- Klein, C.; Lammens, A.; Schafer, W.; Georges, G.; Schwaiger, M.; Mossner, E.; Hopfner, K.P.; Umana, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs 2013, 5, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Kapelski, S.; Cleiren, E.; Attar, R.M.; Philippar, U.; Hasler, J.; Chiu, M.L. Influence of the bispecific antibody IgG subclass on T cell redirection. mAbs 2019, 11, 1012–1024. [Google Scholar] [CrossRef]
- Konitzer, J.D.; Sieron, A.; Wacker, A.; Enenkel, B. Reformatting Rituximab into Human IgG2 and IgG4 Isotypes Dramatically Improves Apoptosis Induction In Vitro. PLoS ONE 2015, 10, e0145633. [Google Scholar] [CrossRef]
- Shields, R.L.; Namenuk, A.K.; Hong, K.; Meng, Y.G.; Rae, J.; Briggs, J.; Xie, D.; Lai, J.; Stadlen, A.; Li, B.; et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2001, 276, 6591–6604. [Google Scholar] [CrossRef]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef]
- Stavenhagen, J.B.; Gorlatov, S.; Tuaillon, N.; Rankin, C.T.; Li, H.; Burke, S.; Huang, L.; Vijh, S.; Johnson, S.; Bonvini, E.; et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 2007, 67, 8882–8890. [Google Scholar] [CrossRef]
- Reddy, M.P.; Kinney, C.A.; Chaikin, M.A.; Payne, A.; Fishman-Lobell, J.; Tsui, P.; Monte, P.R.D.; Doyle, M.L.; Brigham-Burke, M.R.; Anderson, D.; et al. Elimination of Fc receptor-dependent effector functions of a modified IgG4 monoclonal antibody to human CD4. J. Immunol. 2000, 164, 1925–1933. [Google Scholar] [CrossRef]
- Strohl, W.R. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009, 20, 685–691. [Google Scholar] [CrossRef]
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Yogo, R.; Yamaguchi, Y.; Watanabe, H.; Yagi, H.; Satoh, T.; Nakanishi, M.; Onitsuka, M.; Omasa, T.; Shimada, M.; Maruno, T.; et al. The Fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Kilar, F.; Zavodszky, P. Non-covalent interactions between Fab and Fc regions.in immunoglobulin G molecules. Eur. J. Biochem. 1987, 162, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Nussinov, R.; Ma, B. Antigen binding allosterically promotes Fc receptor recognition. mAbs 2019, 11, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.O.; Karki, S.; Lazar, G.A.; Chen, H.; Dang, W.; Desjarlais, J.R. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 2008, 7, 2517–2527. [Google Scholar] [CrossRef]
- Bolt, S.; Routledge, E.; Lloyd, I.; Chatenoud, L.; Pope, H.; Gorman, S.D.; Clark, M.; Waldmann, H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur. J. Immunol. 1993, 23, 403–411. [Google Scholar] [CrossRef]
- Alegre, M.L.; Peterson, L.J.; Xu, D.; Sattar, H.A.; Jeyarajah, D.R.; Kowalkowski, K.; Thistlethwaite, J.R.; Zivin, R.A.; Jolliffe, L.; Bluestone, J.A. A non-activating “humanized” anti-CD3 monoclonal antibody retains immunosuppressive properties in vivo. Transplantation 1994, 57, 1537–1543. [Google Scholar] [CrossRef]
- Cole, M.S.; Stellrecht, K.E.; Shi, J.D.; Homola, M.; Hsu, D.H.; Anasetti, C.; Vasquez, M.; Tso, J.Y. HuM291, a humanized anti-CD3 antibody, is immunosuppressive to T cells while exhibiting reduced mitogenicity in vitro. Transplantation 1999, 68, 563–571. [Google Scholar] [CrossRef]
- Xu, D.; Alegre, M.L.; Varga, S.S.; Rothermel, A.L.; Collins, A.M.; Pulito, V.L.; Hanna, L.S.; Dolan, K.P.; Parren, P.W.; Bluestone, J.A.; et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell. Immunol. 2000, 200, 16–26. [Google Scholar] [CrossRef]
- Tam, S.H.; McCarthy, S.G.; Armstrong, A.A.; Somani, S.; Wu, S.J.; Liu, X.S.; Gervais, A.; Ernst, R.; Saro, D.; Decker, R.; et al. Functional, Biophysical, and Structural Characterization of Human IgG1 and IgG4 Fc Variants with Ablated Immune Functionality. Antibodies 2017, 6, 12. [Google Scholar] [CrossRef]
- Lee, C.H.; Romain, G.; Yan, W.; Watanabe, M.; Charab, W.; Todorova, B.; Lee, J.; Triplett, K.; Donkor, M.; Lungu, O.I.; et al. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat. Immunol. 2017, 18, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Zacharias, M. Atomic resolution model of the antibody Fc interaction with the complement C1q component. Mol. Immunol. 2012, 51, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Idusogie, E.E.; Wong, P.Y.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Ultsch, M.; Mulkerrin, M.G. Engineered antibodies with increased activity to recruit complement. J. Immunol. 2001, 166, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. mAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Kobayashi, Y.; Takahashi, K.; Takahashi, E.; Sakae, S.; Wakitani, M.; Yamano, K.; Suzawa, T.; Yano, K.; Ohta, T.; et al. Fucose content of monoclonal antibodies can be controlled by culture medium osmolality for high antibody-dependent cellular cytotoxicity. Cytotechnology 2012, 64, 249–265. [Google Scholar] [CrossRef]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef]
- Olivier, S.; Jacoby, M.; Brillon, C.; Bouletreau, S.; Mollet, T.; Nerriere, O.; Angel, A.; Danet, S.; Souttou, B.; Guehenneux, F.; et al. EB66 cell line, a duck embryonic stem cell-derived substrate for the industrial production of therapeutic monoclonal antibodies with enhanced ADCC activity. mAbs 2010, 2, 405–415. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Mori, K.; Kuni-Kamochi, R.; Yamane-Ohnuki, N.; Wakitani, M.; Yamano, K.; Imai, H.; Kanda, Y.; Niwa, R.; Iida, S.; Uchida, K.; et al. Engineering Chinese hamster ovary cells to maximize effector function of produced antibodies using FUT8 siRNA. Biotechnol. Bioeng. 2004, 88, 901–908. [Google Scholar] [CrossRef]
