Single-Domain Antibodies as Therapeutic and Imaging Agents for the Treatment of CNS Diseases


Abstract
1. Introduction to sdAbs
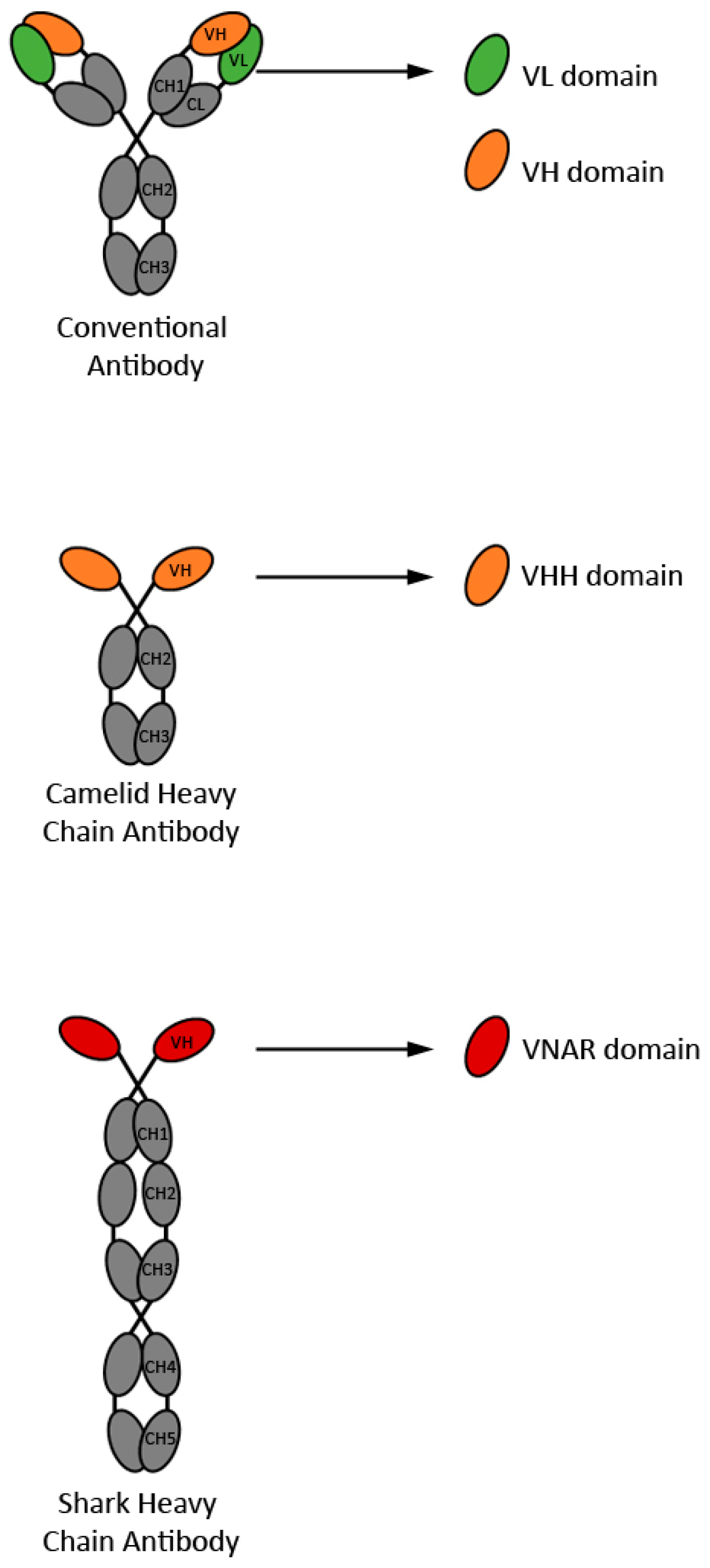
1.1. Structure and Characteristics
1.2. Single-Domain Antibody Libraries and Selection
1.3. Modular Building of Multispecific Molecules
1.4. Developing sdAbs as CNS Diagnostics or Therapeutics
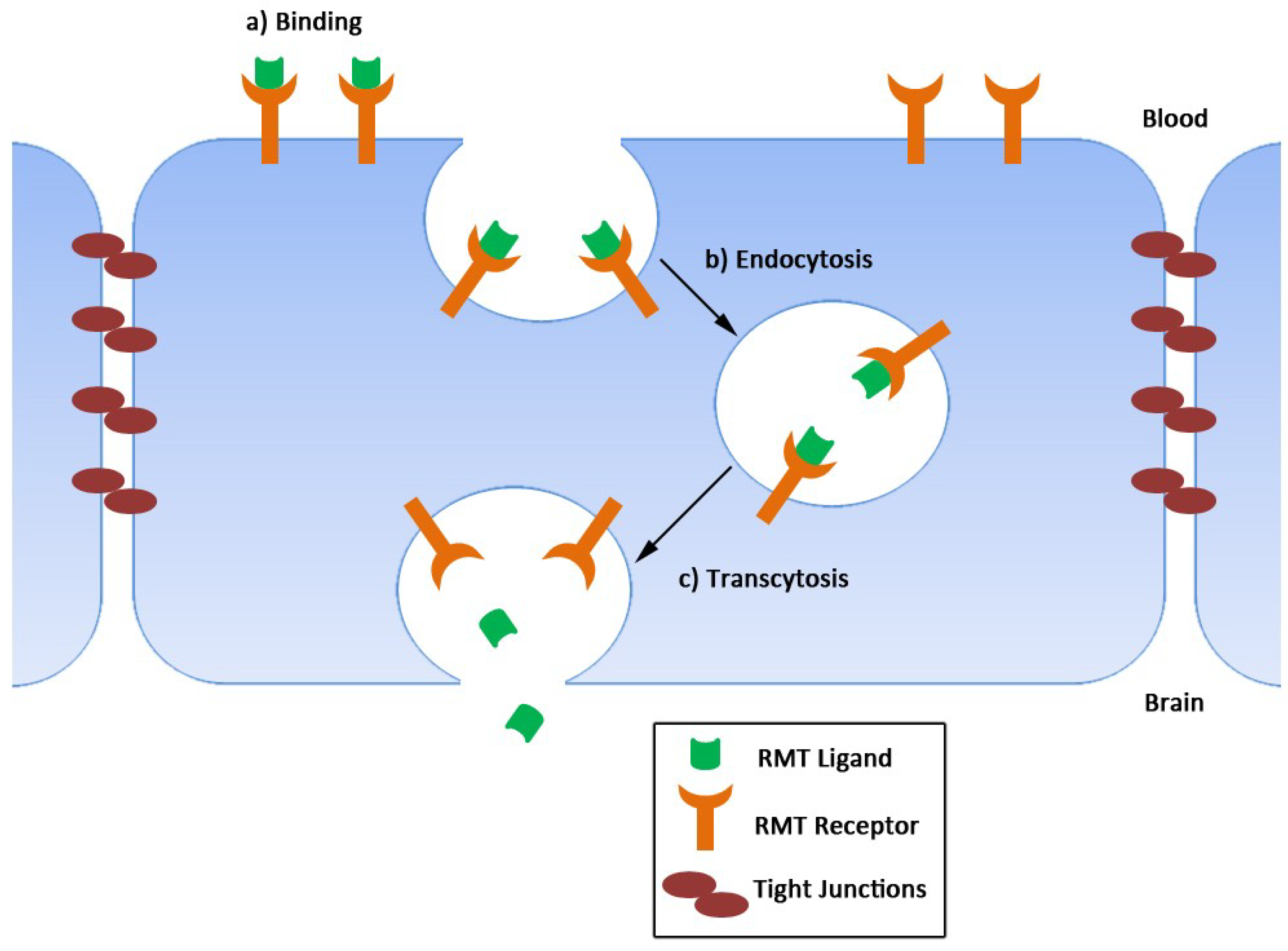
2. Single-Domain Antibodies as Delivery Agents across the BBB
3. Single-Domain Antibodies as Treatments against Neurodegenerative Diseases
3.1. Protein-Misfolding Diseases (PMDs)
3.1.1. Alzheimer’s Disease (AD)
3.1.2. Parkinson’s Disease (PD)
3.1.3. Huntington’s Disease (HD)
3.1.4. Prion Diseases
3.2. Glioblastoma Multiforme (GBM)
4. Single-Domain Antibodies as Neuroimaging Tools
4.1. Single-Domain Antibodies as Targeted Molecular Imaging Agents
4.2. Single-Domain Antibodies for Imaging Brain Tumor Vasculature
4.3. Single-Domain Antibodies for Imaging Brain Targets
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989, 341, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995, 374, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Konning, D.; Zielonka, S.; Grzeschik, J.; Empting, M.; Valldorf, B.; Krah, S.; Schroter, C.; Sellmann, C.; Hock, B.; Kolmar, H. Camelid and shark single domain antibodies: Structural features and therapeutic potential. Curr. Opin. Struct. Boil. 2017, 45, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Hussack, G.; Kandalaft, H.; Tanha, J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim. Biophys. Acta 2014, 1844, 1983–2001. [Google Scholar] [CrossRef]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.A.; Kim, D.Y.; Kandalaft, H.; Lowden, M.J.; Yang, Q.; Schrag, J.D.; Hussack, G.; MacKenzie, C.R.; Tanha, J. Stability-Diversity Tradeoffs Impose Fundamental Constraints on Selection of Synthetic Human VH/VL Single-Domain Antibodies from In Vitro Display Libraries. Front. Immunol. 2017, 8, 1759. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Dekker, S.; Hendriks, R.W.; Panayotou, G.; van Remoortere, A.; San, J.K.; Grosveld, F.; Drabek, D. Generation of heavy-chain-only antibodies in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15130–15135. [Google Scholar] [CrossRef] [PubMed]
- Drabek, D.; Janssens, R.; de Boer, E.; Rademaker, R.; Kloess, J.; Skehel, J.; Grosveld, F. Expression Cloning and Production of Human Heavy-Chain-Only Antibodies from Murine Transgenic Plasma Cells. Front. Immunol. 2016, 7, 619. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.B.a.A. VNARS: An ancient and unique repertoire of small molecules that deliver small, soluble, stable and high affinity binders of proteins. Antibodies 2015, 4, 240–258. [Google Scholar] [CrossRef]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, S.; Empting, M.; Grzeschik, J.; Konning, D.; Barelle, C.J.; Kolmar, H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. mAbs 2015, 7, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, M.; Ferguson, L.; Steven, J.; Porter, A.; Barelle, C. Shark variable new antigen receptor biologics—A novel technology platform for therapeutic drug development. Expert Opin. Boil. Ther. 2014, 14, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N.; MacKenzie, R.; Arbabi Ghahroudi, M. Single-domain antibodies and their utility. Curr. Protoc. Immunol. 2013, 103, 2–17. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Hu, P.; Wu, D.; Chen, W.; Ying, T.; Zhu, Z.; Dimitrov, D.S.; Dropulic, B.; Orentas, R.J. A Unique Human Immunoglobulin Heavy Chain Variable Domain-Only CD33 CAR for the Treatment of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 539. [Google Scholar] [CrossRef]
- Henry, K.A.; Tanha, J. Performance evaluation of phage-displayed synthetic human single-domain antibody libraries: A retrospective analysis. J. Immunol. Methods 2018, 456, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hussack, G.; Riazi, A.; Ryan, S.; van Faassen, H.; MacKenzie, R.; Tanha, J.; Arbabi-Ghahroudi, M. Protease-resistant single-domain antibodies inhibit Campylobacter jejuni motility. Protein Eng. Des. Sel. PEDS 2014, 27, 191–198. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002, 16, 240–242. [Google Scholar] [CrossRef]
- Iezzi, M.E.; Policastro, L.; Werbajh, S.; Podhajcer, O.; Canziani, G.A. Single-Domain Antibodies and the Promise of Modular Targeting in Cancer Imaging and Treatment. Front. Immunol. 2018, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Ubah, O.C.; Buschhaus, M.J.; Ferguson, L.; Kovaleva, M.; Steven, J.; Porter, A.J.; Barelle, C.J. Next-generation flexible formats of VNAR domains expand the drug platform’s utility and developability. Biochem. Soc. Trans. 2018, 46, 1559–1565. [Google Scholar] [CrossRef]
