Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Monoclonal Antibody Based Therapy
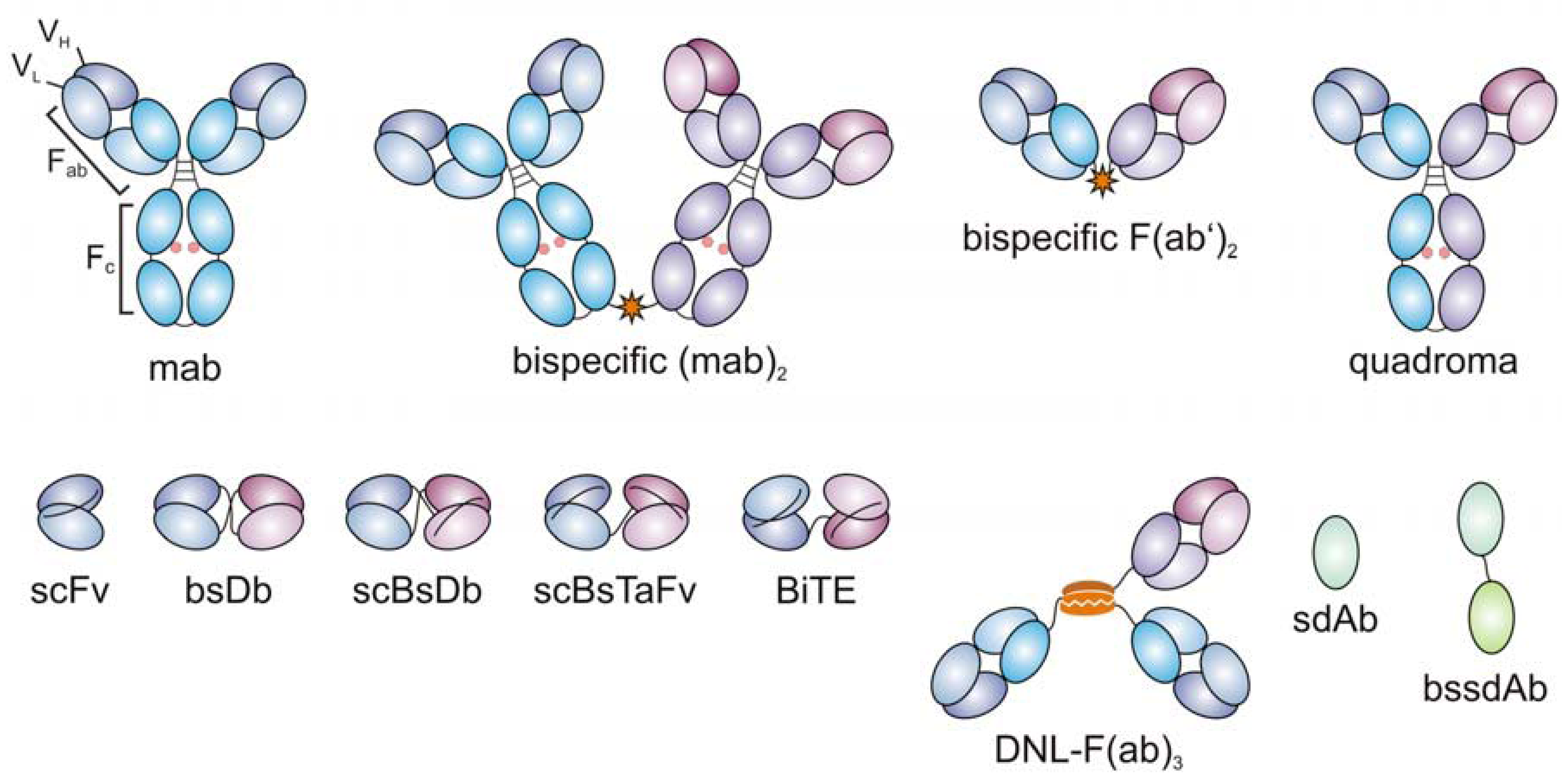
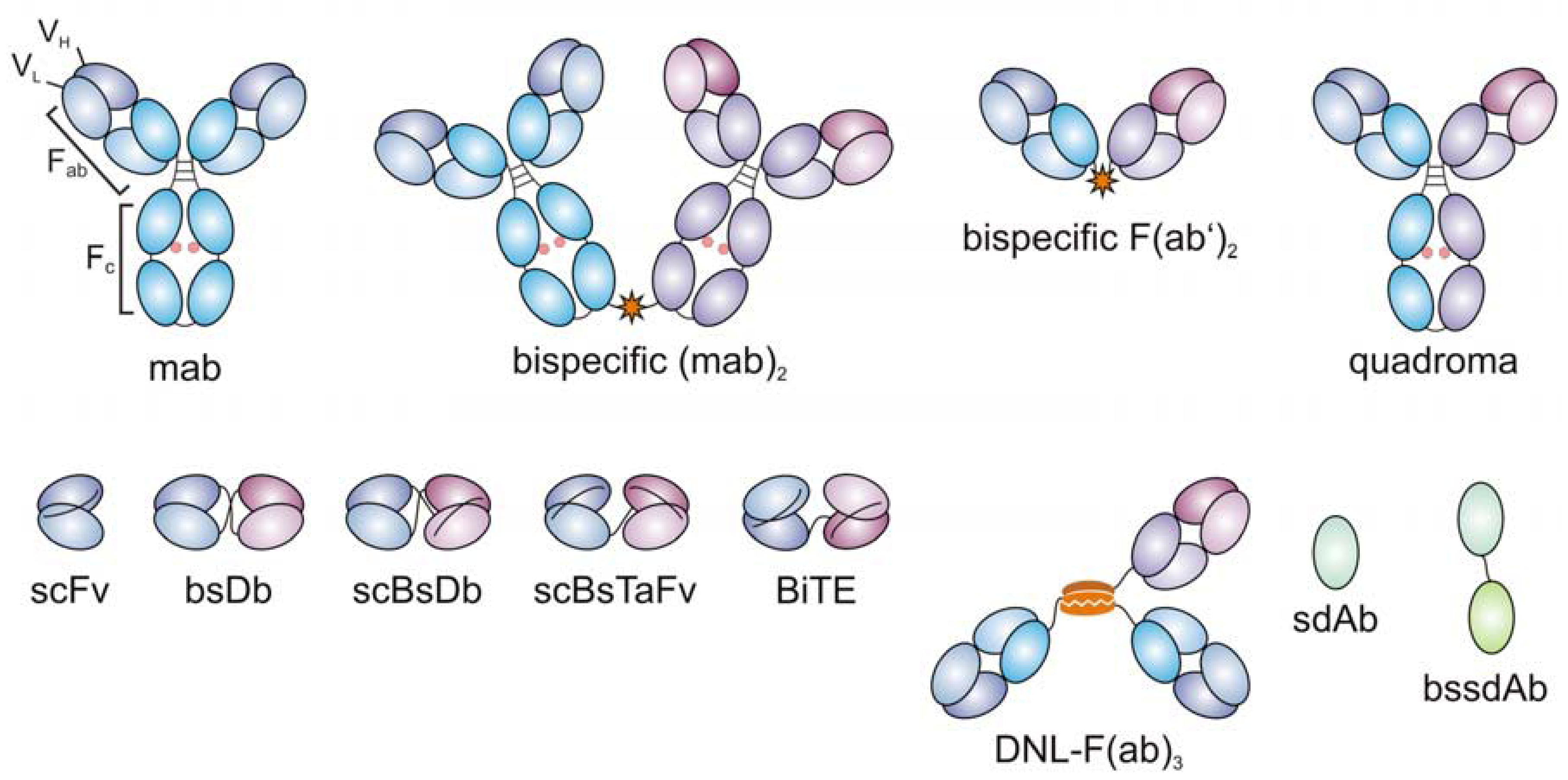