- Ferrara, C.; Stuart, F.; Sondermann, P.; Brunker, P.; Umana, P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 2006, 281, 5032–5036. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Brunker, P.; Suter, T.; Moser, S.; Puntener, U.; Umana, P. Modulation of therapeutic antibody effector functions by glycosylation engineering: Influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol. Bioeng. 2006, 93, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Shankara, S.; Roy, A.; Qiu, H.W.; Estes, S.; McVie-Wylie, A.; Culm-Merdek, K.; Park, A.; Pan, C. Edmunds, Development of a simple and rapid method for producing non-fucosylated oligomannose containing antibodies with increased effector function. Biotechnol. Bioeng. 2007, 99, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J. Pharm. Sci. 2015, 104, 1866–1884. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Pollreisz, A.; Assinger, A.; Hacker, S.; Hoetzenecker, K.; Schmid, W.; Lang, G.; Wolfsberger, M.; Steinlechner, B.; Bielek, E.; Lalla, E.; et al. Intravenous immunoglobulins induce CD32-mediated platelet aggregation in vitro. Br. J. Dermatol. 2008, 159, 578–584. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Aalberse, R.C.; Schuurman, J. When binding is enough: Nonactivating antibody formats. Curr. Opin. Immunol. 2008, 20, 479–485. [Google Scholar] [CrossRef]
- Vafa, O.; Gilliland, G.L.; Brezski, R.J.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W.R. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef]
- Kinder, M.; Greenplate, A.R.; Strohl, W.R.; Jordan, R.E.; Brezski, R.J. An Fc engineering approach that modulates antibody-dependent cytokine release without altering cell-killing functions. mAbs 2015, 7, 494–504. [Google Scholar] [CrossRef]
- Jones, E.A.; Waldmann, T.A. The mechanism of intestinal uptake and transcellular transport of IgG in the neonatal rat. J. Clin. Investig. 1972, 51, 2916–2927. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Jones, E.A. The role of cell-surface receptors in the transport and catabolism of immunoglobulins. Ciba Found. Symp. 1972, 9, 5–23. [Google Scholar] [PubMed]
- Leach, J.L.; Sedmak, D.D.; Osborne, J.M.; Rahill, B.; Lairmore, M.D.; Anderson, C.L. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: Implications for maternal-fetal antibody transport. J. Immunol. 1996, 157, 3317–3322. [Google Scholar] [PubMed]
- Brambell, F.W.; Hemmings, W.A.; Morris, I.G. A Theoretical Model of Gamma-Globulin Catabolism. Nature 1964, 203, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Junghans, R.P.; Anderson, C.L. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 5512–5516. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, W.P.; Huber, A.H.; Bjorkman, P.J. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994, 372, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, W.P.; Gastinel, L.N.; Simister, N.E.; Blum, M.L.; Bjorkman, P.J. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature 1994, 372, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Tsen, M.F.; Ghetie, V.; Ward, E.S. Identifying amino acid residues that influence plasma clearance of murine IgG1 fragments by site-directed mutagenesis. Eur. J. Immunol. 1994, 24, 542–548. [Google Scholar] [CrossRef]
- Huber, A.H.; RKelley, F.; Gastinel, L.N.; Bjorkman, P.J. Crystallization and stoichiometry of binding of a complex between a rat intestinal Fc receptor and Fc. J. Mol. Biol. 1993, 230, 1077–1083. [Google Scholar] [CrossRef]
- Ghetie, V.; Popov, S.; Borvak, J.; Radu, C.; Matesoi, D.; Medesan, C.; Ober, R.J.; Ward, E.S. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat. Biotechnol. 1997, 15, 637–640. [Google Scholar] [CrossRef]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar]
- Dall’Acqua, W.F.; Kiener, P.A.; Wu, H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 2006, 281, 23514–23524. [Google Scholar] [CrossRef] [PubMed]
- Robbie, G.J.; Criste, R.; Dall’acqua, W.F.; Jensen, K.; Patel, N.K.; Losonsky, G.A.; Griffin, M.P. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob. Agents Chemother. 2013, 57, 6147–6153. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, Y.J.; Lewis, G.K.; Montefiori, D.C.; LaBranche, C.C.; Lewis, M.G.; Urban, L.A.; Lees, J.P.; Mao, L.; Jiang, X. Introduction of the YTE mutation into the non-immunogenic HIV bnAb PGT121 induces anti-drug antibodies in macaques. PLoS ONE 2019, 14, e0212649. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.; Nishimura, Y.; Gaughan, N.; Gazumyan, A.; Schoofs, T.; Buckler-White, A.; Seaman, M.S.; Swihart, B.J.; Follmann, D.A.; Nussenzweig, M.C.; et al. A single injection of crystallizable fragment domain-modified antibodies elicits durable protection from SHIV infection. Nat. Med. 2018, 24, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Datta-Mannan, A.; Witcher, D.R.; Tang, Y.; Watkins, J.; Jiang, W.; Wroblewski, V.J. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: Relationship to pharmacokinetics in mice and primates. Drug Metab. Dispos. 2007, 35, 86–94. [Google Scholar] [CrossRef]
- Borrok, M.J.; Wu, Y.; Beyaz, N.; Yu, X.Q.; Oganesyan, V.; Dall’Acqua, W.F.; Tsui, P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J. Biol. Chem. 2015, 290, 4282–4290. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Sun, V.Z. Clinical ramifications of the MHC family Fc receptor FcRn. J. Clin. Immunol. 2010, 30, 790–797. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Christianson, G.J.; Sproule, T.J. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol. Biol. 2010, 602, 93–104. [Google Scholar]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Rath, T.; Kuo, T.T.; Baker, K.; Qiao, S.W.; Kobayashi, K.; Yoshida, M.; Roopenian, D.; Fiebiger, E.; Lencer, W.I.; Blumberg, R.S. The immunologic functions of the neonatal Fc receptor for IgG. J. Clin. Immunol. 2013, 33 (Suppl. 1), S9–S17. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lu, P.; Fang, Y.; Hamuro, L.; Pittman, T.; Carr, B.; Hochman, J.; Prueksaritanont, T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab. Dispos. 2011, 39, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Firan, M.; Radu, C.G.; Kim, C.H.; Ghetie, V.; Ward, E.S. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur. J. Immunol. 1999, 29, 2819–2825. [Google Scholar] [CrossRef]
