Canine CD117-Specific Antibodies with Diverse Binding Properties Isolated from a Phage Display Library Using Cell-Based Biopanning

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning Protocol for Canine CD117-GFP and SCF
2.2. Constructing a pcDNA 3.1 (+)-ECD-CD117-hFc Plasmid for Producing Recombinant Canine CD117-ECD-Human-Fc Fused Protein
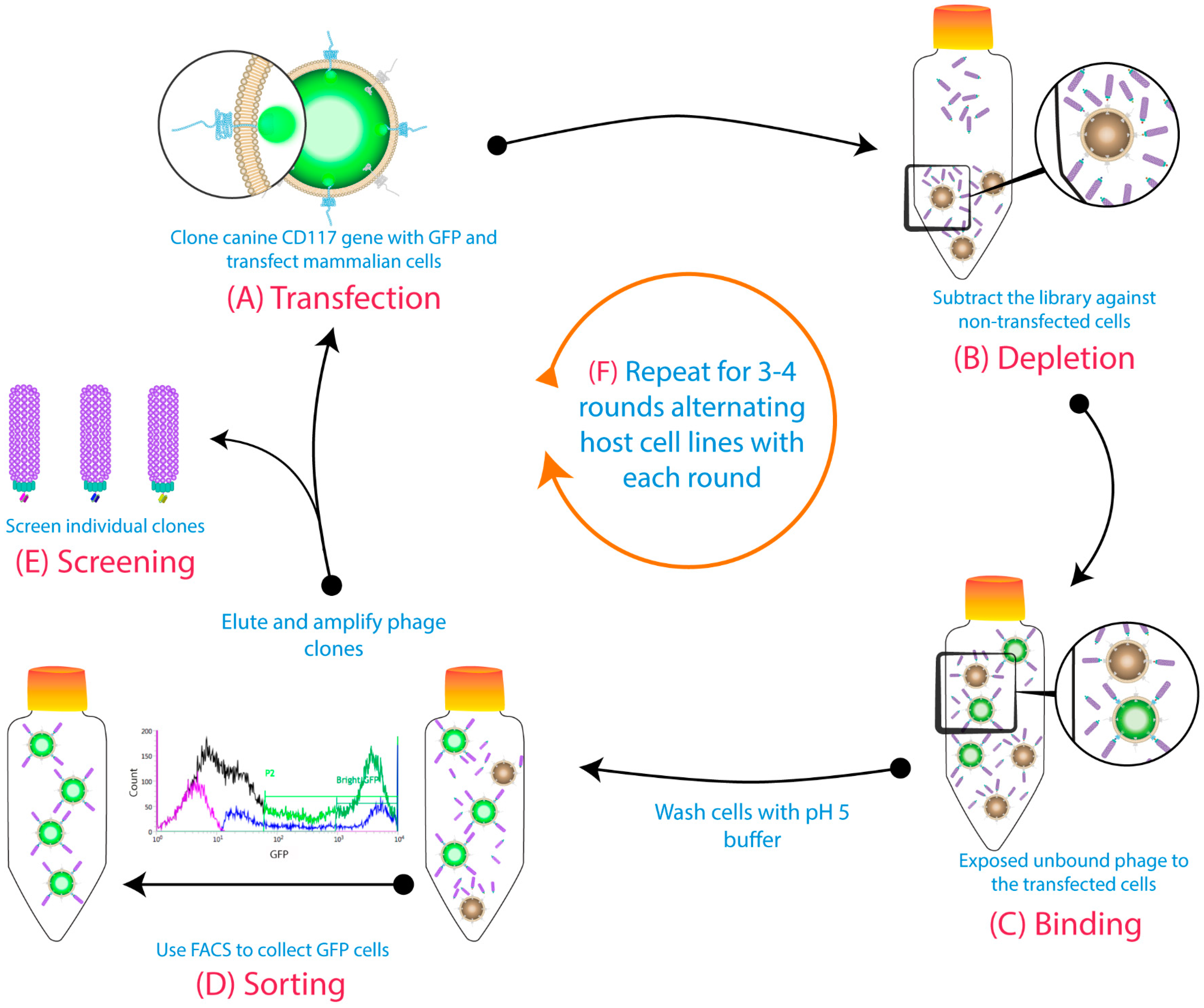
2.3. Biopanning
2.4. Reformatting of Phage Clones
2.5. Protein Expression in CHO Cells
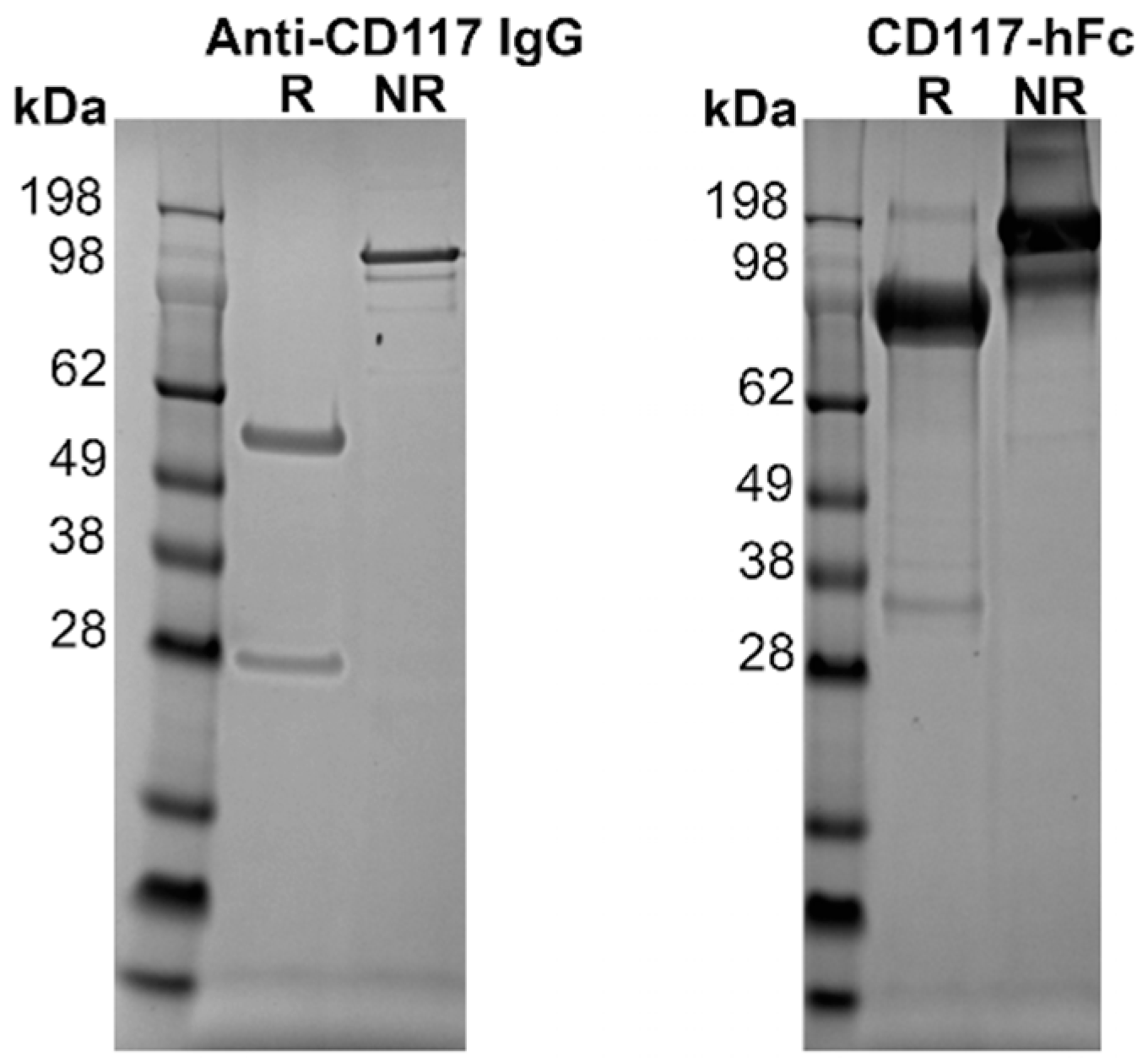
2.6. Protein Purification
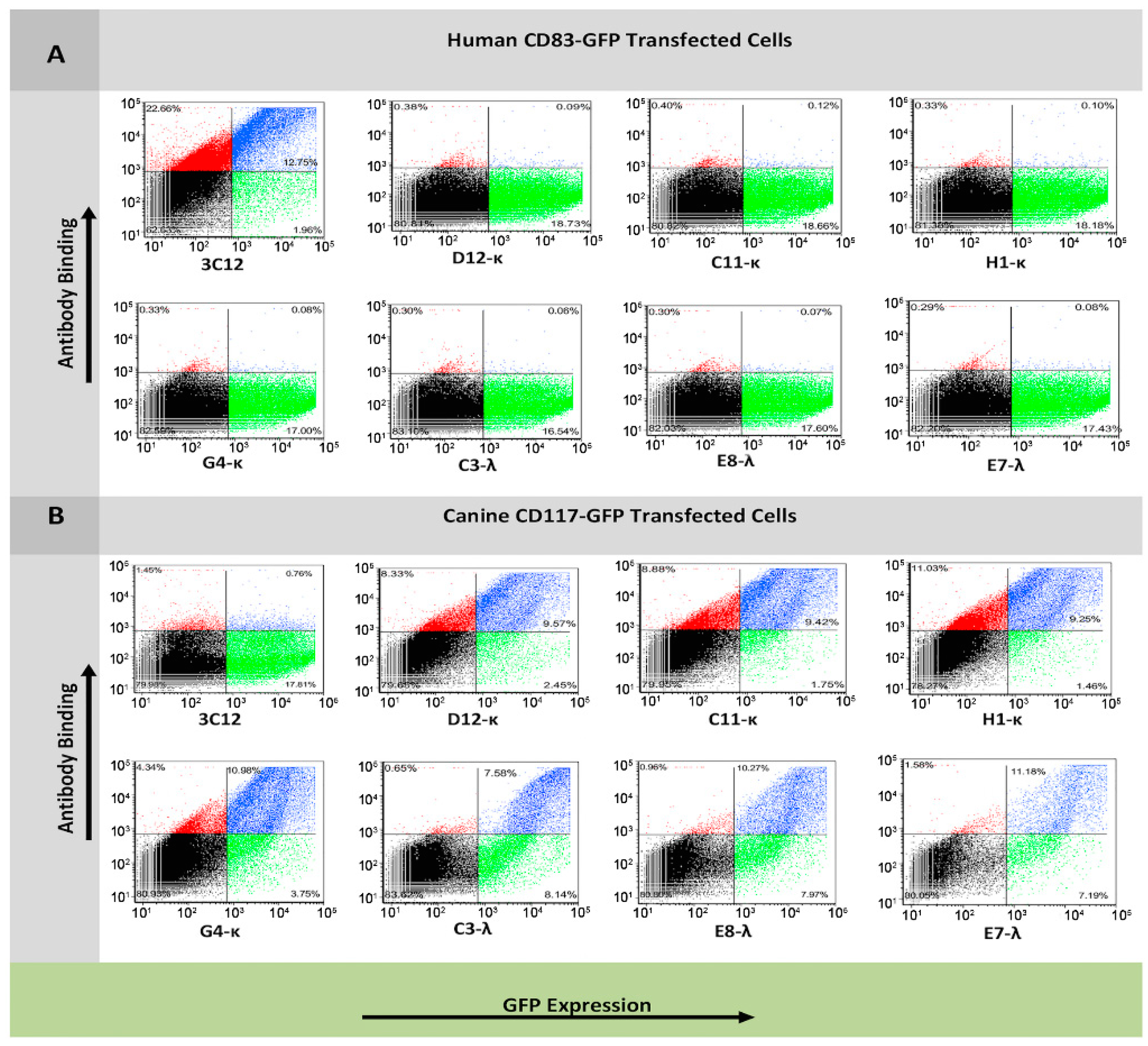
2.7. Flow Cytometry Analysis of Anti-CD83 and Anti-CD117 Reformatted IgGs
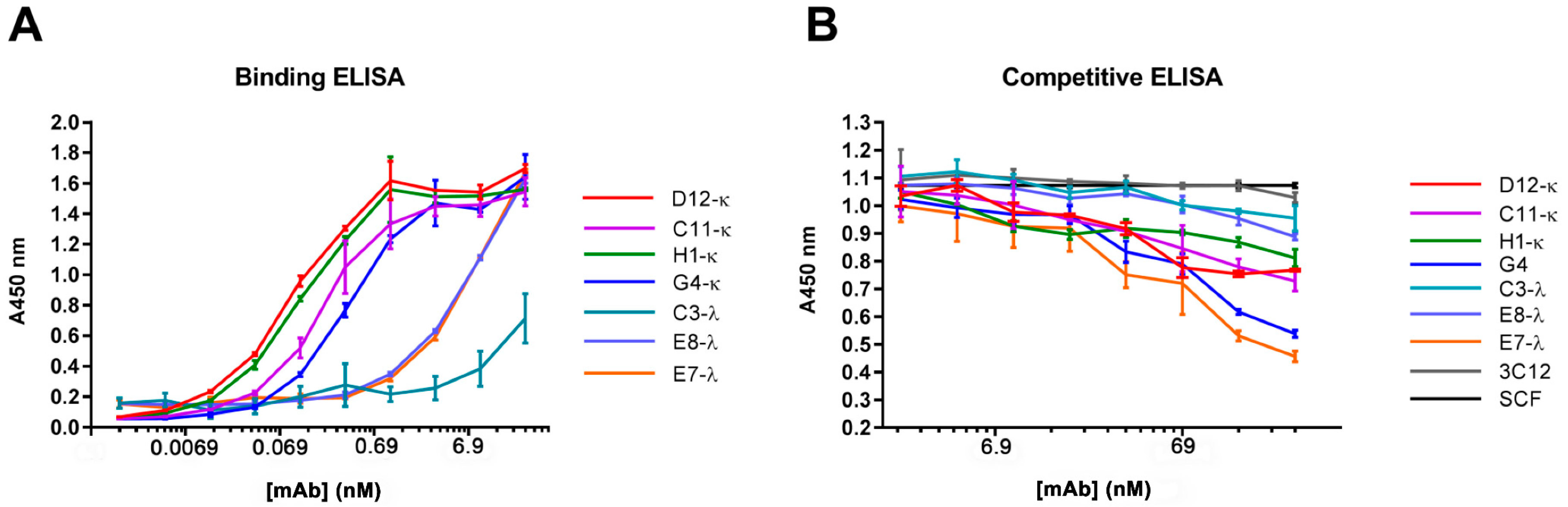
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Affinity and Kinetics Analysis Using Surface Plasmon Resonance
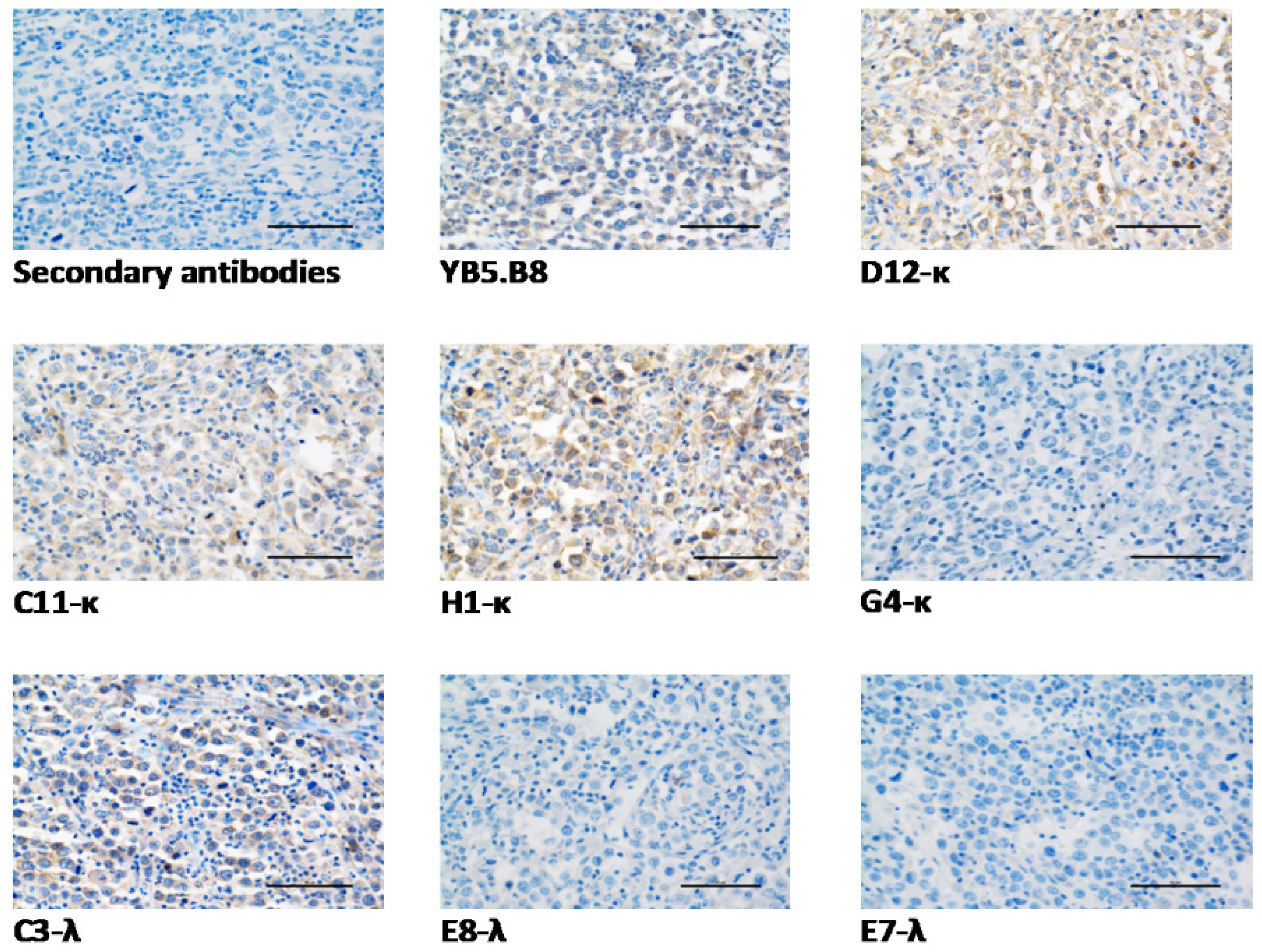
2.10. Immunohistochemistry (IHC)
2.11. Adherent and Non-Adherent Spheres Culture
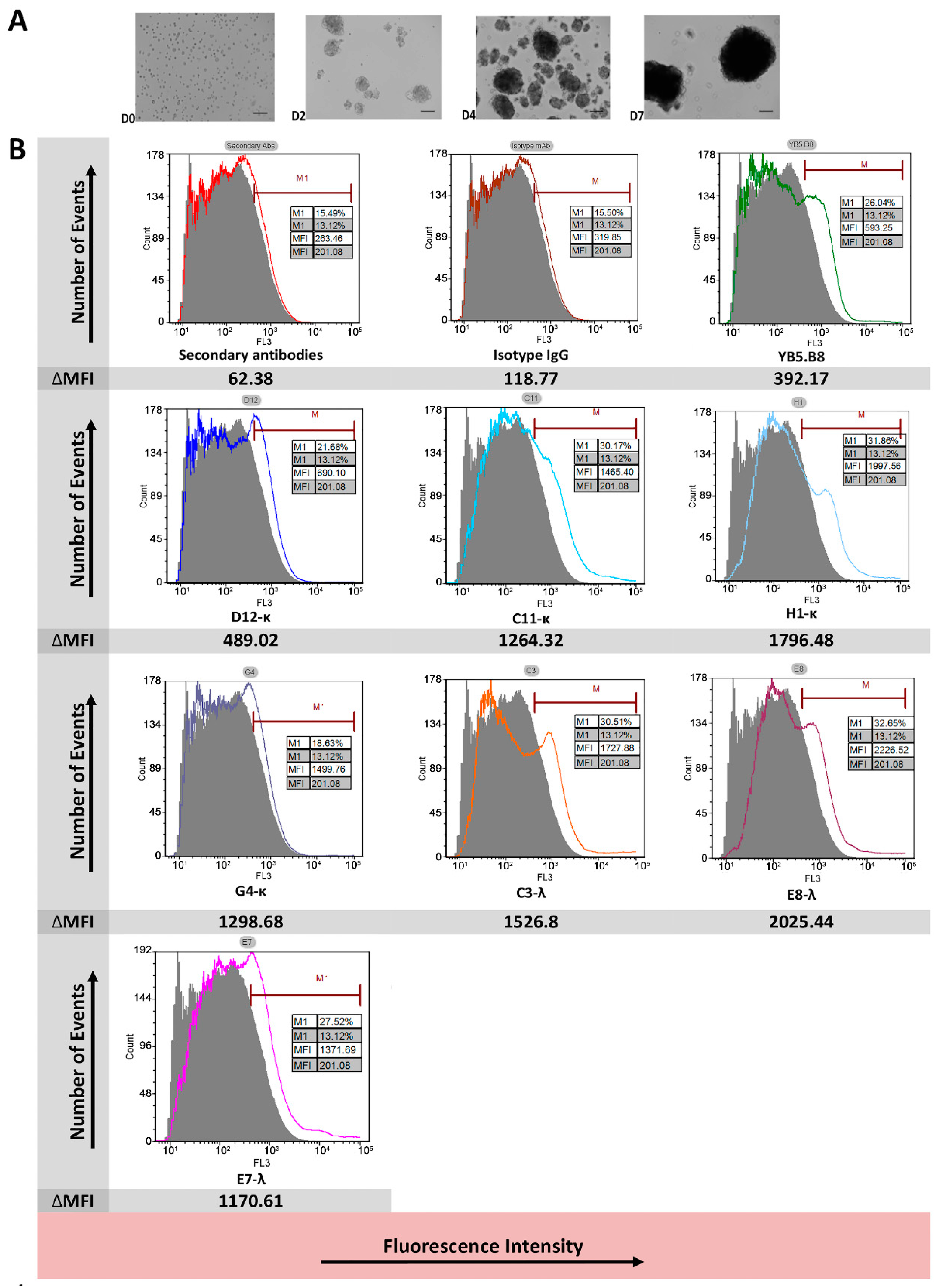
2.12. Flow Cytometry of Canine Hemangiospheres Cell Line DD1
3. Results
3.1. Biopanning
3.2. Expression of Proteins
3.3. Flow Cytometry Analysis of Antibody Binding to Recombinant Cell Surface Canine CD117
3.4. Binding and Competition Assays Using ELISA
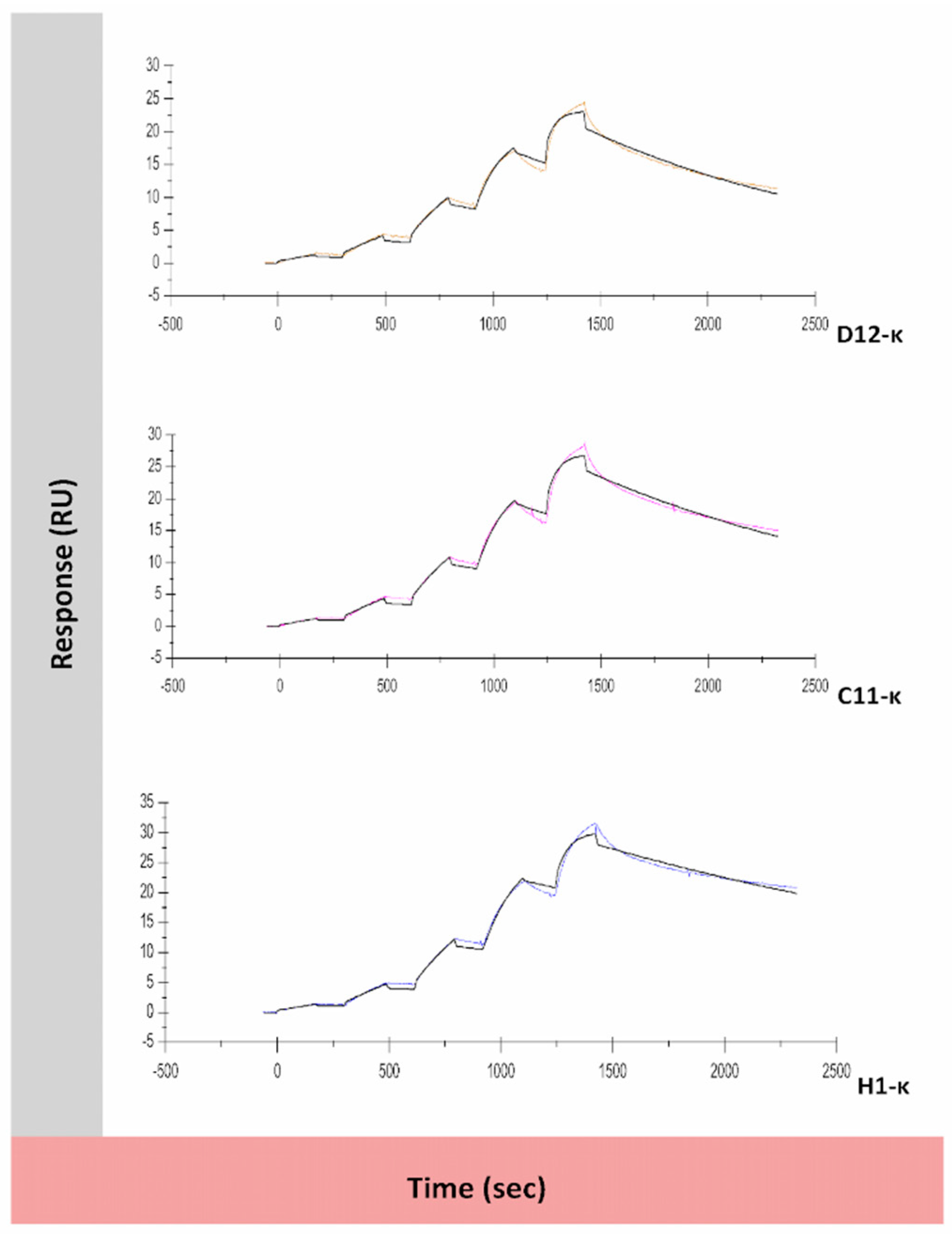
3.5. Binding Assessment of Canine-Specific Anti-CD117 mAbs to Recombinant Canine CD117 Target Using Surface Plasmon Resonance (SPR)
3.6. Anti-Canine CD117 mAbs Binding to Native Canine CD117
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Arce, J.; Sturgis, J.N.; Duneau, J.P. Dissecting membrane protein architecture: An annotation of structural complexity. Biopolymers 2009, 91, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Trail, P. Antibody drug conjugates as cancer therapeutics. Antibodies 2013, 2, 113–129. [Google Scholar] [CrossRef]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Goodall, S.; Jones, M.L.; Mahler, S. Monoclonal antibody-targeted polymeric nanoparticles for cancer therapy—Future prospects. J. Chem. Technol. Biotechnol. 2015, 90, 1169–1176. [Google Scholar] [CrossRef]
- Alfaleh, M.A.; Howard, C.B.; Sedliarou, I.; Jones, M.L.; Gudhka, R.; Vanegas, N.; Weiss, J.; Suurbach, J.H.; de Bakker, C.J.; Milne, M.R.; et al. Targeting mesothelin receptors with drug-loaded bacterial nanocells suppresses human mesothelioma tumour growth in mouse xenograft models. PLoS ONE 2017, 12, e0186137. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Winter, G.; Milstein, C. Man-made antibodies. Nature 1991, 349, 293–299. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Forood, B.; Houghten, R.A.; Perez-Paya, E. Secondary structure induction in aqueous vs membrane-like environments. Biopolymers 1997, 42, 489–498. [Google Scholar] [CrossRef]