3. Evolution of Bispecific Antibodies
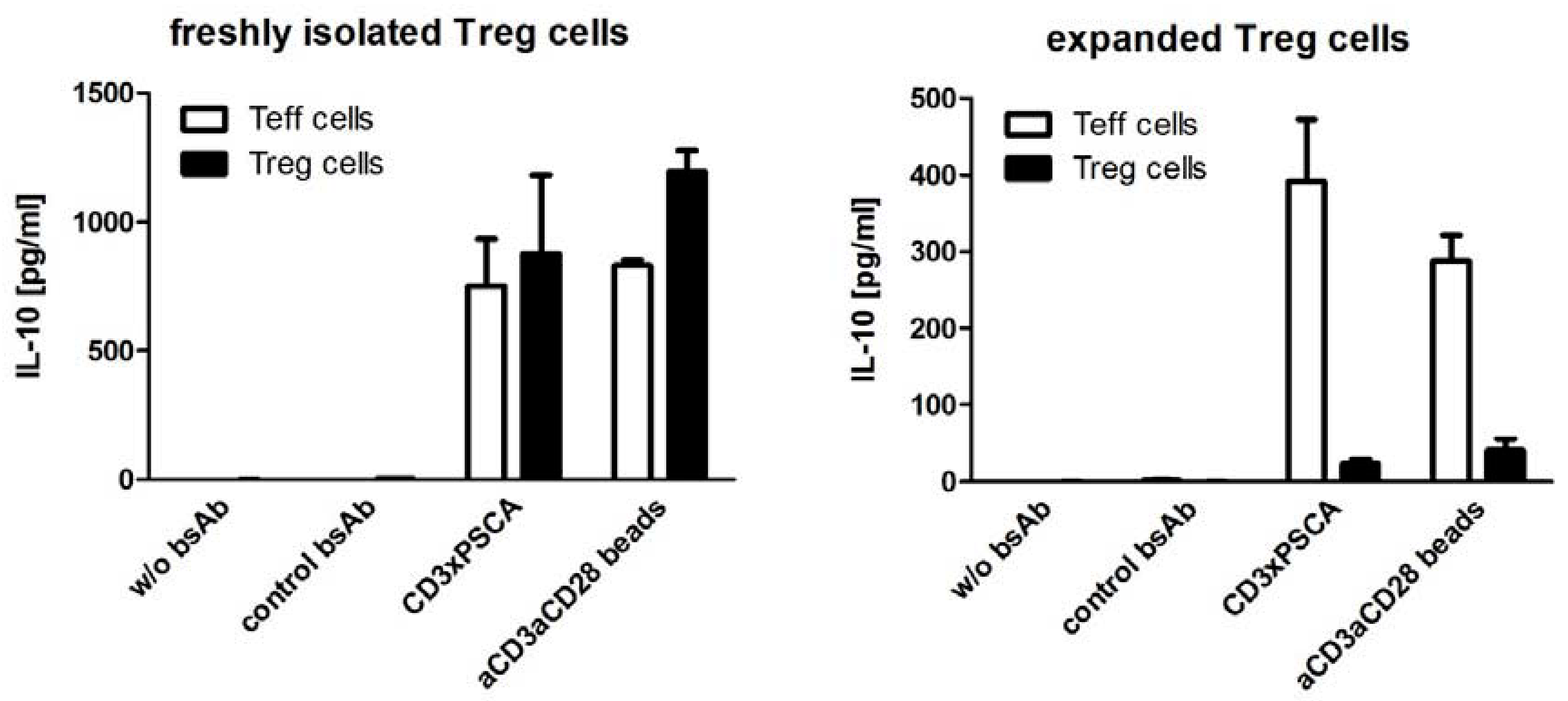
4. Recombinant