- Vaccaro, C.; Zhou, J.; Ober, R.J.; Ward, E.S. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat. Biotechnol. 2005, 23, 1283–1288. [Google Scholar] [CrossRef]
- Hinton, P.R.; Xiong, J.M.; Johlfs, M.G.; Tang, M.T.; Keller, S.; Tsurushita, N. An engineered human IgG1 antibody with longer serum half-life. J. Immunol. 2006, 176, 346–356. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Chow, C.K.; Dickinson, C.; Driver, D.; Lu, J.; Witcher, D.R.; Wroblewski, V.J. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab. Dispos. 2012, 40, 1545–1555. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Witcher, D.R.; Lu, J.; Wroblewski, V.J. Influence of improved FcRn binding on the subcutaneous bioavailability of monoclonal antibodies in cynomolgus monkeys. mAbs 2012, 4, 267–273. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Wu, X.; Reyes, A.E.; Vernes, J.M.; Lien, S.; Lowe, J.; Maia, M.; Forrest, W.F.; Meng, Y.G.; Damico, L.A.; et al. A therapeutic anti-VEGF antibody with increased potency independent of pharmacokinetic half-life. Cancer Res. 2010, 70, 3269–3277. [Google Scholar] [CrossRef]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef]
- Igawa, T.; Tsunoda, H.; Tachibana, T.; Maeda, A.; Mimoto, F.; Moriyama, C.; Nanami, M.; Sekimori, Y.; Nabuchi, Y.; Aso, Y.; et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng. Des. Sel. 2010, 23, 385–392. [Google Scholar] [CrossRef]
- Li, B.; Tesar, D.; Boswell, C.A.; Cahaya, H.S.; Wong, A.; Zhang, J.; Meng, Y.G.; Eigenbrot, C.; Pantua, H.; Diao, J.; et al. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 2014, 6, 1255–1264. [Google Scholar] [CrossRef]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef]
- Igawa, T.; Tsunoda, H.; Kuramochi, T.; Sampei, Z.; Ishii, S.; Hattori, K. Engineering the variable region of therapeutic IgG antibodies. mAbs 2011, 3, 243–252. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Thangaraju, A.; Leung, D.; Tang, Y.; Witcher, D.R.; Lu, J.; Wroblewski, V.J. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015, 7, 483–493. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Lu, J.; Witcher, D.R.; Leung, D.; Tang, Y.; Wroblewski, V.J. The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs 2015, 7, 1084–1093. [Google Scholar] [CrossRef]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef]
- Suresh, T.; Lee, L.X.; Joshi, J.; Barta, S.K. New antibody approaches to lymphoma therapy. J. Hematol. Oncol. 2014, 7, 58. [Google Scholar] [CrossRef]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef]
- Vauquelin, G.; Bricca, G.; van Liefde, I. Avidity and positive allosteric modulation/cooperativity act hand in hand to increase the residence time of bivalent receptor ligands. Fundam. Clin. Pharmacol. 2014, 28, 530–543. [Google Scholar] [CrossRef]
- Ruf, P.; Lindhofer, H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001, 98, 2526–2534. [Google Scholar] [CrossRef]
- Fossati, M.; Buzzonetti, A.; Monego, G.; Catzola, V.; Scambia, G.; Fattorossi, A.; Battaglia, A. Immunological changes in the ascites of cancer patients after intraperitoneal administration of the bispecific antibody catumaxomab (anti-EpCAMxanti-CD3). Gynecol. Oncol. 2015, 138, 343–351. [Google Scholar] [CrossRef]
- Ott, M.G.; Marme, F.; Moldenhauer, G.; Lindhofer, H.; Hennig, M.; Spannagl, R.; Essing, M.M.; Linke, R.; Seimetz, D. Humoral response to catumaxomab correlates with clinical outcome: Results of the pivotal phase II/III study in patients with malignant ascites. Int. J. Cancer 2012, 130, 2195–2203. [Google Scholar] [CrossRef]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results from a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Bargou, R. Blinatumomab: A CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma 2016, 57, 1021–1032. [Google Scholar] [CrossRef]
- Kitazawa, T.; Igawa, T.; Sampei, Z.; Muto, A.; Kojima, T.; Soeda, T.; Yoshihashi, K.; Okuyama-Nishida, Y.; Saito, H.; Tsunoda, H.; et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med. 2012, 18, 1570–1574. [Google Scholar] [CrossRef]
- Lenting, P.J.; Denis, C.V.; Christophe, O.D. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: How does it actually compare to factor VIII? Blood 2017, 130, 2463–2468. [Google Scholar] [CrossRef]
- Knight, T.; Callaghan, M.U. The role of emicizumab, a bispecific factor IXa- and factor X-directed antibody, for the prevention of bleeding episodes in patients with hemophilia A. Ther. Adv. Hematol. 2018, 9, 319–334. [Google Scholar] [CrossRef]
- Sampei, Z.; Igawa, T.; Soeda, T.; Okuyama-Nishida, Y.; Moriyama, C.; Wakabayashi, T.; Tanaka, E.; Muto, A.; Kojima, T.; Kitazawa, T.; et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE 2013, 8, e57479. [Google Scholar] [CrossRef]
- Jarantow, S.W.; Bushey, B.S.; Pardinas, J.R.; Boakye, K.; Lacy, E.R.; Sanders, R.; Sepulveda, M.A.; Moores, S.L.; Chiu, M.L. Impact of Cell-surface Antigen Expression on Target Engagement and Function of an Epidermal Growth Factor Receptor x c-MET Bispecific Antibody. J. Biol. Chem. 2015, 290, 24689–24704. [Google Scholar] [CrossRef]