- Cattaneo, A. Tanezumab, a recombinant humanized mab against nerve growth factor for the treatment of acute and chronic pain. Curr. Opin. Mol. 2010, 12, 94–106. [Google Scholar]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Monne, M.; Chan, K.W.; Slotboom, D.J.; Kunji, E.R. Functional expression of eukaryotic membrane proteins in lactococcus lactis. Protein Sci. 2005, 14, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Alfaleh, M.; Jones, M.; Howard, C.; Mahler, S. Strategies for selecting membrane protein-specific antibodies using phage display with cell-based panning. Antibodies 2017, 6, 10. [Google Scholar] [CrossRef]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Edling, C.E.; Hallberg, B. C-kit—A hematopoietic cell essential receptor tyrosine kinase. Int. J. Biochem. Cell Biol. 2007, 39, 1995–1998. [Google Scholar] [CrossRef]
- Letard, S.; Yang, Y.; Hanssens, K.; Palmerini, F.; Leventhal, P.S.; Guery, S.; Moussy, A.; Kinet, J.P.; Hermine, O.; Dubreuil, P. Gain-of-function mutations in the extracellular domain of kit are common in canine mast cell tumors. Mol. Cancer Res. MCR 2008, 6, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, S.; Ganesan, T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Zwick, E.; Bange, J.; Ullrich, A. Receptor tyrosine kinases as targets for anticancer drugs. Trends Mol. Med. 2002, 8, 17–23. [Google Scholar] [CrossRef]
- Shchemelinin, I.; Sefc, L.; Necas, E. Protein kinases, their function and implication in cancer and other diseases. Folia Biol. 2006, 52, 81–100. [Google Scholar]
- Kim, J.H.; Graef, A.J.; Dickerson, E.B.; Modiano, J.F. Pathobiology of hemangiosarcoma in dogs: Research advances and future perspectives. Vet. Sci. 2015, 2, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Alves, C.E.; Kobayashi, P.E.; Palmieri, C.; Laufer-Amorim, R. Investigation of c-kit and ki67 expression in normal, preneoplastic and neoplastic canine prostate. BMC Vet. Res. 2017, 13, 380. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Kisseberth, W.C.; Galli, S.J.; Geissler, E.N.; Helfand, S.C. Expression of stem cell factor receptor (c-kit) by the malignant mast cells from spontaneous canine mast cell tumours. J. Comp. Pathol. 1996, 115, 399–414. [Google Scholar] [CrossRef]