- Els Conrath, K.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Boil. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. PEDS 2008, 21, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.; Dreier, T.; Silence, K.; de Haard, H.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Van de Wiele, C.; Staelens, L.; et al. Formatted anti-tumor necrosis factor alpha VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.P.; Abregu, F.A.; Krishnan, U.V.; Proll, D.F.; Streltsov, V.A.; Doughty, L.; Hattarki, M.K.; Nuttall, S.D. Dimerisation strategies for shark IgNAR single domain antibody fragments. J. Immunol. Methods 2006, 315, 171–184. [Google Scholar] [CrossRef] [PubMed]
- De Bernardis, F.; Liu, H.; O’Mahony, R.; La Valle, R.; Bartollino, S.; Sandini, S.; Grant, S.; Brewis, N.; Tomlinson, I.; Basset, R.C.; et al. Human domain antibodies against virulence traits of Candida albicans inhibit fungus adherence to vaginal epithelium and protect against experimental vaginal candidiasis. J. Infect. Dis. 2007, 195, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.R.; Saunders, K.; Grace, C.; Jin, M.; Piche-Nicholas, N.; Steven, J.; O’Dwyer, R.; Wu, L.; Khetemenee, L.; Vugmeyster, Y.; et al. Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain. mAbs 2012, 4, 673–685. [Google Scholar] [CrossRef]
- Steven, J.; Muller, M.R.; Carvalho, M.F.; Ubah, O.C.; Kovaleva, M.; Donohoe, G.; Baddeley, T.; Cornock, D.; Saunders, K.; Porter, A.J.; et al. In Vitro Maturation of a Humanized Shark VNAR Domain to Improve Its Biophysical Properties to Facilitate Clinical Development. Front. Immunol. 2017, 8, 1361. [Google Scholar] [CrossRef]
- Nosenko, M.A.; Atretkhany, K.N.; Mokhonov, V.V.; Efimov, G.A.; Kruglov, A.A.; Tillib, S.V.; Drutskaya, M.S.; Nedospasov, S.A. VHH-Based Bispecific Antibodies Targeting Cytokine Production. Front. Immunol. 2017, 8, 1073. [Google Scholar] [CrossRef]
- Beirnaert, E.; Desmyter, A.; Spinelli, S.; Lauwereys, M.; Aarden, L.; Dreier, T.; Loris, R.; Silence, K.; Pollet, C.; Cambillau, C.; et al. Bivalent Llama Single-Domain Antibody Fragments against Tumor Necrosis Factor Have Picomolar Potencies due to Intramolecular Interactions. Front. Immunol. 2017, 8, 867. [Google Scholar] [CrossRef] [PubMed]
- Darling, T.L.; Sherwood, L.J.; Hayhurst, A. Intracellular Crosslinking of Filoviral Nucleoproteins with Xintrabodies Restricts Viral Packaging. Front. Immunol. 2017, 8, 1197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Schmidt, D.; Liu, W.; Li, S.; Shi, L.; Sheng, J.; Chen, K.; Yu, H.; Tremblay, J.M.; Chen, X.; et al. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J. Infect. Dis. 2014, 210, 964–972. [Google Scholar] [CrossRef]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; t Hart, B.; Poelmans, S.; Bontinck, L.; et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody(R) ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Desmyter, A.; Spinelli, S.; Boutton, C.; Saunders, M.; Blachetot, C.; de Haard, H.; Denecker, G.; Van Roy, M.; Cambillau, C.; Rommelaere, H. Neutralization of Human Interleukin 23 by Multivalent Nanobodies Explained by the Structure of Cytokine-Nanobody Complex. Front. Immunol. 2017, 8, 884. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Fijten, H.P.; Dekker, A.; Eble, P.L. Passive immunization of pigs with bispecific llama single-domain antibody fragments against foot-and-mouth disease and porcine immunoglobulin. Vet. Microbiol. 2008, 132, 56–64. [Google Scholar] [CrossRef]
- Zhang, J.; Tanha, J.; Hirama, T.; Khieu, N.H.; To, R.; Tong-Sevinc, H.; Stone, E.; Brisson, J.R.; MacKenzie, C.R. Pentamerization of single-domain antibodies from phage libraries: A novel strategy for the rapid generation of high-avidity antibody reagents. J. Mol. Boil. 2004, 335, 49–56. [Google Scholar] [CrossRef]
- Stone, E.; Hirama, T.; Tanha, J.; Tong-Sevinc, H.; Li, S.; MacKenzie, C.R.; Zhang, J. The assembly of single domain antibodies into bispecific decavalent molecules. J. Immunol. Methods 2007, 318, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Bell, A.; To, R.; Baral, T.N.; Azizi, A.; Li, J.; Cass, B.; Durocher, Y. Transient expression and purification of chimeric heavy chain antibodies. Protein Expr. Purif. 2009, 65, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Ubah, O.C.; Steven, J.; Kovaleva, M.; Ferguson, L.; Barelle, C.; Porter, A.J.R.; Barelle, C.J. Novel, Anti-hTNF-alpha Variable New Antigen Receptor Formats with Enhanced Neutralizing Potency and Multifunctionality, Generated for Therapeutic Development. Front. Immunol. 2017, 8, 1780. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, M.; Johnson, K.; Steven, J.; Barelle, C.J.; Porter, A. Therapeutic Potential of Shark Anti-ICOSL VNAR Domains is Exemplified in a Murine Model of Autoimmune Non-Infectious Uveitis. Front. Immunol. 2017, 8, 1121. [Google Scholar] [CrossRef]