- Zheng, S.; Moores, S.; Jarantow, S.; Pardinas, J.; Chiu, M.; Zhou, H.; Wang, W. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. mAbs 2016, 8, 551–561. [Google Scholar] [CrossRef]
- Moores, S.L.; Chiu, M.L.; Bushey, B.S.; Chevalier, K.; Luistro, L.; Dorn, K.; Brezski, R.J.; Haytko, P.; Kelly, T.; Wu, S.J.; et al. A Novel Bispecific Antibody Targeting EGFR and cMet Is Effective against EGFR Inhibitor-Resistant Lung Tumors. Cancer Res. 2016, 76, 3942–3953. [Google Scholar] [CrossRef]
- Yang, F.; Wen, W.; Qin, W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2017, 18, 48. [Google Scholar] [CrossRef]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014, 28, 4764–4778. [Google Scholar] [CrossRef]
- Stanimirovic, D.; Kemmerich, K.; Haqqani, A.S.; Farrington, G.K. Engineering and pharmacology of blood-brain barrier-permeable bispecific antibodies. Adv. Pharmacol. 2014, 71, 301–335. [Google Scholar]
- Couch, J.A.; Yu, Y.J.; Zhang, Y.; Tarrant, J.M.; Fuji, R.N.; Meilandt, W.J.; Solanoy, H.; Tong, R.K.; Hoyte, K.; Luk, W.; et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 2013, 5, 1–12. [Google Scholar] [CrossRef]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef]
- Atwal, J.K.; Chen, Y.; Chiu, C.; Mortensen, D.L.; Meilandt, W.J.; Liu, Y.; Heise, C.E.; Hoyte, K.; Luk, W.; Lu, Y.; et al. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci. Transl. Med. 2011, 3, 84ra43. [Google Scholar] [CrossRef]
- Liu, Z.; Gunasekaran, K.; Wang, W.; Razinkov, V.; Sekirov, L.; Leng, E.; Sweet, H.; Foltz, I.; Howard, M.; Rousseau, A.M.; et al. Asymmetrical Fc engineering greatly enhances antibody-dependent cellular cytotoxicity (ADCC) effector function and stability of the modified antibodies. J. Biol. Chem. 2014, 289, 3571–3590. [Google Scholar] [CrossRef]
- Escobar-Carbrera, E.; Lario, P.I.; Shrag, J.; Durocher, Y.; Dixit, S.B. Asymmetric Fc Engineering for Bispecific Antibodies with Reduced Effector Function. Antibodies 2017, 6, 7. [Google Scholar] [CrossRef]
- Riethmuller, G. Symmetry breaking: Bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 2012, 12, 12. [Google Scholar] [PubMed]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67 Pt A, 95–106. [Google Scholar] [CrossRef]
- Ha, J.H.; Kim, J.E.; Kim, Y.S. Immunoglobulin Fc Heterodimer Platform Technology: From Design to Applications in Therapeutic Antibodies and Proteins. Front. Immunol. 2016, 7, 394. [Google Scholar] [CrossRef] [PubMed]
- Godar, M.; de Haard, H.; Blanchetot, C.; Rasser, J. Therapeutic bispecific antibody formats: A patent applications review (1994–2017). Expert Opin. Ther. Pat. 2018, 28, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein-protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef]
- Xenaki, K.T.; Oliveira, S.; van Bergen, P.M.P. En henegouwen. Antibody or antibody fragments: Implications for molecular imaging and targeted therapy of solid tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Dave, E.; Adams, R.; Zaccheo, O.; Carrington, B.; Compson, J.E.; Dugdale, S.; Airey, M.; Malcolm, S.; Hailu, H.; Wild, G.; et al. Fab-dsFv: A bispecific antibody format with extended serum half-life through albumin binding. mAbs 2016, 8, 1319–1335. [Google Scholar] [CrossRef]
- Wu, X.; Sereno, A.J.; Huang, F.; Lewis, S.M.; Lieu, R.L.; Weldon, C.; Torres, C.; Fine, C.; Batt, M.A.; Fitchett, J.R.; et al. Fab-based bispecific antibody formats with robust biophysical properties and biological activity. mAbs 2015, 7, 470–482. [Google Scholar] [CrossRef]
- Schoonjans, R.; Willems, A.; Schoonooghe, S.; Fiers, W.; Grooten, J.; Mertens, N. Fab chains as an efficient heterodimerization scaffold for the production of recombinant bispecific and trispecific antibody derivatives. J. Immunol. 2000, 165, 7050–7057. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, P.; Zhou, C.; Jing, L.; Dong, B.; Chen, S.Y.; Zhang, N.; Liu, Y.; Maio, J.; Wang, Z.; et al. A Novel Bispecific Antibody, S-Fab, Induces Potent Cancer Cell Killing. J. Immunother. 2015, 38, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Linsley, P.S.; Gayle, M.A.; Bajorath, J.; Brady, W.A.; Norris, N.A.; Fell, H.P.; Ledbetter, J.A.; Gilliland, L.K. Single-chain mono- and bispecific antibody derivatives with novel biological properties and antitumour activity from a COS cell transient expression system. Ther. Immunol. 1994, 1, 3–15. [Google Scholar] [PubMed]
- Mallender, W.D.; Voss, E.W., Jr. Construction, expression, and activity of a bivalent bispecific single-chain antibody. J. Biol. Chem. 1994, 269, 199–206. [Google Scholar] [PubMed]
- Mack, M.; Riethmuller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Brusselbach, S.; Korn, T.; Volkel, T.; Muller, R.; Kontermann, R.E. Enzyme recruitment and tumor cell killing in vitro by a secreted bispecific single chain diabody. Tumor Target. 1999, 4, 115–123. [Google Scholar]
- Zhu, Z.; Presta, L.G.; Zapata, G.; Carter, P. Remodeling domain interfaces to enhance heterodimer formation. Protein Sci. 1997, 6, 781–788. [Google Scholar] [CrossRef]
- FitzGerald, K.; Holliger, P.; Winter, G. Improved tumour targeting by disulphide stabilized diabodies expressed in Pichia pastoris. Protein Eng. 1997, 10, 1221–1225. [Google Scholar] [CrossRef]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; von der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Cochlovius, B.; Kipriyanov, S.M.; Stassar, M.J.; Schuhmacher, J.; Benner, A.; Moldenhauer, G.; Little, M. Cure of Burkitt’s lymphoma in severe combined immunodeficiency mice by T cells, tetravalent CD3 × CD19 tandem diabody, and CD28 costimulation. Cancer Res. 2000, 60, 4336–4341. [Google Scholar] [PubMed]