- Reguera, M.J.; Rabanal, R.M.; Puigdemont, A.; Ferrer, L. Canine mast cell tumors express stem cell factor receptor. Am. J. Dermatopathol. 2000, 22, 49–54. [Google Scholar] [CrossRef]
- Komuro, T. Comparative morphology of interstitial cells of cajal: Ultrastructural characterization. Microsc. Res. Tech. 1999, 47, 267–285. [Google Scholar] [CrossRef]
- Bettini, G.; Morini, M.; Marcato, P.S. Gastrointestinal spindle cell tumours of the dog: Histological and immunohistochemical study. J. Comp. Pathol. 2003, 129, 283–293. [Google Scholar] [CrossRef]
- Gillespie, V.; Baer, K.; Farrelly, J.; Craft, D.; Luong, R. Canine gastrointestinal stromal tumors: Immunohistochemical expression of cd34 and examination of prognostic indicators including proliferation markers ki67 and agnor. Vet. Pathol. 2011, 48, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Yuzbasiyan-Gurkan, V.; Kaneene, J.B.; Miller, R.; Resau, J.H.; Kiupel, M. The role of c-kit in tumorigenesis: Evaluation in canine cutaneous mast cell tumors. Neoplasia 2006, 8, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Yuzbasiyan-Gurkan, V.; Miller, R.A.; Kaneene, J.B.; Kiupel, M. Cellular proliferation in canine cutaneous mast cell tumors: Associations with c-kit and its role in prognostication. Vet. Pathol. 2007, 44, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R. The gist paradigm: Lessons for other kinase-driven cancers. J. Pathol. 2011, 223, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Blanke, C.D.; Druker, B.J.; Corless, C.L. Inhibition of kit tyrosine kinase activity: A novel molecular approach to the treatment of kit-positive malignancies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.Q.; Tang, Y.M.; Yang, S.L.; Qian, B.Q.; Song, H.; Shi, S.W.; Xu, W.Q. Analysis of cd117 expression on leukemia cells. Zhonghua Xue Ye Xue Za Zhi Zhonghua Xueyexue Zazhi 2003, 24, 228–230. [Google Scholar] [PubMed]
- Frost, D.; Lasota, J.; Miettinen, M. Gastrointestinal stromal tumors and leiomyomas in the dog: A histopathologic, immunohistochemical, and molecular genetic study of 50 cases. Vet. Pathol. 2003, 40, 42–54. [Google Scholar] [CrossRef]
- Gregory-Bryson, E.; Bartlett, E.; Kiupel, M.; Hayes, S.; Yuzbasiyan-Gurkan, V. Canine and human gastrointestinal stromal tumors display similar mutations in c-kit exon 11. BMC Cancer 2010, 10, 559. [Google Scholar] [CrossRef]