- D’Eall, C.; Pon, R.A.; Rossotti, M.A.; Krahn, N.; Spearman, M.; Callaghan, D.; van Faassen, H.; Hussack, G.; Stetefeld, J.; Butler, M.; et al. Modulating antibody-dependent cellular cytotoxicity of epidermal growth factor receptor-specific heavy-chain antibodies through hinge engineering. Immunol. Cell Boil. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rossotti, M.A.; Henry, K.A.; van Faassen, H.; Tanha, J.; Callaghan, D.; Hussack, G.; Arbabi-Ghahroudi, M.; MacKenzie, C.R. Camelid single-domain antibodies raised by DNA immunization are potent inhibitors of EGFR signaling. Biochem. J. 2019, 476, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.A.; Kandalaft, H.; Lowden, M.J.; Rossotti, M.A.; van Faassen, H.; Hussack, G.; Durocher, Y.; Kim, D.Y.; Tanha, J. A disulfide-stabilized human VL single-domain antibody library is a source of soluble and highly thermostable binders. Mol. Immunol. 2017, 90, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Schutze, K.; Petry, K.; Hambach, J.; Schuster, N.; Fumey, W.; Schriewer, L.; Rockendorf, J.; Menzel, S.; Albrecht, B.; Haag, F.; et al. CD38-Specific Biparatopic Heavy Chain Antibodies Display Potent Complement-Dependent Cytotoxicity Against Multiple Myeloma Cells. Front. Immunol. 2018, 9, 2553. [Google Scholar] [CrossRef] [PubMed]
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D’Angelo, I.; Brault, K.; Kelly, J.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. mAbs 2013, 5, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N.; Magez, S.; Stijlemans, B.; Conrath, K.; Vanhollebeke, B.; Pays, E.; Muyldermans, S.; De Baetselier, P. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat. Med. 2006, 12, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, J.; Zhu, X.; Tang, X.; Bao, Y.; Sun, X.; Huang, Y.; Tian, F.; Liu, X.; Yang, L. Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lymphoblastic leukemia potential. Int. J. Nanomed. 2017, 12, 1969–1983. [Google Scholar] [CrossRef] [PubMed]
- Behdani, M.; Zeinali, S.; Karimipour, M.; Khanahmad, H.; Schoonooghe, S.; Aslemarz, A.; Seyed, N.; Moazami-Godarzi, R.; Baniahmad, F.; Habibi-Anbouhi, M.; et al. Development of VEGFR2-specific Nanobody Pseudomonas exotoxin A conjugated to provide efficient inhibition of tumor cell growth. New Biotechnol. 2013, 30, 205–209. [Google Scholar] [CrossRef]
- Tian, B.; Wong, W.Y.; Hegmann, E.; Gaspar, K.; Kumar, P.; Chao, H. Production and characterization of a camelid single domain antibody-urease enzyme conjugate for the treatment of cancer. Bioconj. Chem. 2015, 26, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Wong, W.Y.; Uger, M.D.; Wisniewski, P.; Chao, H. Development and Characterization of a Camelid Single Domain Antibody-Urease Conjugate That Targets Vascular Endothelial Growth Factor Receptor 2. Front. Immunol. 2017, 8, 956. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Wolak, D.J.; Thorne, R.G. Diffusion of macromolecules in the brain: Implications for drug delivery. Mol. Pharm. 2013, 10, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, M.E.; Wolak, D.J.; Kumar, N.N.; Brunette, E.; Brunnquell, C.L.; Hannocks, M.J.; Abbott, N.J.; Meyerand, M.E.; Sorokin, L.; Stanimirovic, D.B.; et al. Intrathecal antibody distribution in the rat brain: Surface diffusion, perivascular transport and osmotic enhancement of delivery. J. Physiol. 2018, 596, 445–475. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Buciak, J.L.; Friden, P.M. Selective transport of an anti-transferrin receptor antibody through the blood-brain barrier in vivo. J. Pharmacol. Exp. Ther. 1991, 259, 66–70. [Google Scholar] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Delaney, C.E.; Brunette, E.; Baumann, E.; Farrington, G.K.; Sisk, W.; Eldredge, J.; Ding, W.; Tremblay, T.L.; Stanimirovic, D.B. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood brain barrier. J. Cereb. Blood Flow Metab. 2018, 38, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Coloma, M.J.; Lee, H.J.; Kurihara, A.; Landaw, E.M.; Boado, R.J.; Morrison, S.L.; Pardridge, W.M. Transport across the primate blood-brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm. Res. 2000, 17, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhou, Q.H.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. Pharmacokinetics and brain uptake of a genetically engineered bifunctional fusion antibody targeting the mouse transferrin receptor. Mol. Pharm. 2010, 7, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Giugliani, L.; de Oliveira Poswar, F.; Donis, K.C.; Corte, A.D.; Schmidt, M.; Boado, R.J.; Nestrasil, I.; Nguyen, C.; Chen, S.; et al. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): An open label phase 1-2 trial. Orphanet J. Rare Dis. 2018, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Boado, R.J.; Patrick, D.J.; Ka-Wai Hui, E.; Lu, J.Z. Blood-Brain Barrier Transport, Plasma Pharmacokinetics, and Neuropathology Following Chronic Treatment of the Rhesus Monkey with a Brain Penetrating Humanized Monoclonal Antibody Against the Human Transferrin Receptor. Mol. Pharm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Walsh, F.S.; Wicher, K.; Szary, J.; Stocki, P.; Demydchuk, M.; Rutkowski, L. Delivery of a CD20 transferrin receptor VNAR bispecific antibody to the brain for CNS lymphoma. In Proceedings of the AACR Annual Meeting, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Bahney, J.; von Bartheld, C.S. The Cellular Composition and Glia-Neuron Ratio in the Spinal Cord of a Human and a Nonhuman Primate: Comparison with Other Species and Brain Regions. Anat. Rec. 2018, 301, 697–710. [Google Scholar] [CrossRef]