- Cochlovius, B.; Kipriyanov, S.M.; Stassar, M.J.; Christ, O.; Schuhmacher, J.; Strauss, G.; Moldenhauer, G.; Little, M. Treatment of human B cell lymphoma xenografts with a CD3 × CD19 diabody and T cells. J. Immunol. 2000, 165, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Els Conrath, K.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [PubMed]
- De Bernardis, F.; Liu, H.; O’Mahony, R.; la Valle, R.; Bartollino, S.; Sandini, S.; Grant, S.; Brewis, N.; Tomlinson, I.; Basset, R.C.; et al. Human domain antibodies against virulence traits of Candida albicans inhibit fungus adherence to vaginal epithelium and protect against experimental vaginal candidiasis. J. Infect. Dis. 2007, 195, 149–157. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies as Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef]
- Moore, G.L.; Bernett, M.J.; Rashid, R.; Pong, E.W.; Nguyen, D.T.; Jacinto, J.; Eivazi, A.; Nisthal, A.; Diaz, J.E.; Chu, S.Y.; et al. A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019, 154, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; Priem, P.; de Jong, R.N.; van den Bremer, E.T.; van Kampen, M.D.; Gerritsen, A.F.; Schuurman, J.; Parren, P.W. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat. Protoc. 2014, 9, 2450–2463. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.; van den Bremer, E.T.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Goulet, D.R.; Orcutt, S.J.; Zwolak, A.; Rispens, T.; Labrijn, A.F.; de Jong, R.N.; Atkins, W.M.; Chiu, M.L. Kinetic mechanism of controlled Fab-arm exchange for the formation of bispecific immunoglobulin G1 antibodies. J. Biol. Chem. 2018, 293, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Ho, W.H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219. [Google Scholar] [CrossRef]
- Zwolak, A.; Leettola, C.N.; Tam, S.H.; Goulet, D.R.; Derebe, M.G.; Pardinas, J.R.; Zheng, S.; Decker, R.; Emmell, E.; Chiu, M.L. Rapid Purification of Human Bispecific Antibodies via Selective Modulation of Protein a Binding. Sci. Rep. 2017, 7, 15521. [Google Scholar] [CrossRef]
- Zwolak, A.; Armstrong, A.A.; Tam, S.H.; Pardinas, J.R.; Goulet, D.R.; Zheng, S.; Brosnan, K.; Emmell, E.; Luo, J.; Gilliland, G.L.; et al. Modulation of protein A binding allows single-step purification of mouse bispecific antibodies that retain FcRn binding. mAbs 2017, 9, 1306–1316. [Google Scholar] [CrossRef]
- Eigenbrot, C.; Fuh, G. Two-in-One antibodies with dual action Fabs. Curr. Opin. Chem. Biol. 2013, 17, 400–405. [Google Scholar] [CrossRef]
- Lee, C.V.; Koenig, P.; Fuh, G. A two-in-one antibody engineered from a humanized interleukin 4 antibody through mutation in heavy chain complementarity-determining regions. mAbs 2014, 6, 622–627. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Bose, S.; Miller, R.; Medina, L.; Santora, L.; Ghayur, T. Molecular construction and optimization of anti-human IL-1alpha/beta dual variable domain immunoglobulin (DVD-Ig) molecules. mAbs 2009, 1, 339–347. [Google Scholar] [CrossRef]
- Lacy, S.E.; Wu, C.; Ambrosi, D.J.; Hsieh, C.M.; Bose, S.; Miller, R.; Conlon, D.M.; Tarcsa, E.; Chari, R.; Ghayur, T.; et al. Generation and characterization of ABT-981, a dual variable domain immunoglobulin (DVD-Ig(TM)) molecule that specifically and potently neutralizes both IL-1alpha and IL-1beta. mAbs 2015, 7, 605–619. [Google Scholar] [CrossRef]
- Jakob, C.G.; Edalji, R.; Judge, R.A.; DiGiammarino, E.; Li, Y.; Gu, J.; Ghayur, T. Structure reveals function of the dual variable domain immunoglobulin (DVD-Ig) molecule. mAbs 2013, 5, 358–363. [Google Scholar] [CrossRef]
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113. [Google Scholar] [CrossRef]
- Krah, S.; Sellmann, C.; Rhiel, L.; Schroter, C.; Dickgiesser, S.; Beck, J.; Zielonka, S.; Toleikis, L.; Hock, B.; Kolmar, H.; et al. Engineering bispecific antibodies with defined chain pairing. New Biotechnol. 2017, 39, 167–173. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umana, P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef]
- Bonisch, M.; Sellmann, C.; Maresch, D.; Halbig, C.; Becker, S.; Toleikis, L.; Hock, B.; Ruker, F. Novel CH1:CL interfaces that enhance correct light chain pairing in heterodimeric bispecific antibodies. Protein Eng. Des. Sel. 2017, 30, 685–696. [Google Scholar] [CrossRef]
- Dillon, M.; Yin, Y.; Zhou, J.; McCarty, L.; Ellerman, D.; Slaga, D.; Junttila, T.T.; Han, G.; Sandoval, W.; Ovacik, M.A.; et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. mAbs 2017, 9, 213–230. [Google Scholar] [CrossRef]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Liu, Z.; Leng, E.C.; Gunasekaran, K.; Pentony, M.; Shen, M.; Howard, M.; Stoops, J.; Manchulenko, K.; Razinkov, V.; Liu, H.; et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. J. Biol. Chem. 2015, 290, 7535–7562. [Google Scholar] [CrossRef]
- Woods, R.J.; Xie, M.H.; von Kreudenstein, T.S.; Ng, G.Y.; Dixit, S.B. LC-MS characterization and purity assessment of a prototype bispecific antibody. mAbs 2013, 5, 711–722. [Google Scholar] [CrossRef]
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D’Angelo, I.; Brault, K.; Kelly, J.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. mAbs 2013, 5, 646–654. [Google Scholar] [CrossRef]
- Stephenson, R.P. A Modification of Receptor Theory. Br. J. Pharmacol. 1956, 11, 379–393. [Google Scholar] [CrossRef]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Zhukovsky, E.A.; Morse, R.J.; Maus, M.V. Bispecific antibodies and CARs: Generalized immunotherapeutics harnessing T cell redirection. Curr. Opin. Immunol. 2016, 40, 24–35. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. mAbs 2019, 11, 639–652. [Google Scholar] [CrossRef]