- London, C.A.; Galli, S.J.; Yuuki, T.; Hu, Z.Q.; Helfand, S.C.; Geissler, E.N. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp. Hematol. 1999, 27, 689–697. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Breen, M. Comparative oncology: What dogs and other species can teach us about humans with cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 19, 370. [Google Scholar] [CrossRef]
- Alvarez, C.E. Naturally occurring cancers in dogs: Insights for translational genetics and medicine. ILAR J./Natl. Res. Counc. Inst. Lab. Anim. Resour. 2014, 55, 16–45. [Google Scholar] [CrossRef] [PubMed]
- Kiupel, M.; Webster, J.D.; Kaneene, J.B.; Miller, R.; Yuzbasiyan-Gurkan, V. The use of kit and tryptase expression patterns as prognostic tools for canine cutaneous mast cell tumors. Vet. Pathol. 2004, 41, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.C.; Khanna, C. Comparative oncology today. Vet. Clin. N. Am. Small Anim. Pract. 2007, 37, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.K.; Mazcko, C.N.; Khanna, C. Defining the value of a comparative approach to cancer drug development. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Seldon, T.; Smede, M.; Linville, A.; Chin, D.Y.; Barnard, R.; Mahler, S.M.; Munster, D.; Hart, D.; Gray, P.P.; et al. A method for rapid, ligation-independent reformatting of recombinant monoclonal antibodies. J. Immunol. Methods 2010, 354, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kiupel, M.; Webster, J.D.; Bailey, K.L.; Best, S.; DeLay, J.; Detrisac, C.J.; Fitzgerald, S.D.; Gamble, D.; Ginn, P.E.; Goldschmidt, M.H.; et al. Proposal of a 2-tier histologic grading system for canine cutaneous mast cell tumors to more accurately predict biological behavior. Vet. Pathol. 2011, 48, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Lamerato-Kozicki, A.R.; Helm, K.M.; Jubala, C.M.; Cutter, G.C.; Modiano, J.F. Canine hemangiosarcoma originates from hematopoietic precursors with potential for endothelial differentiation. Exp. Hematol. 2006, 34, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Fosmire, S.P.; Dickerson, E.B.; Scott, A.M.; Bianco, S.R.; Pettengill, M.J.; Meylemans, H.; Padilla, M.; Frazer-Abel, A.A.; Akhtar, N.; Getzy, D.M.; et al. Canine malignant hemangiosarcoma as a model of primitive angiogenic endothelium. Lab. Investig. 2004, 84, 562–572. [Google Scholar] [CrossRef]