- Couch, J.A.; Yu, Y.J.; Zhang, Y.; Tarrant, J.M.; Fuji, R.N.; Meilandt, W.J.; Solanoy, H.; Tong, R.K.; Hoyte, K.; Luk, W.; et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 2013, 5, 183ra57. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.I.; Stanimirovic, D.B. A gateway to the brain: Shuttles for brain delivery of macromolecules. Ther. Deliv. 2015, 6, 1321–1324. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Caram-Salas, N.; Ding, W.; Brunette, E.; Delaney, C.E.; Baumann, E.; Boileau, E.; Stanimirovic, D. Multiplexed evaluation of serum and CSF pharmacokinetics of brain-targeting single-domain antibodies using a NanoLC-SRM-ILIS method. Mol. Pharm. 2013, 10, 1542–1556. [Google Scholar] [CrossRef]
- Abulrob, A.; Sprong, H.; Van Bergen en Henegouwen, P.; Stanimirovic, D. The blood-brain barrier transmigrating single domain antibody: Mechanisms of transport and antigenic epitopes in human brain endothelial cells. J. Neurochem. 2005, 95, 1201–1214. [Google Scholar] [CrossRef]
- Albulrob, A.; Stanimirovic, D.; Muruganandam, A. Blood-Brain Barrier Epitopes and Uses Thereof. Patent WO2007036021A1, 5 April 2007. [Google Scholar]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014, 28, 4764–4778. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.I.; Caram-Salas, N.; Haqqani, A.S.; Thom, G.; Brown, L.; Rennie, K.; Yogi, A.; Costain, W.; Brunette, E.; Stanimirovic, D.B. Brain penetration, target engagement, and disposition of the blood-brain barrier-crossing bispecific antibody antagonist of metabotropic glutamate receptor type 1. FASEB J. 2016, 30, 1927–1940. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, D.; Kemmerich, K.; Haqqani, A.S.; Sulea, T.; Arbabi-Ghahroudi, M.; Massie, B.; Gilbert, R. Insulin-Like Growth Factor 1 Receptor-Specific Antibodies and Uses Thereof. U.S. Patent US20170015748A1, 19 January 2017. [Google Scholar]
- Ribecco-Lutkiewicz, M.; Sodja, C.; Haukenfrers, J.; Haqqani, A.S.; Ly, D.; Zachar, P.; Baumann, E.; Ball, M.; Huang, J.; Rukhlova, M.; et al. A novel human induced pluripotent stem cell blood-brain barrier model: Applicability to study antibody-triggered receptor-mediated transcytosis. Sci. Rep. 2018, 8, 1873. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.S.; Cherian, L.; Shah, M.; Garcia, R.; Navarro, J.C.; Grill, R.J.; Hand, C.C.; Tian, T.S.; Hannay, H.J. Neuroprotection with an erythropoietin mimetic peptide (pHBSP) in a model of mild traumatic brain injury complicated by hemorrhagic shock. J. Neurotrauma 2012, 29, 1156–1166. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Boil. 2004, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Katzman, R. Editorial: The prevalence and malignancy of Alzheimer disease. A major killer. Arch. Neurol. 1976, 33, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002, 16, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Wang, D.S.; Dickson, D.W.; Malter, J.S. Tissue transglutaminase, protein cross-linking and Alzheimer’s disease: Review and views. Int. J. Clin. Exp. Pathol. 2008, 1, 5–18. [Google Scholar]
- Habicht, G.; Haupt, C.; Friedrich, R.P.; Hortschansky, P.; Sachse, C.; Meinhardt, J.; Wieligmann, K.; Gellermann, G.P.; Brodhun, M.; Gotz, J.; et al. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Abeta protofibrils. Proc. Natl. Acad. Sci. USA 2007, 104, 19232–19237. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Boil. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef]
- Morgado, I.; Wieligmann, K.; Bereza, M.; Ronicke, R.; Meinhardt, K.; Annamalai, K.; Baumann, M.; Wacker, J.; Hortschansky, P.; Malesevic, M.; et al. Molecular basis of beta-amyloid oligomer recognition with a conformational antibody fragment. Proc. Natl. Acad. Sci. USA 2012, 109, 12503–12508. [Google Scholar] [CrossRef] [PubMed]
- Wacker, J.; Ronicke, R.; Westermann, M.; Wulff, M.; Reymann, K.G.; Dobson, C.M.; Horn, U.; Crowther, D.C.; Luheshi, L.M.; Fandrich, M. Oligomer-targeting with a conformational antibody fragment promotes toxicity in Abeta-expressing flies. Acta Neuropathol. Commun. 2014, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Rutgers, K.S.; van Remoortere, A.; van Buchem, M.A.; Verrips, C.T.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P.; van Duinen, S.G.; Maat-Schieman, M.L.; van der Maarel, S.M. Differential recognition of vascular and parenchymal beta amyloid deposition. Neurobiol. Aging 2011, 32, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Rutgers, K.S.; Nabuurs, R.J.; van den Berg, S.A.; Schenk, G.J.; Rotman, M.; Verrips, C.T.; van Duinen, S.G.; Maat-Schieman, M.L.; van Buchem, M.A.; de Boer, A.G.; et al. Transmigration of beta amyloid specific heavy chain antibody fragments across the in vitro blood-brain barrier. Neuroscience 2011, 190, 37–42. [Google Scholar] [CrossRef]