- Strohl, W.R.; Naso, M.F. Bispecific T-Cell Redirection versus Chimeric Antigen. Receptor (CAR)-T Cells as Approaches to Kill. Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Kellner, C.; Peipp, M.; Valerius, T. Effector Cell Recruitment by Bispecific Antibodies. In Bispecific Antibodies; Kontermann, R.E., Ed.; Springer: Heidelberg/Berlin, Germany, 2011; pp. 217–241. [Google Scholar]
- Peipp, M.; Wesch, D.; Oberg, H.H.; Lutz, S.; Muskulus, A.; van de Winkel, J.G.J.; Parren, P.; Burger, R.; Humpe, A.; Kabelitz, D.; et al. CD20-Specific Immunoligands Engaging NKG2D Enhance gammadelta T Cell-Mediated Lysis of Lymphoma Cells. Scand. J. Immunol. 2017, 86, 196–206. [Google Scholar] [CrossRef]
- Li, B.; Xu, L.; Pi, C.; Yin, Y.; Xie, K.; Tao, F.; Li, R.; Gu, H.; Fang, J. CD89-mediated recruitment of macrophages via a bispecific antibody enhances anti-tumor efficacy. Oncoimmunology 2017, 7, e1380142. [Google Scholar] [CrossRef]
- Dheilly, E.; Moine, V.; Broyer, L.; Salgado-Pires, S.; Johnson, Z.; Papaioannou, A.; Cons, L.; Calloud, S.; Majocchi, S.; Nelson, R.; et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol. Ther. 2017, 25, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, C.; Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongeneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science 2012, 337, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.A.; Friesen, R.H.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246–251. [Google Scholar] [CrossRef]
- Laursen, N.S.; Wilson, I.A. Broadly neutralizing antibodies against influenza viruses. Antivir. Res. 2013, 98, 476–483. [Google Scholar] [CrossRef]
- Tan, G.S.; Lee, P.S.; Hoffman, R.M.; Mazel-Sanchez, B.; Krammer, F.; Leon, P.E.; Ward, A.B.; Wilson, I.A.; Palese, P. Characterization of a broadly neutralizing monoclonal antibody that targets the fusion domain of group 2 influenza A virus hemagglutinin. J. Virol. 2014, 88, 13580–13592. [Google Scholar] [CrossRef]
- Laursen, N.S.; Friesen, R.H.E.; Zhu, X.; Jongeneelen, M.; Blokland, S.; Vermond, J.; van Eijgen, A.; Tang, C.; van Diepen, H.; Obmolova, G.; et al. Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 2018, 362, 598–602. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Tan, G.S.; Palese, P.; Ravetch, J.V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat. Med. 2014, 20, 143–151. [Google Scholar] [CrossRef]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef]
- Balandin, T.G.; Edelweiss, E.; Andronova, N.V.; Treshalina, E.M.; Sapozhnikov, A.M.; Deyev, S.M. Antitumor activity and toxicity of anti-HER2 immunoRNase scFv 4D5-dibarnase in mice bearing human breast cancer xenografts. Investig. New Drugs 2011, 29, 22–32. [Google Scholar] [CrossRef]
- Deyev, S.M.; Waibel, R.; Lebedenko, E.N.; Schubiger, A.P.; Pluckthun, A. Design of multivalent complexes using the barnase*barstar module. Nat. Biotechnol. 2003, 21, 1486–1492. [Google Scholar] [CrossRef]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 2013, 5, 207ra144. [Google Scholar] [CrossRef]
- Tietze, L.F.; Major, F.; Schuberth, I. Antitumor agents: Development of highly potent glycosidic duocarmycin analogues for selective cancer therapy. Angew. Chem. Int. Ed. Eng. 2006, 45, 6574–6577. [Google Scholar] [CrossRef]
- Jin, W.; Trzupek, J.D.; Rayl, T.J.; Broward, M.A.; Vielhauer, G.A.; Weir, S.J.; Hwang, I.; Boger, D.L. A unique class of duocarmycin and CC-1065 analogues subject to reductive activation. J. Am. Chem. Soc. 2007, 129, 15391–15397. [Google Scholar] [CrossRef]
- Bogen, J.P.; Hinz, S.C.; Grzeschik, J.; Ebening, A.; Krah, S.; Zielonka, S.; Kolmar, H. Dual Function pH Responsive Bispecific Antibodies for Tumor Targeting and Antigen Depletion in Plasma. Front. Immunol. 2019, 10, 1892. [Google Scholar] [CrossRef]
- Andrady, C.; Sharma, S.K.; Chester, K.A. Antibody-enzyme fusion proteins for cancer therapy. Immunotherapy 2011, 3, 193–211. [Google Scholar] [CrossRef]
- Lian, X.; Huang, Y.; Zhu, Y.; Fang, Y.; Zhao, R.; Joseph, E.; Li, J.; Pellois, J.P.; Zhou, H.C. Enzyme-MOF Nanoreactor Activates Nontoxic Paracetamol for Cancer Therapy. Angew. Chem. Int. Ed. Eng. 2018, 57, 5725–5730. [Google Scholar] [CrossRef]
- Sharma, S.K.; Bagshawe, K.D. Antibody Directed Enzyme Prodrug Therapy (ADEPT): Trials and tribulations. Adv. Drug Deliv. Rev. 2017, 118, 2–7. [Google Scholar] [CrossRef]
- Sharma, S.K.; Bagshawe, K.D. Translating antibody directed enzyme prodrug therapy (ADEPT) and prospects for combination. Expert Opin. Biol. Ther. 2017, 17, 1–13. [Google Scholar] [CrossRef]
- Kiefer, J.D.; Neri, D. Immunocytokines and bispecific antibodies: Two complementary strategies for the selective activation of immune cells at the tumor site. Immunol. Rev. 2016, 270, 178–192. [Google Scholar] [CrossRef]
- Hutmacher, C.; Neri, D. Antibody-cytokine fusion proteins: Biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv. Drug Deliv. Rev. 2018, 141, 67–91. [Google Scholar] [CrossRef]
- Marschall, A.L.; Zhang, C.; Frenzel, A.; Schirrmann, T.; Hust, M.; Perez, F.; Dubel, S. Delivery of antibodies to the cytosol: Debunking the myths. mAbs 2014, 6, 943–956. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21. [Google Scholar] [CrossRef]
- Weill, C.O.; Biri, S.; Erbacher, P. Cationic lipid-mediated intracellular delivery of antibodies into live cells. Biotechniques 2008, 44, Pvii–Pxi. [Google Scholar] [CrossRef]
- Weill, C.O.; Biri, S.; Adib, A.; Erbacher, P. A practical approach for intracellular protein delivery. Cytotechnology 2008, 56, 41–48. [Google Scholar] [CrossRef][Green Version]