- Gorden, B.H.; Kim, J.H.; Sarver, A.L.; Frantz, A.M.; Breen, M.; Lindblad-Toh, K.; O’Brien, T.D.; Sharkey, L.C.; Modiano, J.F.; Dickerson, E.B. Identification of three molecular and functional subtypes in canine hemangiosarcoma through gene expression profiling and progenitor cell characterization. Am. J. Pathol. 2014, 184, 985–995. [Google Scholar] [CrossRef]
- Chen, G.; Gulbranson, D.R.; Yu, P.; Hou, Z.; Thomson, J.A. Thermal stability of fibroblast growth factor protein is a determinant factor in regulating self-renewal, differentiation, and reprogramming in human pluripotent stem cells. Stem Cells 2012, 30, 623–630. [Google Scholar] [CrossRef]
- Gorden, B.H.; Saha, J.; Khammanivong, A.; Schwartz, G.K.; Dickerson, E.B. Lysosomal drug sequestration as a mechanism of drug resistance in vascular sarcoma cells marked by high csf-1r expression. Vasc. Cell 2014, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Bavcar, S.; Argyle, D.J. Receptor tyrosine kinase inhibitors: Molecularly targeted drugs for veterinary cancer therapy. Vet. Comp. Oncol. 2012, 10, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Rebuzzi, L.; Mayerhofer, M.; Gruze, A.; Hadzijusufovic, E.; Sonneck, K.; Vales, A.; Kneidinger, M.; Samorapoompichit, P.; Thaiwong, T.; et al. Synergistic antiproliferative effects of kit tyrosine kinase inhibitors on neoplastic canine mast cells. Exp. Hematol. 2007, 35, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Kobie, K.; Kawabata, M.; Hioki, K.; Tanaka, A.; Matsuda, H.; Mori, T.; Maruo, K. The tyrosine kinase inhibitor imatinib [sti571] induces regression of xenografted canine mast cell tumors in scid mice. Res. Vet. Sci. 2007, 82, 239–241. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Hannah, A.L.; Zadovoskaya, R.; Chien, M.B.; Kollias-Baker, C.; Rosenberg, M.; Downing, S.; Post, G.; Boucher, J.; Shenoy, N.; et al. Phase i dose-escalating study of su11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin. Cancer Res. 2003, 9, 2755–2768. [Google Scholar] [PubMed]
- London, C.A.; Malpas, P.B.; Wood-Follis, S.L.; Boucher, J.F.; Rusk, A.W.; Rosenberg, M.P.; Henry, C.J.; Mitchener, K.L.; Klein, M.K.; Hintermeister, J.G.; et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (su11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- London, C.A. Tyrosine kinase inhibitors in veterinary medicine. Top. Companion Anim. Med. 2009, 24, 106–112. [Google Scholar] [CrossRef]