- Nabuurs, R.J.; Rutgers, K.S.; Welling, M.M.; Metaxas, A.; de Backer, M.E.; Rotman, M.; Bacskai, B.J.; van Buchem, M.A.; van der Maarel, S.M.; van der Weerd, L. In vivo detection of amyloid-beta deposits using heavy chain antibody fragments in a transgenic mouse model for Alzheimer’s disease. PLoS ONE 2012, 7, e38284. [Google Scholar] [CrossRef] [PubMed]
- Lafaye, P.; Achour, I.; England, P.; Duyckaerts, C.; Rougeon, F. Single-domain antibodies recognize selectively small oligomeric forms of amyloid beta, prevent Abeta-induced neurotoxicity and inhibit fibril formation. Mol. Immunol. 2009, 46, 695–704. [Google Scholar] [CrossRef]
- David, M.A.; Jones, D.R.; Tayebi, M. Potential candidate camelid antibodies for the treatment of protein-misfolding diseases. J. Neuroimmunol. 2014, 272, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Medecigo, M.; Manoutcharian, K.; Vasilevko, V.; Govezensky, T.; Munguia, M.E.; Becerril, B.; Luz-Madrigal, A.; Vaca, L.; Cribbs, D.H.; Gevorkian, G. Novel amyloid-beta specific scFv and VH antibody fragments from human and mouse phage display antibody libraries. J. Neuroimmunol. 2010, 223, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Perchiacca, J.M.; Ladiwala, A.R.; Bhattacharya, M.; Tessier, P.M. Structure-based design of conformation- and sequence-specific antibodies against amyloid beta. Proc. Natl. Acad. Sci. USA 2012, 109, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, A.R.; Bhattacharya, M.; Perchiacca, J.M.; Cao, P.; Raleigh, D.P.; Abedini, A.; Schmidt, A.M.; Varkey, J.; Langen, R.; Tessier, P.M. Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 19965–19970. [Google Scholar] [CrossRef] [PubMed]
- Julian, M.C.; Lee, C.C.; Tiller, K.E.; Rabia, L.A.; Day, E.K.; Schick, A.J., 3rd; Tessier, P.M. Co-evolution of affinity and stability of grafted amyloid-motif domain antibodies. Protein Eng. Des. Sel. PEDS 2015, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Julian, M.C.; Tiller, K.E.; Meng, F.; DuConge, S.E.; Akter, R.; Raleigh, D.P.; Tessier, P.M. Design and Optimization of Anti-amyloid Domain Antibodies Specific for beta-Amyloid and Islet Amyloid Polypeptide. J. Boil. Chem. 2016, 291, 2858–2873. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression, and mortality. Neurology 1998, 50, 318. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Swallow, D.; Grosset, K.A.; Anichtchik, O.; Spillantini, M.; Grosset, D.G. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease—A systematic review. Acta Neurol. Scand. 2014, 130, 59–72. [Google Scholar] [CrossRef]
- De Genst, E.J.; Guilliams, T.; Wellens, J.; O’Day, E.M.; Waudby, C.A.; Meehan, S.; Dumoulin, M.; Hsu, S.T.; Cremades, N.; Verschueren, K.H.; et al. Structure and properties of a complex of alpha-synuclein and a single-domain camelid antibody. J. Mol. Boil. 2010, 402, 326–343. [Google Scholar] [CrossRef]
- Guilliams, T.; El-Turk, F.; Buell, A.K.; O’Day, E.M.; Aprile, F.A.; Esbjorner, E.K.; Vendruscolo, M.; Cremades, N.; Pardon, E.; Wyns, L.; et al. Nanobodies raised against monomeric alpha-synuclein distinguish between fibrils at different maturation stages. J. Mol. Boil. 2013, 425, 2397–2411. [Google Scholar] [CrossRef]
- Iljina, M.; Hong, L.; Horrocks, M.H.; Ludtmann, M.H.; Choi, M.L.; Hughes, C.D.; Ruggeri, F.S.; Guilliams, T.; Buell, A.K.; Lee, J.E.; et al. Nanobodies raised against monomeric a-synuclein inhibit fibril formation and destabilize toxic oligomeric species. BMC Boil. 2017, 15, 57. [Google Scholar] [CrossRef]
- Vuchelen, A.; O’Day, E.; De Genst, E.; Pardon, E.; Wyns, L.; Dumoulin, M.; Dobson, C.M.; Christodoulou, J.; Hsu, S.T. 1H, 13C and 15N assignments of a camelid nanobody directed against human alpha-synuclein. Biomol. NMR Assign. 2009, 3, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Zhou, C.; Messer, A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J. Mol. Boil. 2008, 377, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.C.; Joshi, S.N.; Genst, E.; Baghel, A.S.; Dobson, C.M.; Messer, A. Bifunctional Anti-Non-Amyloid Component alpha-Synuclein Nanobodies Are Protective In Situ. PLoS ONE 2016, 11, e0165964. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an alpha-synuclein-based Parkinson’s disease model. NPJ Park. Dis. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.C.; McLear, J.A.; Messer, A. Engineered antibody therapies to counteract mutant huntingtin and related toxic intracellular proteins. Prog. Neurobiol. 2012, 97, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Garg, P.; Holden, T.; Chao, G.; Webster, J.M.; Messer, A.; Ingram, V.M.; Wittrup, K.D. Development of a human light chain variable domain (V(L)) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J. Mol. Boil. 2004, 342, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Chu, Y.; Cassady, J.P.; Duennwald, M.; Zazulak, H.; Webster, J.M.; Messer, A.; Lindquist, S.; Ingram, V.M.; Wittrup, K.D. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc. Natl. Acad. Sci. USA 2004, 101, 17616–17621. [Google Scholar] [CrossRef]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Khoshnan, A.; Dunn, D.E.; Bugg, C.W.; Lo, D.C.; Patterson, P.H. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J. Neurosci. 2008, 28, 9013–9020. [Google Scholar] [CrossRef] [PubMed]