- Freund, G.; Sibler, A.P.; Desplancq, D.; Oulad-Abdelghani, M.; Vigneron, M.; Gannon, J.; van Regenmortel, M.H.; Weiss, E. Targeting endogenous nuclear antigens by electrotransfer of monoclonal antibodies in living cells. mAbs 2013, 5, 518–522. [Google Scholar] [CrossRef]
- Fowler, D.A.; Filla, M.B.; Little, C.D.; Rongish, B.J.; Larsson, H.C.E. Live tissue antibody injection: A novel method for imaging ECM in limb buds and other tissues. Methods Cell. Biol. 2018, 143, 41–56. [Google Scholar]
- Becerril, B.; Poul, M.A.; Marks, J.D. Toward selection of internalizing antibodies from phage libraries. Biochem. Biophys. Res. Commun. 1999, 255, 386–393. [Google Scholar] [CrossRef]
- Poul, M.A.; Becerril, B.; Nielsen, U.B.; Morisson, P.; Marks, J.D. Selection of tumor-specific internalizing human antibodies from phage libraries. J. Mol. Biol. 2000, 301, 1149–1161. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, L.; Marks, J.D. Selection and characterization of cell binding and internalizing phage antibodies. Arch. Biochem. Biophys. 2012, 526, 107–113. [Google Scholar] [CrossRef]
- Zhou, Y.; Marks, J.D. Discovery of internalizing antibodies to tumor antigens from phage libraries. Methods Enzymol. 2012, 502, 43–66. [Google Scholar]
- Goenaga, A.L.; Zhou, Y.; Legay, C.; Bougherara, H.; Huang, L.; Liu, B.; Drummond, D.C.; Kirpotin, D.B.; Auclair, C.; Marks, J.D.; et al. Identification and characterization of tumor antigens by using antibody phage display and intrabody strategies. Mol. Immunol. 2007, 44, 3777–3788. [Google Scholar] [CrossRef]
- Shin, S.M.; Choi, D.K.; Jung, K.; Bae, J.; Kim, J.S.; Park, S.W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090. [Google Scholar] [CrossRef]
- Beyer, I.; van Rensburg, R.; Strauss, R.; Li, Z.; Wang, H.; Persson, J.; Yumul, R.; Feng, Q.; Song, H.; Bartek, J.; et al. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res. 2011, 71, 7080–7090. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1035. [Google Scholar] [CrossRef]
- Avignolo, C.; Bagnasco, L.; Biasotti, B.; Melchiori, A.; Tomati, V.; Bauer, I.; Salis, A.; Chiossone, L.; Mingari, M.C.; Orecchia, P.; et al. Internalization via Antennapedia protein transduction domain of an scFv antibody toward c-Myc protein. FASEB J. 2008, 22, 1237–1245. [Google Scholar] [CrossRef]
- Montrose, K.; Yang, Y.; Sun, X.; Wiles, S.; Krissansen, G.W. Xentry, a new class of cell-penetrating peptide uniquely equipped for delivery of drugs. Sci. Rep. 2013, 3, 1661. [Google Scholar] [CrossRef]
- Halaby, R. Role of lysosomes in cancer therapy. Res. Rep. Biol. 2015, 6, 147–155. [Google Scholar] [CrossRef]
- Shin, T.H.; Sung, E.S.; Kim, Y.J.; Kim, K.S.; Kim, S.H.; Kim, S.K.; Lee, Y.D.; Kim, Y.S. Enhancement of the tumor penetration of monoclonal antibody by fusion of a neuropilin-targeting peptide improves the antitumor efficacy. Mol. Cancer Ther. 2014, 13, 651–661. [Google Scholar] [CrossRef]
- Kim, Y.J.; Bae, J.; Shin, T.H.; Kang, S.H.; Jeong, M.; Han, Y.; Park, J.H.; Kim, S.K.; Kim, Y.S. Immunoglobulin Fc-fused, neuropilin-1-specific peptide shows efficient tumor tissue penetration and inhibits tumor growth via anti-angiogenesis. J. Control. Release 2015, 216, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Choi, D.K.; Shin, J.Y.; Shin, S.M.; Park, S.W.; Cho, H.S.; Kim, Y.S. Endosomal acidic pH-induced conformational changes of a cytosol-penetrating antibody mediate endosomal escape. J. Control. Release 2016, 235, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Park, J.Y.; Shin, S.M.; Park, S.W.; Jun, S.Y.; Hong, J.S.; Choi, D.K.; Kim, Y.S. Engineering of a tumor cell-specific, cytosol-penetrating antibody with high endosomal escape efficacy. Biochem. Biophys. Res. Commun. 2018, 503, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Boldicke, T. Single domain antibodies for the knockdown of cytosolic and nuclear proteins. Protein Sci. 2017, 26, 925–945. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K.; Bae, J.; Shin, S.M.; Shin, J.Y.; Kim, S.; Kim, Y.S. A general strategy for generating intact, full-length IgG antibodies that penetrate into the cytosol of living cells. mAbs 2014, 6, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Biocca, S. The potential of intracellular antibodies for therapeutic targeting of protein-misfolding diseases. Trends Mol. Med. 2008, 14, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Biocca, S. Combating protein misfolding and aggregation by intracellular antibodies. Curr. Mol. Med. 2008, 8, 2–11. [Google Scholar]
- Cohen, P.A.; Mani, J.C.; Lane, D.P. Characterization of a new intrabody directed against the N-terminal region of human p53. Oncogene 1998, 17, 2445–2456. [Google Scholar] [CrossRef][Green Version]
- Cohen, P.A. Intrabodies. Targeting scFv expression to eukaryotic intracellular compartments. Methods Mol. Biol. 2002, 178, 367–378. [Google Scholar]
- Wheeler, Y.Y.; Kute, T.E.; Willingham, M.C.; Chen, S.Y.; Sane, D.C. Intrabody-based strategies for inhibition of vascular endothelial growth factor receptor-2: Effects on apoptosis, cell growth, and angiogenesis. FASEB J. 2003, 17, 1733–1735. [Google Scholar] [CrossRef][Green Version]
- Tanaka, T.; Lobato, M.N.; Rabbitts, T.H. Single domain intracellular antibodies: A minimal fragment for direct in vivo selection of antigen-specific intrabodies. J. Mol. Biol. 2003, 331, 1109–1120. [Google Scholar] [CrossRef]
- Auf der Maur, A.; Escher, D.; Barberis, A. Antigen-independent selection of stable intracellular single-chain antibodies. FEBS Lett. 2001, 508, 407–412. [Google Scholar] [CrossRef]