- Reber, L.; Da Silva, C.A.; Frossard, N. Stem cell factor and its receptor c-kit as targets for inflammatory diseases. Eur. J. Pharmacol. 2006, 533, 327–340. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M. A new twist in the transmembrane signaling tool-kit. Cell 2007, 130, 213–215. [Google Scholar] [CrossRef]
- Hassan, H.T. C-kit expression in human normal and malignant stem cells prognostic and therapeutic implications. Leuk. Res. 2009, 33, 5–10. [Google Scholar] [CrossRef]
- Philo, J.S.; Wen, J.; Wypych, J.; Schwartz, M.G.; Mendiaz, E.A.; Langley, K.E. Human stem cell factor dimer forms a complex with two molecules of the extracellular domain of its receptor, kit. J. Biol. Chem. 1996, 271, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Kusakabe, M.; Yoshinaga, K.; Ogawa, M.; Hayashi, S.; Kunisada, T.; Era, T.; Sakakura, T.; Nishikawa, S. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: Two distinct waves of c-kit-dependency during melanocyte development. EMBO J. 1991, 10, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, J.D.; Thuneberg, L.; Kluppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/kit gene required for interstitial cells of cajal and for intestinal pacemaker activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Foote, J.; Eisen, H.N. Kinetic and affinity limits on antibodies produced during immune responses. Proc. Natl. Acad. Sci. USA 1995, 92, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.R.; Jensen, A.; Haurum, J.S.; Andersen, P.S. Limits for antibody affinity maturation and repertoire diversification in hypervaccinated humans. J. Immunol. 2011, 187, 4229–4235. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Chen, G.; Iverson, B.L.; Georgiou, G. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain fv antibodies. Proc. Natl. Acad. Sci. USA 2000, 97, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Sela, M.; Schechter, B.; Schechter, I.; Borek, F. Antibodies to sequential and conformational determinants. Cold Spring Harb. Symp. Quant. Biol. 1967, 32, 537–545. [Google Scholar] [CrossRef]
- Barlow, D.J.; Edwards, M.S.; Thornton, J.M. Continuous and discontinuous protein antigenic determinants. Nature 1986, 322, 747–748. [Google Scholar] [CrossRef]
- Huang, J.; Honda, W. Ced: A conformational epitope database. BMC Immunol. 2006, 7, 7. [Google Scholar] [CrossRef]