- Schut, M.H.; Pepers, B.A.; Klooster, R.; van der Maarel, S.M.; El Khatabi, M.; Verrips, T.; den Dunnen, J.T.; van Ommen, G.J.; van Roon-Mom, W.M. Selection and characterization of llama single domain antibodies against N-terminal huntingtin. Neurol. Sci. 2015, 36, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A. Cellular biology of prion diseases. Clin. Microbiol. Rev. 1999, 12, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Ladogana, A.; Puopolo, M.; Croes, E.A.; Budka, H.; Jarius, C.; Collins, S.; Klug, G.M.; Sutcliffe, T.; Giulivi, A.; Alperovitch, A.; et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 2005, 64, 1586–1591. [Google Scholar] [CrossRef]
- Jones, D.R.; Taylor, W.A.; Bate, C.; David, M.; Tayebi, M. A camelid anti-PrP antibody abrogates PrP replication in prion-permissive neuroblastoma cell lines. PLoS ONE 2010, 5, e9804. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-terminal beta-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Veliz, I.; Loo, Y.; Castillo, O.; Karachaliou, N.; Nigro, O.; Rosell, R. Advances and challenges in the molecular biology and treatment of glioblastoma-is there any hope for the future? Ann. Transl. Med. 2015, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Shen, M.M. The roots of cancer: Stem cells and the basis for tumor heterogeneity. BioEssays News Rev. Mol. Cell. Dev. Boil. 2013, 35, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Bourkoula, E.; Mangoni, D.; Ius, T.; Pucer, A.; Isola, M.; Musiello, D.; Marzinotto, S.; Toffoletto, B.; Sorrentino, M.; Palma, A.; et al. Glioma-associated stem cells: A novel class of tumor-supporting cells able to predict prognosis of human low-grade gliomas. Stem Cells 2014, 32, 1239–1253. [Google Scholar] [CrossRef]
- Jovcevska, I.; Zupanec, N.; Kocevar, N.; Cesselli, D.; Podergajs, N.; Stokin, C.L.; Myers, M.P.; Muyldermans, S.; Ghassabeh, G.H.; Motaln, H.; et al. TRIM28 and beta-actin identified via nanobody-based reverse proteomics approach as possible human glioblastoma biomarkers. PLoS ONE 2014, 9, e113688. [Google Scholar] [CrossRef]
- Jovcevska, I.; Zupanec, N.; Urlep, Z.; Vranic, A.; Matos, B.; Stokin, C.L.; Muyldermans, S.; Myers, M.P.; Buzdin, A.A.; Petrov, I.; et al. Differentially expressed proteins in glioblastoma multiforme identified with a nanobody-based anti-proteome approach and confirmed by OncoFinder as possible tumor-class predictive biomarker candidates. Oncotarget 2017, 8, 44141–44158. [Google Scholar] [CrossRef]
- Samec, N.; Jovcevska, I.; Stojan, J.; Zottel, A.; Liovic, M.; Myers, M.P.; Muyldermans, S.; Sribar, J.; Krizaj, I.; Komel, R. Glioblastoma-specific anti-TUFM nanobody for in-vitro immunoimaging and cancer stem cell targeting. Oncotarget 2018, 9, 17282–17299. [Google Scholar] [CrossRef]
- Zorniak, M.; Clark, P.A.; Umlauf, B.J.; Cho, Y.; Shusta, E.V.; Kuo, J.S. Yeast display biopanning identifies human antibodies targeting glioblastoma stem-like cells. Sci. Rep. 2017, 7, 15840. [Google Scholar] [CrossRef] [PubMed]
- van Lith, S.A.; Roodink, I.; Verhoeff, J.J.; Makinen, P.I.; Lappalainen, J.P.; Yla-Herttuala, S.; Raats, J.; van Wijk, E.; Roepman, R.; Letteboer, S.J.; et al. In vivo phage display screening for tumor vascular targets in glioblastoma identifies a llama nanobody against dynactin-1-p150Glued. Oncotarget 2016, 7, 71594–71607. [Google Scholar] [CrossRef] [PubMed]
- van de Water, J.A.; Bagci-Onder, T.; Agarwal, A.S.; Wakimoto, H.; Roovers, R.C.; Zhu, Y.; Kasmieh, R.; Bhere, D.; Van Bergen en Henegouwen, P.M.; Shah, K. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proc. Natl. Acad. Sci. USA 2012, 109, 16642–16647. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef] [PubMed]