- Shaki-Loewenstein, S.; Zfania, R.; Hyland, S.; Wels, W.S.; Benhar, I. A universal strategy for stable intracellular antibodies. J. Immunol. Methods 2005, 303, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Natraj, U.; Karande, A.A.; Gulati, A.; Murthy, M.R.; Murugesan, S.; Mukunda, P.; Savithri, H.S. Intracellular delivery of antibodies by chimeric Sesbania mosaic virus (SeMV) virus like particles. Sci. Rep. 2016, 6, 21803. [Google Scholar] [CrossRef] [PubMed]
- Strebe, N.; Guse, A.; Schungel, M.; Schirrmann, T.; Hafner, M.; Jostock, T.; Hust, M.; Muller, W.; Dubel, S. Functional knockdown of VCAM-1 at the posttranslational level with ER retained antibodies. J. Immunol. Methods 2009, 341, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Nordin, M.A.C.; Teow, S.-Y. Review of Current Cell-Penetrating Antibody Developments for HIV-1 Therapy. Molecules 2018, 23, 335. [Google Scholar] [CrossRef]
- Serruys, B.; van Houtte, F.; Verbrugghe, P.; Leroux-Roels, G.; Vanlandschoot, P. Llama-derived single-domain intrabodies inhibit secretion of hepatitis B virions in mice. Hepatology 2009, 49, 39–49. [Google Scholar] [CrossRef]
- Serruys, B.; van Houtte, F.; Farhoudi-Moghadam, A.; Leroux-Roels, G.; Vanlandschoot, P. Production, characterization and in vitro testing of HBcAg-specific VHH intrabodies. J. Gen. Virol. 2010, 91, 643–652. [Google Scholar] [CrossRef]
- Behar, G.; Chames, P.; Teulon, I.; Cornillon, A.; Alshoukr, F.; Roquet, F.; Pugniere, M.; Teillaud, J.L.; Gruaz-Guyon, A.; Pelegrin, A.; et al. Llama single-domain antibodies directed against nonconventional epitopes of tumor-associated carcinoembryonic antigen absent from nonspecific cross-reacting antigen. FEBS J. 2009, 276, 3881–3893. [Google Scholar] [CrossRef]











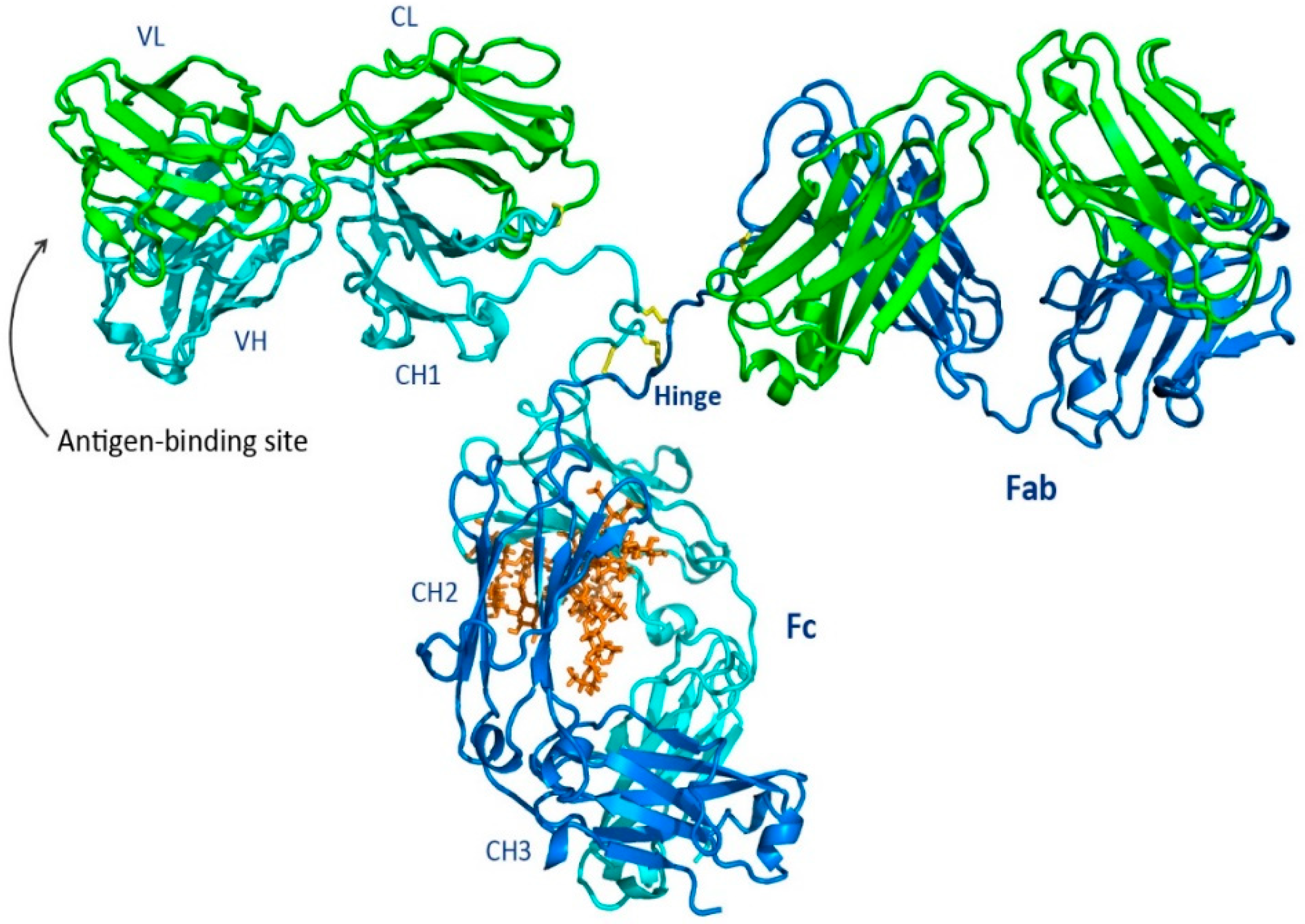{kind=link}
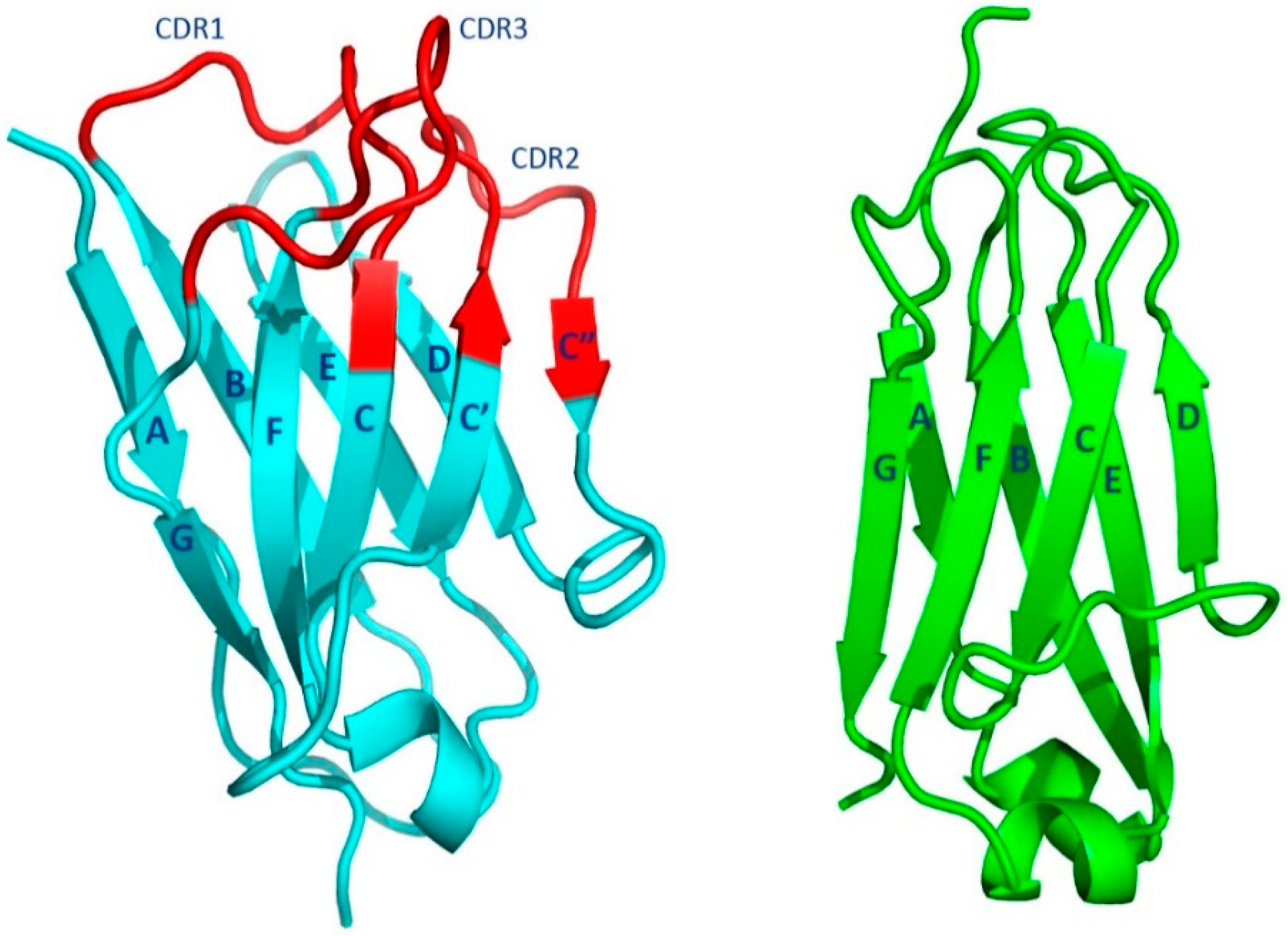{kind=link}
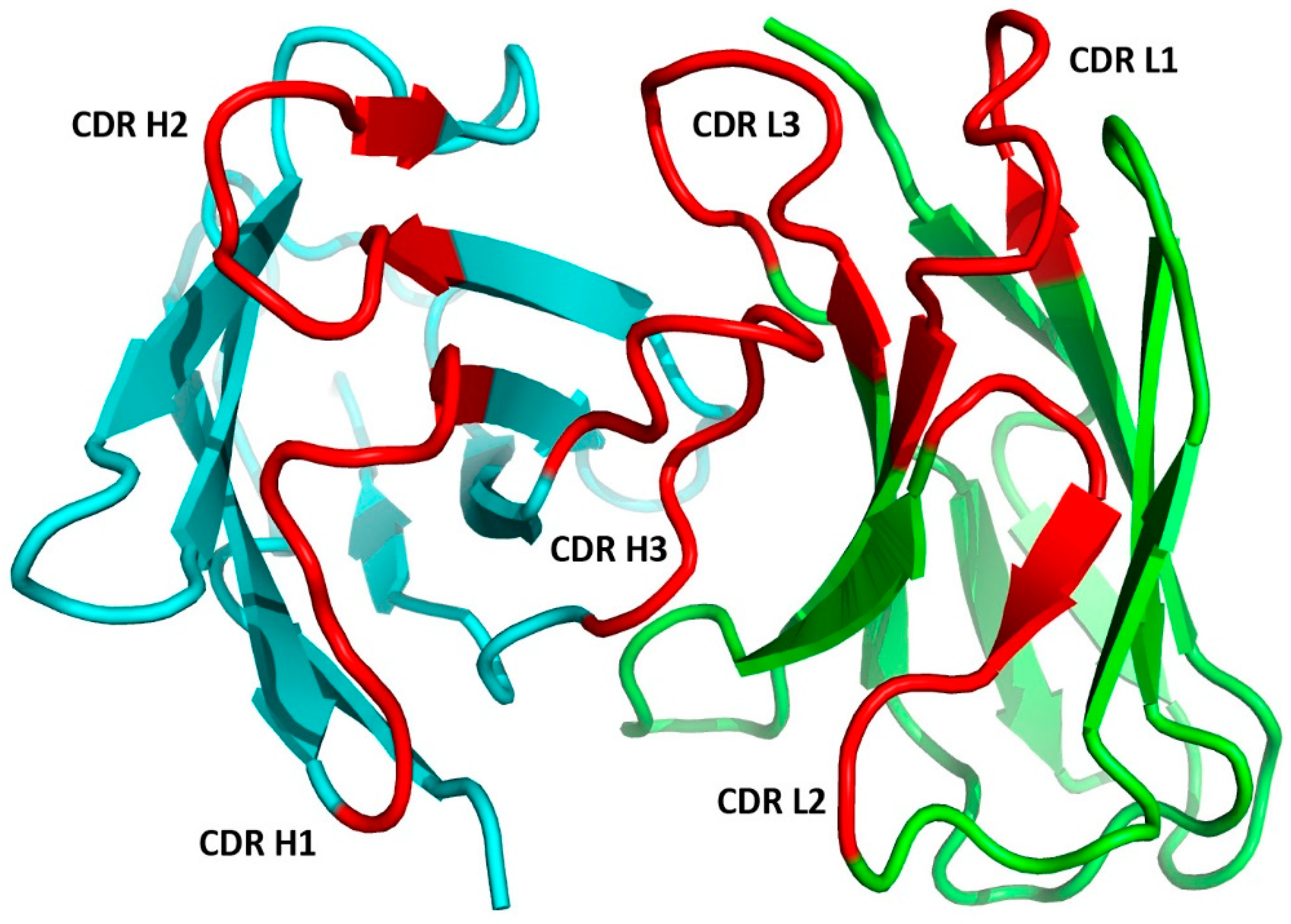{kind=link}
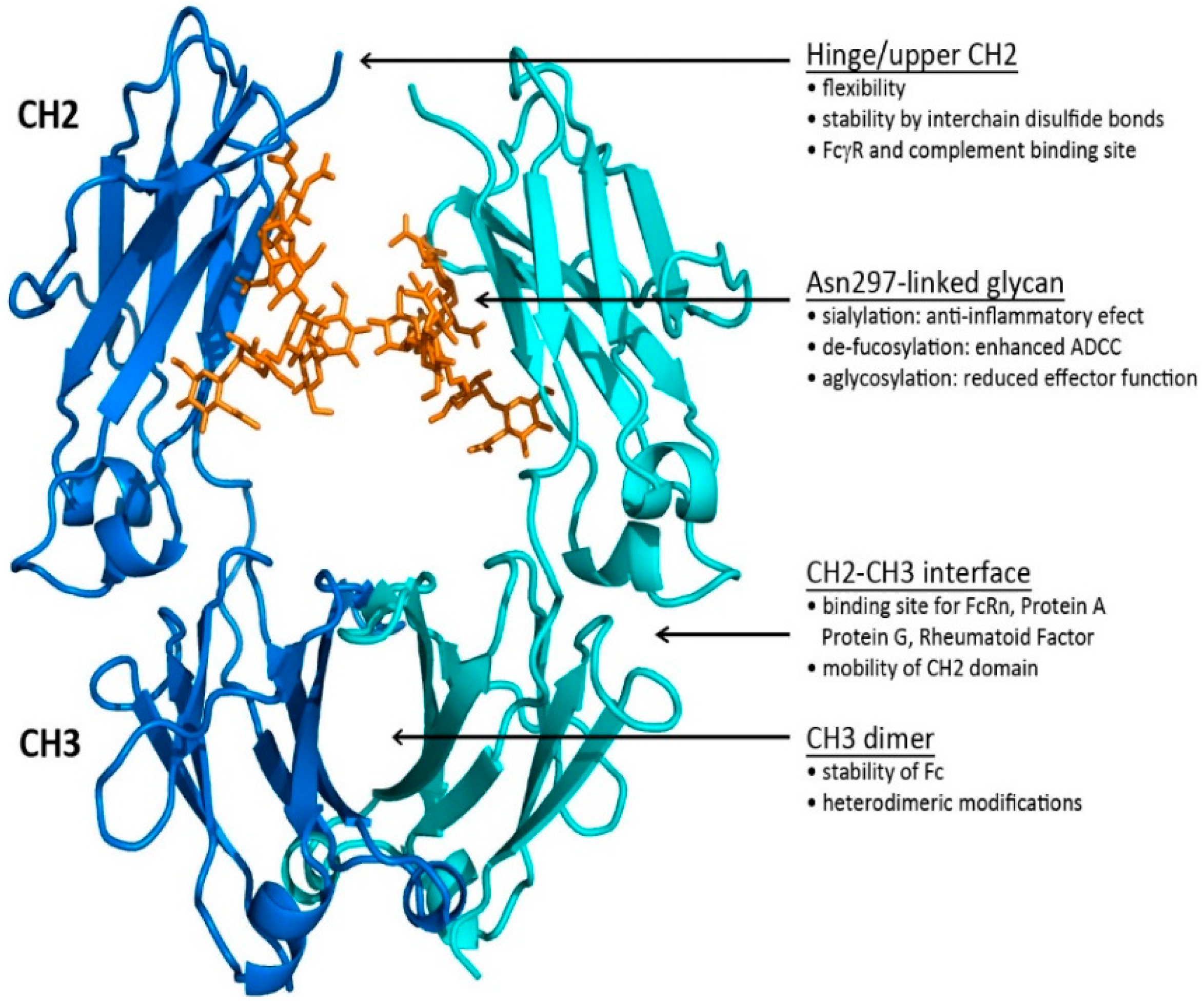{kind=link}
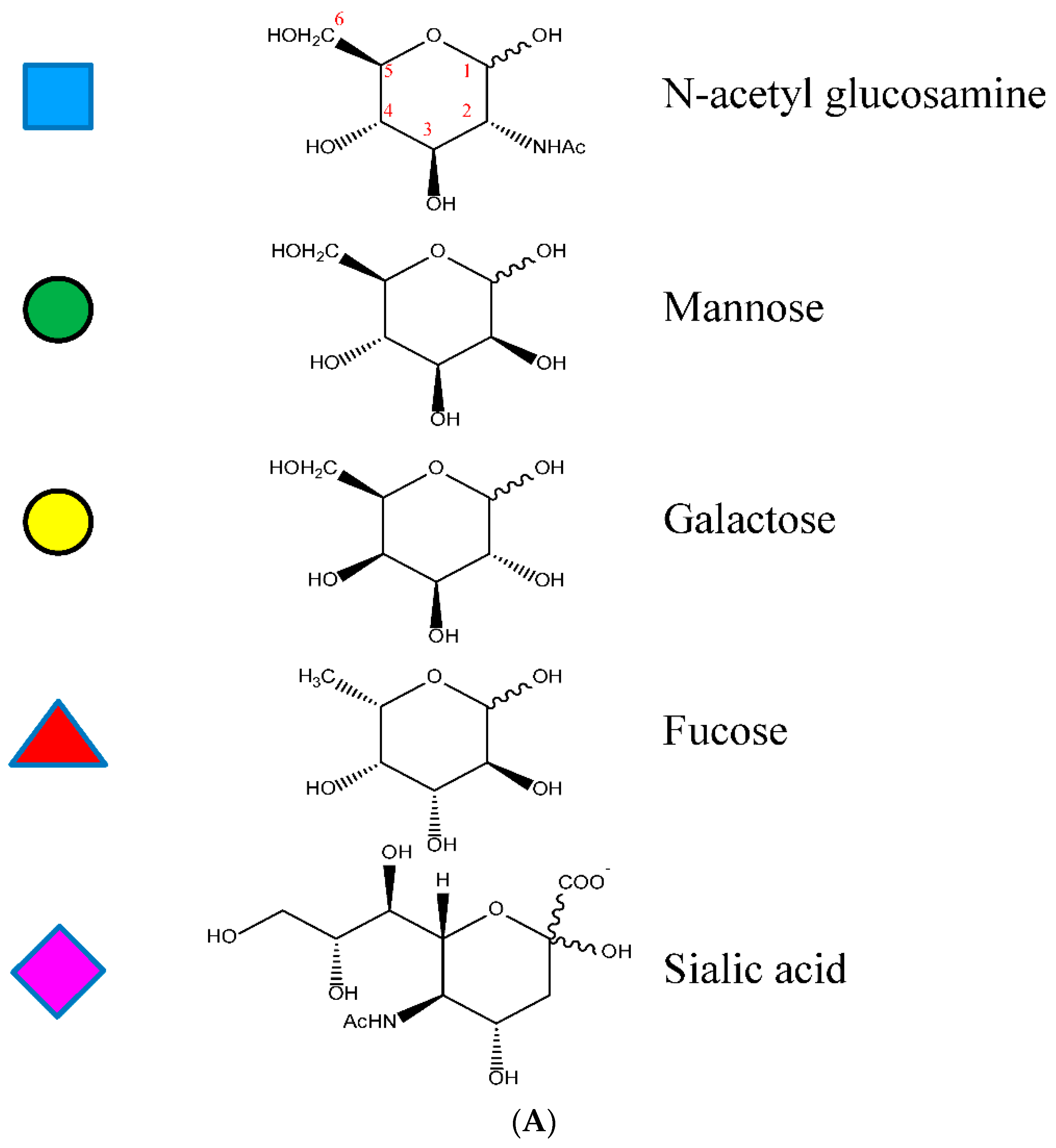{kind=link}
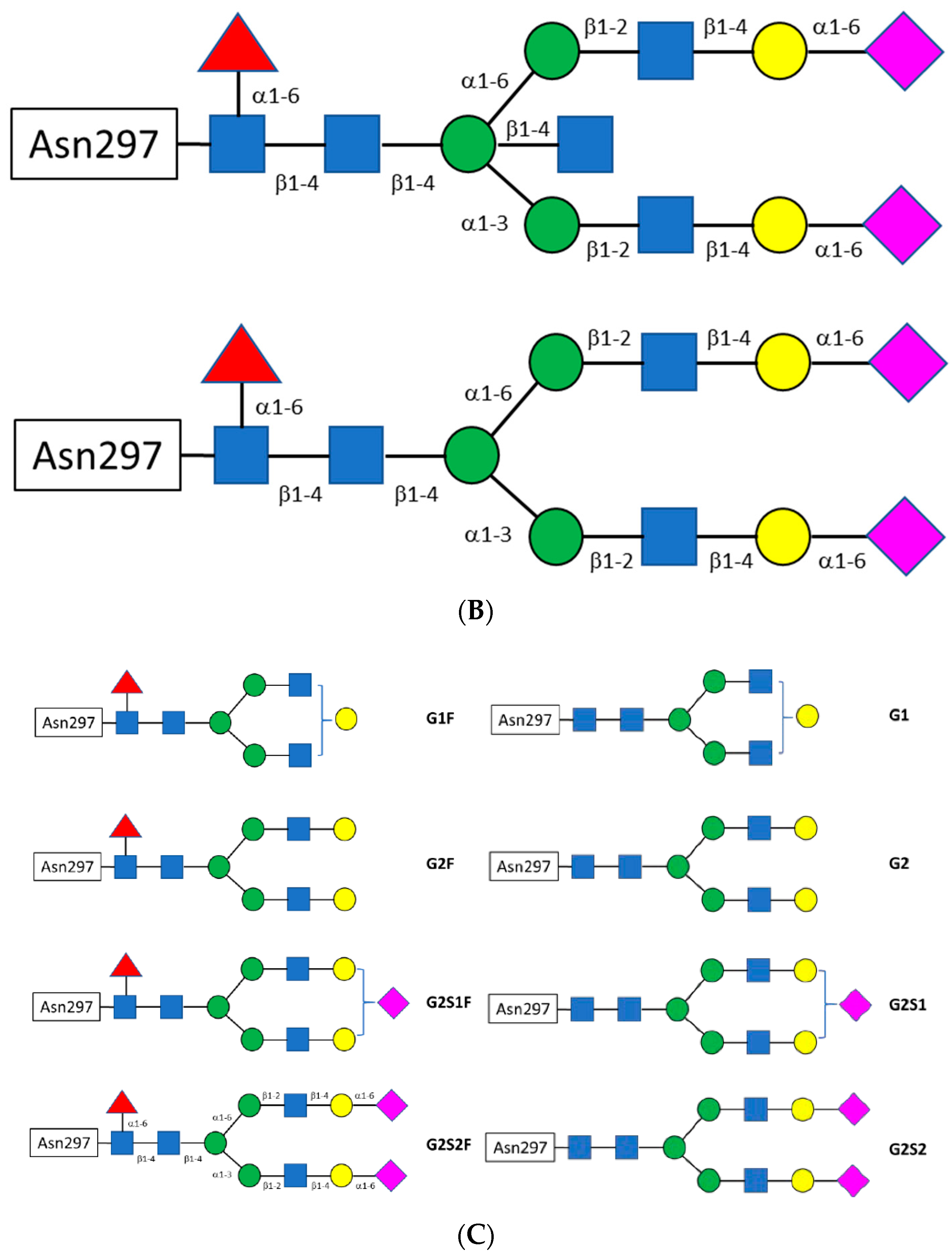{kind=link}
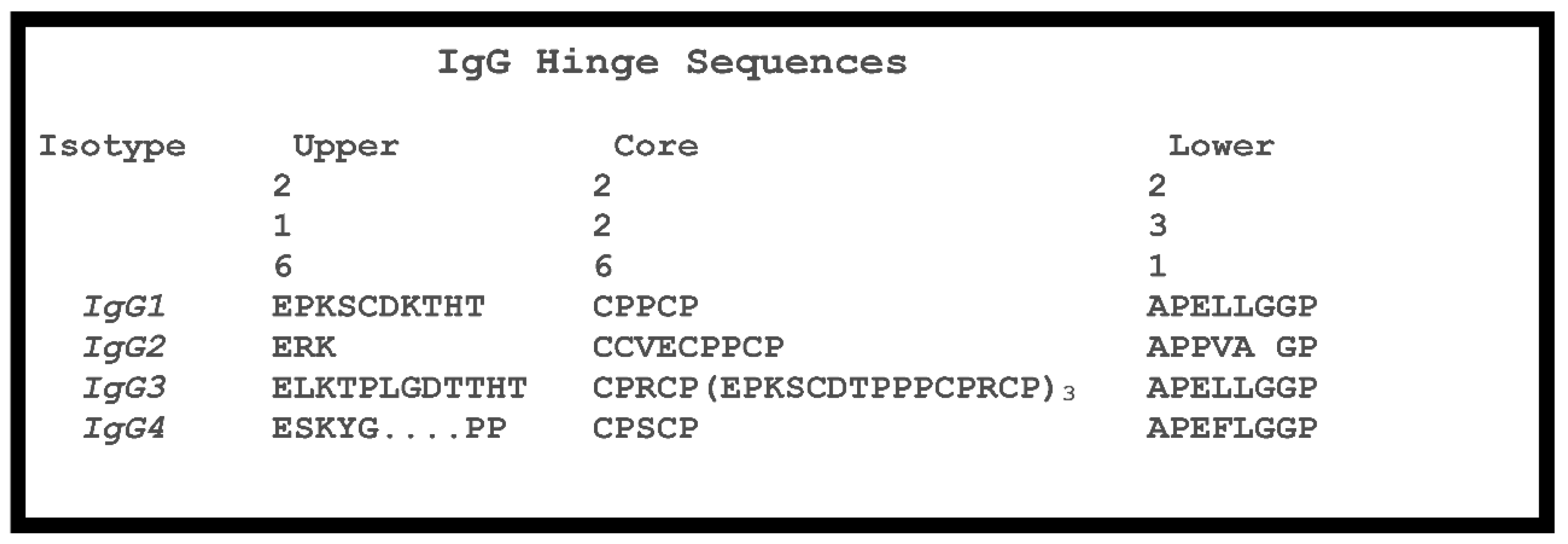{kind=link}
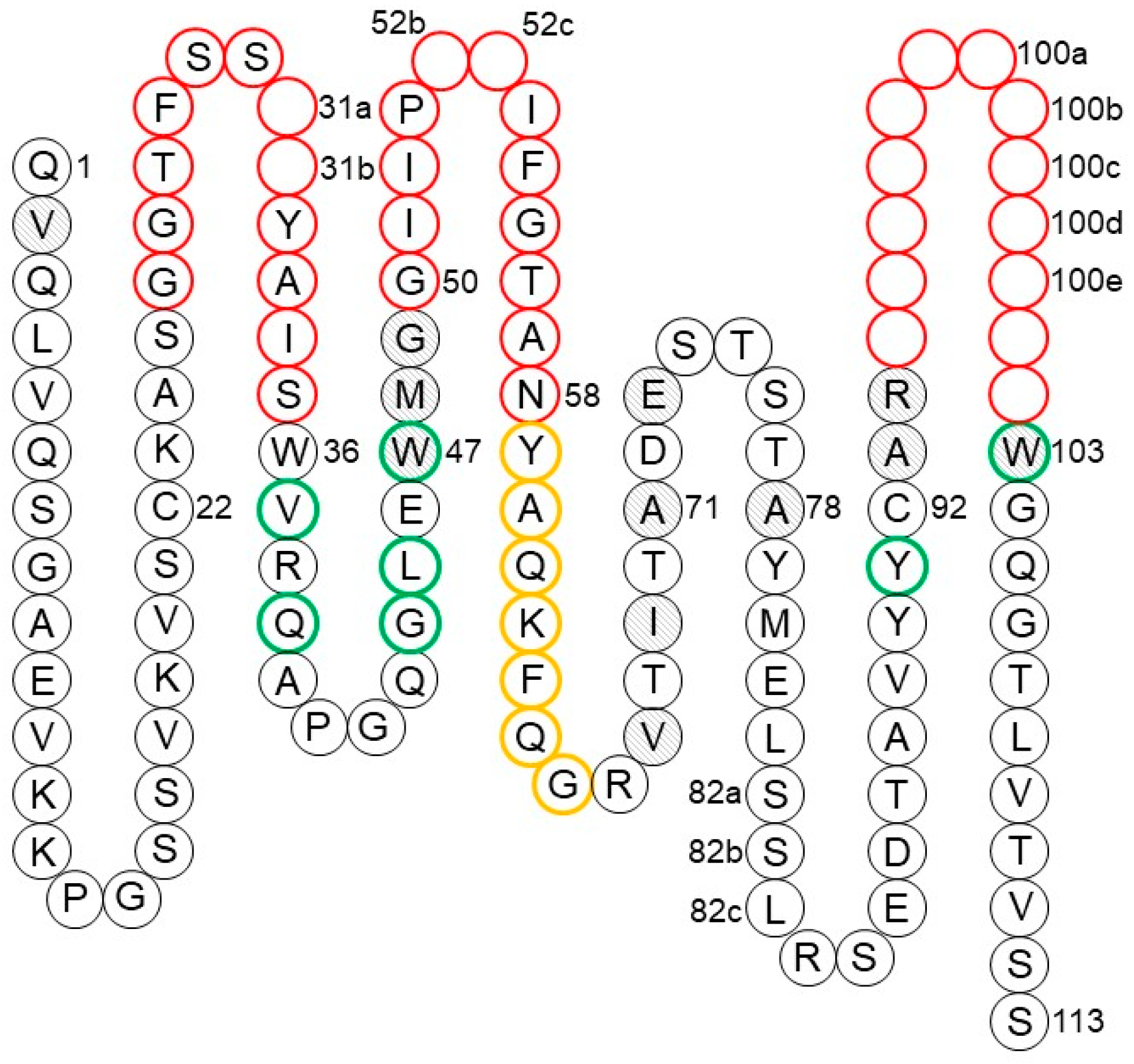{kind=link}
{kind=link}
{kind=link}
| CDR | Kabat | Chothia | Martin * | PyIgClassify ** | IMGT ** |
|---|---|---|---|---|---|
| L1 | 24–34 | 24–34 | 24–34 | 24–34 | 27–32 (M − 5) |
| L2 | 50–56 | 50–56 | 50–56 | 49–56 (M + 1) | 50–52 (M − 4) |
| L3 | 89–97 | 89–97 | 89–97 | 89–97 | 89–97 |
| H1 | 31–35 | 26–32 | 26–35 (K + C) | 23–35 (M + 3) | 26–33 (M − 2) |
| H2 | 50–65 | 52–56 | 50–58 (K − 7) | 50–58 | 51–57 (M − 2) |
| H3 | 95–102 | 95–102 | 95–102 | 93–102 (M + 2) | 93–102 (M + 2) |
| Amino Acid Changes | Chemistry | Effect on Protein | Effect on Biology |
|---|---|---|---|
| Asn-(Gly/Ser); Asp-(Gly/Ser) | Asn deamidation, Aspartic acid isomerization | Protein degradation [220,221,222]; Tertiary changes to Ab structure [223]; Isoaspartic acid [224]; Aggregation [225] | Isomerization can affect IgG avidity [226]; Deamidation affects binding [227]; Deamidation affects PK [216] |
| Gln | Gln deamidation | Slower deamidation than Asn, heterogeneity and stability [228] | Biological activity on Fab and Fc * |
| Met | Oxidation | Presence of oxidized methionine affects charged state of proteins [229,230,231]; Methionine oxidation decreases affinity to protein A and FcRn [232] | Methionine oxidation on Fc region can modulate FcγRIIa engagement [233]; FcRn and Fcγ receptors [234]; PK [235,236] |
| Trp | Oxidation | Changes in Trp aromaticity [237]; color changes [238]; Effects on detergent excipients for Ab formulation [239]; Higher order structure [240] | Biological activity on Fab and Fc [241,242] |
| Cys | Oxidation | Cysteinylation; Hinge disulfide chemistry with Cu2+ ion results in hydrolysis or oxidation that can lead to cleavage of the mAb [243,244,245] | Cysteinylation in CDRs leads to loss of potency [246,247]; Changing disulfide patterns in IgG subtypes [248] |
| His | Oxidation | Oxidized histidine react with intact histidine, lysine, and free cysteine to crosslink IgG [249]. Oxidized histidine [250,251] | Biological activity on Fab and Fc * |
| Asp-(Pro/Gly) | Amide bond hydrolysis | Cleavage at aspartic acid under acidic conditions [252,253]; Clipping at CH2 domain leads to aggregation [254] | Biological activity on Fab and Fc * |
| N terminal Glu/Gln | Pyroglutamate formation | Cyclized N terminal glutamine [255]; Challenges with molecule comparability [256] | Biological activity [256] |
| C terminal truncation | Carboxypeptidase substrate | Human IgG is produced with C-terminal Lysines that are cleaved off in circulation. There can be changes in charge variation | C terminal lysine loss can enhance complement activation [257] |
| Glycation | Reducing sugar reaction with Lysines | Charge variants [218]; Structural heterogeneity [258] | Biological activity on Fab and Fc * |
| Glycosylation changes | Changes in glycosylation profiles | Glycan structure [67,69,259,260,261,262,263]; High mannose and afucosylation affect stability [264]; Sialylation [265]; Fucosylation [266] | Biological activity [267]; PK and PD [268]; Clearance [269] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. https://doi.org/10.3390/antib8040055
Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies. 2019; 8(4):55. https://doi.org/10.3390/antib8040055
Chicago/Turabian StyleChiu, Mark L., Dennis R. Goulet, Alexey Teplyakov, and Gary L. Gilliland. 2019. "Antibody Structure and Function: The Basis for Engineering Therapeutics" Antibodies 8, no. 4: 55. https://doi.org/10.3390/antib8040055
APA StyleChiu, M. L., Goulet, D. R., Teplyakov, A., & Gilliland, G. L. (2019). Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies, 8(4), 55. https://doi.org/10.3390/antib8040055