- Sompuram, S.R.; Vani, K.; Hafer, L.J.; Bogen, S.A. Antibodies immunoreactive with formalin-fixed tissue antigens recognize linear protein epitopes. Am. J. Clin. Pathol. 2006, 125, 82–90. [Google Scholar] [CrossRef]
- Scalia, C.R.; Gendusa, R.; Basciu, M.; Riva, L.; Tusa, L.; Musaro, A.; Veronese, S.; Formenti, A.; D’Angelo, D.; Ronzio, A.G.; et al. Epitope recognition in the human-pig comparison model on fixed and embedded material. J. Histochem. Cytochem. 2015, 63, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Stradleigh, T.W.; Ishida, A.T. Fixation strategies for retinal immunohistochemistry. Prog. Retin. Eye Res. 2015, 48, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Mok, P.; Balakrishnan, T.; Rahmat, S.N.; Zweigerdt, R. Up-scaling single cell-inoculated suspension culture of human embryonic stem cells. Stem Cell Res. 2010, 4, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Liao, J.W.; Hsu, W.L.; Chang, S.C. Identification of the two kit isoforms and their expression status in canine hemangiosarcomas. BMC Vet. Res. 2016, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Preziosi, R.; Morini, M.; Sarli, G. Expression of the kit protein (cd117) in primary cutaneous mast cell tumors of the dog. J. Vet. Diagn. Investig. Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc. 2004, 16, 554–561. [Google Scholar] [CrossRef]
- Sailasuta, A.; Ketpun, D.; Piyaviriyakul, P.; Theerawatanasirikul, S.; Theewasutrakul, P.; Rungsipipat, A. The relevance of cd117-immunocytochemistry staining patterns to mutational exon-11 in c-kit detected by pcr from fine-needle aspirated canine mast cell tumor cells. Vet. Med. Int. 2014, 2014, 787498. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Fazekas, J.; Wang, W.; Weichselbaumer, M.; Matz, M.; Mader, A.; Steinfellner, W.; Meitz, S.; Mechtcheriakova, D.; Sobanov, Y.; et al. Generation of a canine anti-egfr (erbb-1) antibody for passive immunotherapy in dog cancer patients. Mol. Cancer Ther. 2014, 13, 1777–1790. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selection Round | Cell Line | Transfected Cells (cells/mL) | FACS High-GFP Sorted Cells | Phage Input Titer (cfu) | Phage Output Titer (cfu) |
|---|---|---|---|---|---|
| Round 1 | CHOs | 1.3 × 106 | ~3.0 × 106 | 1 × 1013 | 1.8 × 105 |
| Round 2 | HEK293 | 1.0 × 106 | ~3.0 × 106 | 6.8 × 1012 | 2.1 × 105 |
| Round 3 | CHOs | 2 × 106 | ~2.5 × 106 | 5.2 × 1012 | 2.3 × 105 |