- Roovers, R.C.; Laeremans, T.; Huang, L.; De Taeye, S.; Verkleij, A.J.; Revets, H.; de Haard, H.J.; van Bergen en Henegouwen, P.M. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol. Immunother. CII 2007, 56, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J. Nucl. Med. 2016, 57, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Freise, A.C.; Wu, A.M. In vivo imaging with antibodies and engineered fragments. Mol. Immunol. 2015, 67, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.; Vaneycken, I.; D’Huyvetter, M.; Heemskerk, J.; Keyaerts, M.; Vincke, C.; Devoogdt, N.; Muyldermans, S.; Lahoutte, T.; Caveliers, V. Synthesis, preclinical validation, dosimetry, and toxicity of 68Ga-NOTA-anti-HER2 Nanobodies for iPET imaging of HER2 receptor expression in cancer. J. Nucl. Med. 2013, 54, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Krippendorff, B.F.; Sharma, S.; Walz, A.C.; Lave, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. mAbs 2016, 8, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Vandesquille, M.; Koukouli, F.; Dudeffant, C.; Youssef, I.; Lenormand, P.; Ganneau, C.; Maskos, U.; Czech, C.; Grueninger, F.; et al. Camelid single-domain antibodies: A versatile tool for in vivo imaging of extracellular and intracellular brain targets. J. Control. Release 2016, 243, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, U.; Trojahn, U.; Albaghdadi, H.; Zhang, J.; O’Connor-McCourt, M.; Stanimirovic, D.; Tomanek, B.; Sutherland, G.; Abulrob, A. Kinetic analysis of novel mono- and multivalent VHH-fragments and their application for molecular imaging of brain tumours. Br. J. Pharmacol. 2010, 160, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Daumas-Duport, C.; Varlet, P.; Tucker, M.L.; Beuvon, F.; Cervera, P.; Chodkiewicz, J.P. Oligodendrogliomas. Part I: Patterns of growth, histological diagnosis, clinical and imaging correlations: A study of 153 cases. J. Neuro-Oncol. 1997, 34, 37–59. [Google Scholar] [CrossRef]
- Daumas-Duport, C.; Tucker, M.L.; Kolles, H.; Cervera, P.; Beuvon, F.; Varlet, P.; Udo, N.; Koziak, M.; Chodkiewicz, J.P. Oligodendrogliomas. Part II: A new grading system based on morphological and imaging criteria. J. Neuro-Oncol. 1997, 34, 61–78. [Google Scholar] [CrossRef]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Boil. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Varner, J.A.; Cheresh, D.A. Integrins and cancer. Curr. Opin. Cell Boil. 1996, 8, 724–730. [Google Scholar] [CrossRef]
- Pen, A.; Moreno, M.J.; Martin, J.; Stanimirovic, D.B. Molecular markers of extracellular matrix remodeling in glioblastoma vessels: Microarray study of laser-captured glioblastoma vessels. Glia 2007, 55, 559–572. [Google Scholar] [CrossRef]
- Iqbal, U.; Albaghdadi, H.; Luo, Y.; Arbabi, M.; Desvaux, C.; Veres, T.; Stanimirovic, D.; Abulrob, A. Molecular imaging of glioblastoma multiforme using anti-insulin-like growth factor-binding protein-7 single-domain antibodies. Br. J. Cancer 2010, 103, 1606–1616. [Google Scholar] [CrossRef]
- Nagakubo, D.; Murai, T.; Tanaka, T.; Usui, T.; Matsumoto, M.; Sekiguchi, K.; Miyasaka, M. A high endothelial venule secretory protein, mac25/angiomodulin, interacts with multiple high endothelial venule-associated molecules including chemokines. J. Immunol. 2003, 171, 553–561. [Google Scholar] [CrossRef]
- Iqbal, U.; Albaghdadi, H.; Nieh, M.P.; Tuor, U.I.; Mester, Z.; Stanimirovic, D.; Katsaras, J.; Abulrob, A. Small unilamellar vesicles: A platform technology for molecular imaging of brain tumors. Nanotechnology 2011, 22, 195102. [Google Scholar] [CrossRef] [PubMed]
- Tomanek, B.; Iqbal, U.; Blasiak, B.; Abulrob, A.; Albaghdadi, H.; Matyas, J.R.; Ponjevic, D.; Sutherland, G.R. Evaluation of brain tumor vessels specific contrast agents for glioblastoma imaging. Neuro-Oncology 2012, 14, 53–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pardridge, W.M. Blood-brain barrier drug targeting: The future of brain drug development. Mol. Interv. 2003, 3, 90–105, 151. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Keliher, E.J.; Bilate, A.M.; Duarte, J.N.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Cragnolini, J.; Swee, L.K.; Victora, G.D.; Weissleder, R.; et al. Noninvasive imaging of immune responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6146–6151. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, U.; Abulrob, A.; Stanimirovic, D.B. Integrated platform for brain imaging and drug delivery across the blood-brain barrier. Methods Mol. Boil. 2011, 686, 465–481. [Google Scholar] [CrossRef]
- Herve, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Bickel, U.; Yoshikawa, T.; Pardridge, W.M. Delivery of peptides and proteins through the blood-brain barrier. Adv. Drug Deliv. Rev. 2001, 46, 247–279. [Google Scholar] [CrossRef]
- Li, T.; Bourgeois, J.P.; Celli, S.; Glacial, F.; Le Sourd, A.M.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Rougeon, F.; et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012, 26, 3969–3979. [Google Scholar] [CrossRef]
- Traenkle, B.; Emele, F.; Anton, R.; Poetz, O.; Haeussler, R.S.; Maier, J.; Kaiser, P.D.; Scholz, A.M.; Nueske, S.; Buchfellner, A.; et al. Monitoring interactions and dynamics of endogenous beta-catenin with intracellular nanobodies in living cells. Mol. Cell. Proteom. MCP 2015, 14, 707–723. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Product Name | Target | sdAb Type | Source | References |
|---|---|---|---|---|
| BBB Shuttles | ||||
| TBX4 | TfR1 | VNAR | Synthetic phage-displayed VNAR library | [66] |