| Round 4 | HEK293 | 1.2× 106 | ~3.0 × 106 | 9.2 × 1010 | 8.8 × 105 |
| mAb | ka (M−1 S−1) | kd (S−1) | KD (M) | Rmax (RU) | Chi² (RU²) | Theoretical Rmax (RU) |
|---|---|---|---|---|---|---|
| D12-κ | 8.1 × 105 | 7.5 × 10−4 | 9.3 × 10−10 | 21.4 | 0.382 | 77.9 |
| C11-κ | 7 × 105 | 6.1 × 10−4 | 8.8 × 10−10 | 25.5 | 0.487 | 77.9 |
| H1-κ | 6.6 × 105 | 3.9 × 10−4 | 5.9 × 10−10 | 29.0 | 0.513 | 77.9 |
| mAb | Recombinant Canine CD117 | Native Canine CD117 | |||||
|---|---|---|---|---|---|---|---|
| Flow Cytometry | ELISA | SPR | Ligand Blocking | IHC | Flow Cytometry | Suggested Epitope Type | |
| D12-κ | +4 | +4 | +4 | +2 | √ | +1 | Linear |
| C11-κ | +4 | +3 | +4 | +2 | √ | +3 | Linear |
| H1-κ | +4 | +4 | +4 | +2 | √ | +3 | Linear |
| G4-κ | +3 | +3 | +1 | +3 | × | +1 | Conformational |
| C3-λ | +2 | +1 | 0 | +1 | √ | +3 | Linear |
| E8-λ | +2 | +2 | +1 | +1 | × | +4 | Conformational |
| E7-λ | +2 | +2 | +1 | +3 | × | +3 | Conformational |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfaleh, M.A.; Arora, N.; Yeh, M.; de Bakker, C.J.; Howard, C.B.; Macpherson, P.; Allavena, R.E.; Chen, X.; Harkness, L.; Mahler, S.M.; et al. Canine CD117-Specific Antibodies with Diverse Binding Properties Isolated from a Phage Display Library Using Cell-Based Biopanning. Antibodies 2019, 8, 15. https://doi.org/10.3390/antib8010015
Alfaleh MA, Arora N, Yeh M, de Bakker CJ, Howard CB, Macpherson P, Allavena RE, Chen X, Harkness L, Mahler SM, et al. Canine CD117-Specific Antibodies with Diverse Binding Properties Isolated from a Phage Display Library Using Cell-Based Biopanning. Antibodies. 2019; 8(1):15. https://doi.org/10.3390/antib8010015
Chicago/Turabian StyleAlfaleh, Mohamed A., Neetika Arora, Michael Yeh, Christopher J. de Bakker, Christopher B. Howard, Philip Macpherson, Rachel E. Allavena, Xiaoli Chen, Linda Harkness, Stephen M. Mahler, and et al. 2019. "Canine CD117-Specific Antibodies with Diverse Binding Properties Isolated from a Phage Display Library Using Cell-Based Biopanning" Antibodies 8, no. 1: 15. https://doi.org/10.3390/antib8010015
APA StyleAlfaleh, M. A., Arora, N., Yeh, M., de Bakker, C. J., Howard, C. B., Macpherson, P., Allavena, R. E., Chen, X., Harkness, L., Mahler, S. M., & Jones, M. L. (2019). Canine CD117-Specific Antibodies with Diverse Binding Properties Isolated from a Phage Display Library Using Cell-Based Biopanning. Antibodies, 8(1), 15. https://doi.org/10.3390/antib8010015