| IGF1R-3 | IGF1R | VHH | Immune phage-displayed VHH library | [75,76] |
| FC5 | TMEM-30A | VHH | Non-immune phage-displayed VHH library | [20,70,71,72,73,74] |
| FC44 | Unknown | VHH | Non-immune phage-displayed VHH library | [20,70,71] |
| Neurodegenerative diseases | ||||
| Alzheimer’s disease | ||||
| B10 | Mature Aβ (1-40) fribrils and protofibrils | VHH | Synthetic phage-displayed VHH library | [84] |
| KW1 | Non-fibrillar Aβ (1-40) oligomers | VHH | Synthetic phage-displayed VHH library | [86,87] |
| ni3A | Aβ (1-42) deposits | VHH | Non-immune phage-displayed VHH library | [88,89,90] |
| V31-1 | Monomers and small Aβ (1-42) oligomers | VHH | Immune phage-displayed VHH library | [91] |
| PrioAD12 | Aβ (1-40) peptide | VHH | Immune phage-displayed VHH library | [92] |
| PrioAD13 | Aβ (1-42) peptide | VHH | Immune phage-displayed VHH library | [92] |
| PrioAD120 | Tau (1-16) peptide | VHH | Immune phage-displayed VHH library | [92] |
| VH1.27, VH1.28, VH2.8 | Aβ (1-42) peptide | VH | Immune phage-displayed mouse VH library | [93] |
| Aβ (1-10), Aβ (3-12), Aβ (6-15), Aβ (9-18), Aβ (12-21), Aβ (15-24), Aβ (18-27), Aβ (21-30), Aβ (24-33), Aβ (27-36), Aβ (30-39), Aβ (33-42) | AB monomers, soluble oligomers or fibrils | VH | Grafted amyloid-motif antibodies (Gammabody) | [94,95,96,97] |
| DesAb-Aβ | Aβ (15-21) peptide | VH | Gammabody | [98] |
| Parkinson’s disease | ||||
| αSyn (69-78) | αSyn fibrils | VH | Gammabody | [95] |
| DesAb-D, DesAb-E, DesAb-F | αSyn (61-67) or αSyn (70-76) peptide | VH | Gammabody | [98] |
| NbSyn2 | Monomeric αSyn and mature fibrils | VHH | Immune phage-displayed VHH library | [101,102,104] |
| NbSyn87 | Monomeric αSyn(A53T) and mature fibrils | VHH | Immune phage-displayed VHH library | [102,103,107,108] |
| VH14 | Monomeric αSyn | VH | Non-immune yeast-displayed human scFv library | [106,107,108] |
| Huntington’s disease | ||||
| VL12.3 | Htt protein | VL | Non-immune yeast-displayed human scFv library | [110,111,112,113] |
| Happ1, Happ3 | Htt protein | VL | Non-immune phage-displayed human scFv library | [112,113] |
| iVHH1, iVHH2, iVHH3, iVHH4 | Htt protein | VHH | Immune phage-displayed VHH library | [114] |
| Prion diseases | ||||
| PrioV3 | PrPc and PrPsc | VHH | Immune phage-displayed VHH library | [92,117] |
| Nb484 | MoPrP (23-230) | VHH | Immune phage-displayed VHH library | [118] |
| Glioblastoma multiforme | ||||
| Nb237 | TRIM28 | VHH | Immune phage-displayed VHH library | [122] |
| Nb141 | β-actin | VHH | Immune phage-displayed VHH library | [122] |
| Nb10 | ACTB/NUCL complex | VHH | Immune phage-displayed VHH library | [123] |
| Nb79 | VIM | VHH | Immune phage-displayed VHH library | [123] |
| Nb179 | NAP1L1 | VHH | Immune phage-displayed VHH library | [123] |
| Nb225 | TUFM | VHH | Immune phage-displayed VHH library | [123] |
| Nb314 | DPYSL2 and MTHFD1 | VHH | Immune phage-displayed VHH library | [123] |
| Nb394 | CRMP1 | VHH | Immune phage-displayed VHH library | [123] |
| Nb395 | ALYREF | VHH | Immune phage-displayed VHH library | [123] |
| Nb206 | TUFM | VHH | Immune phage-displayed VHH library | [124] |
| VH-9.7 | GSC | VH | Non-immune yeast-displayed human scFv library | [125] |
| C-C7 | Dynactin-1-p150Glued | VHH | Non-immune phage-displayed VHH library | [126] |
| ENb1, ENb2 | EGFR | VHH | Immune phage-displayed VHH library | [127,130] |
| Neuroimaging | ||||
| EG(2) | EGFR | VHH | Immune phage-displayed VHH library | [136] |
| sdAb 4.43 | IGFBP7 | VHH | Immune phage-displayed VHH library | [142,144,145] |
| FC5 | TMEM-30A | VHH | Non-immune phage-displayed VHH library | [148] |
| R3VQ | Aβ (1-42) peptide | VHH | Immune phage-displayed VHH library | [135] |
| A2 | Phospho-Tau protein | VHH | Immune phage-displayed VHH library | [135] |
| mVHH A10, mVHH E9, mVHH E3 | GFAP | VHH | Immune ribosome-displayed VHH library | [151] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bélanger, K.; Iqbal, U.; Tanha, J.; MacKenzie, R.; Moreno, M.; Stanimirovic, D. Single-Domain Antibodies as Therapeutic and Imaging Agents for the Treatment of CNS Diseases. Antibodies 2019, 8, 27. https://doi.org/10.3390/antib8020027
Bélanger K, Iqbal U, Tanha J, MacKenzie R, Moreno M, Stanimirovic D. Single-Domain Antibodies as Therapeutic and Imaging Agents for the Treatment of CNS Diseases. Antibodies. 2019; 8(2):27. https://doi.org/10.3390/antib8020027
Chicago/Turabian StyleBélanger, Kasandra, Umar Iqbal, Jamshid Tanha, Roger MacKenzie, Maria Moreno, and Danica Stanimirovic. 2019. "Single-Domain Antibodies as Therapeutic and Imaging Agents for the Treatment of CNS Diseases" Antibodies 8, no. 2: 27. https://doi.org/10.3390/antib8020027
APA StyleBélanger, K., Iqbal, U., Tanha, J., MacKenzie, R., Moreno, M., & Stanimirovic, D. (2019). Single-Domain Antibodies as Therapeutic and Imaging Agents for the Treatment of CNS Diseases. Antibodies, 8(2), 27. https://doi.org/10.3390/antib8020027