Bispecific Antibodies for Targeted Tumor Therapy

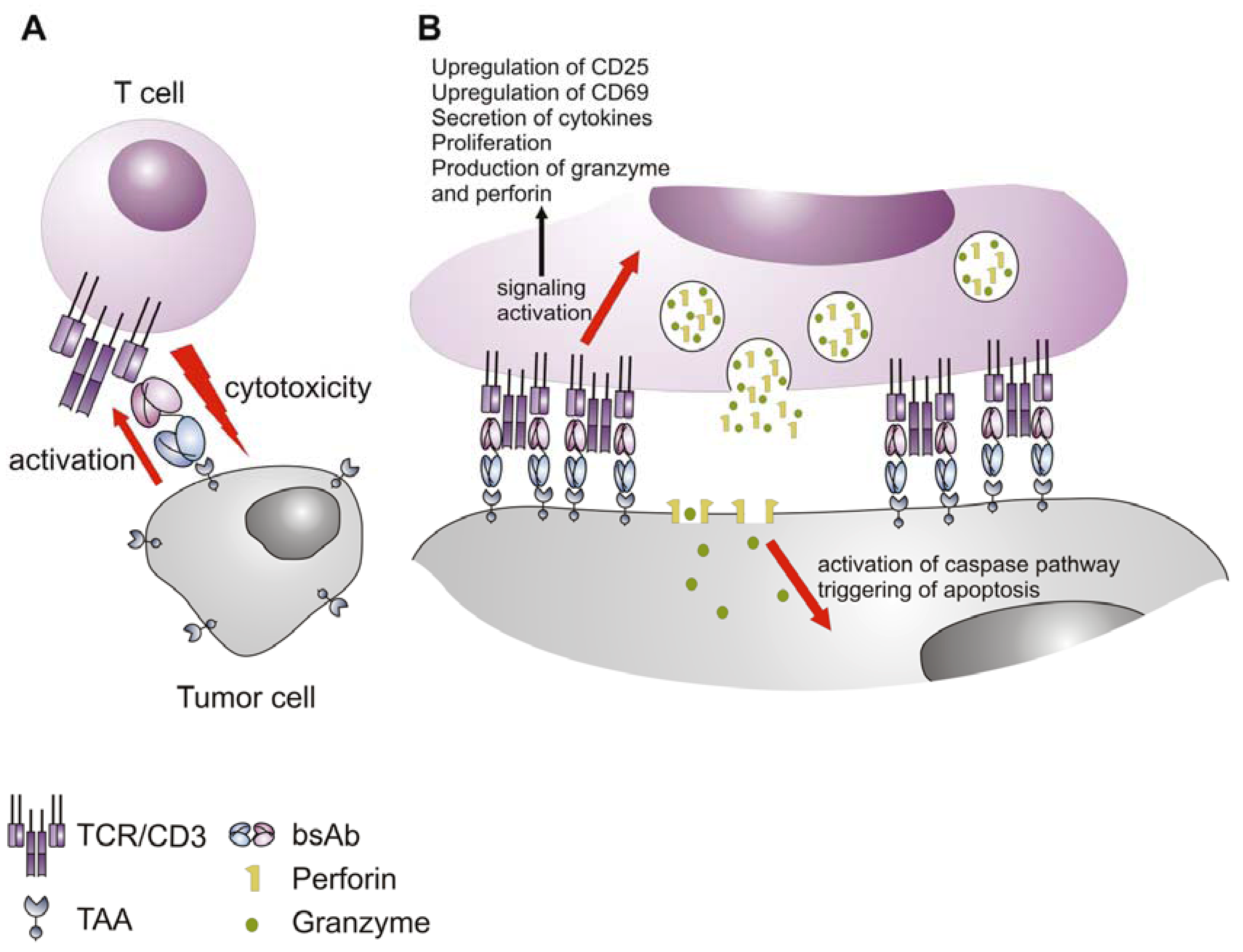
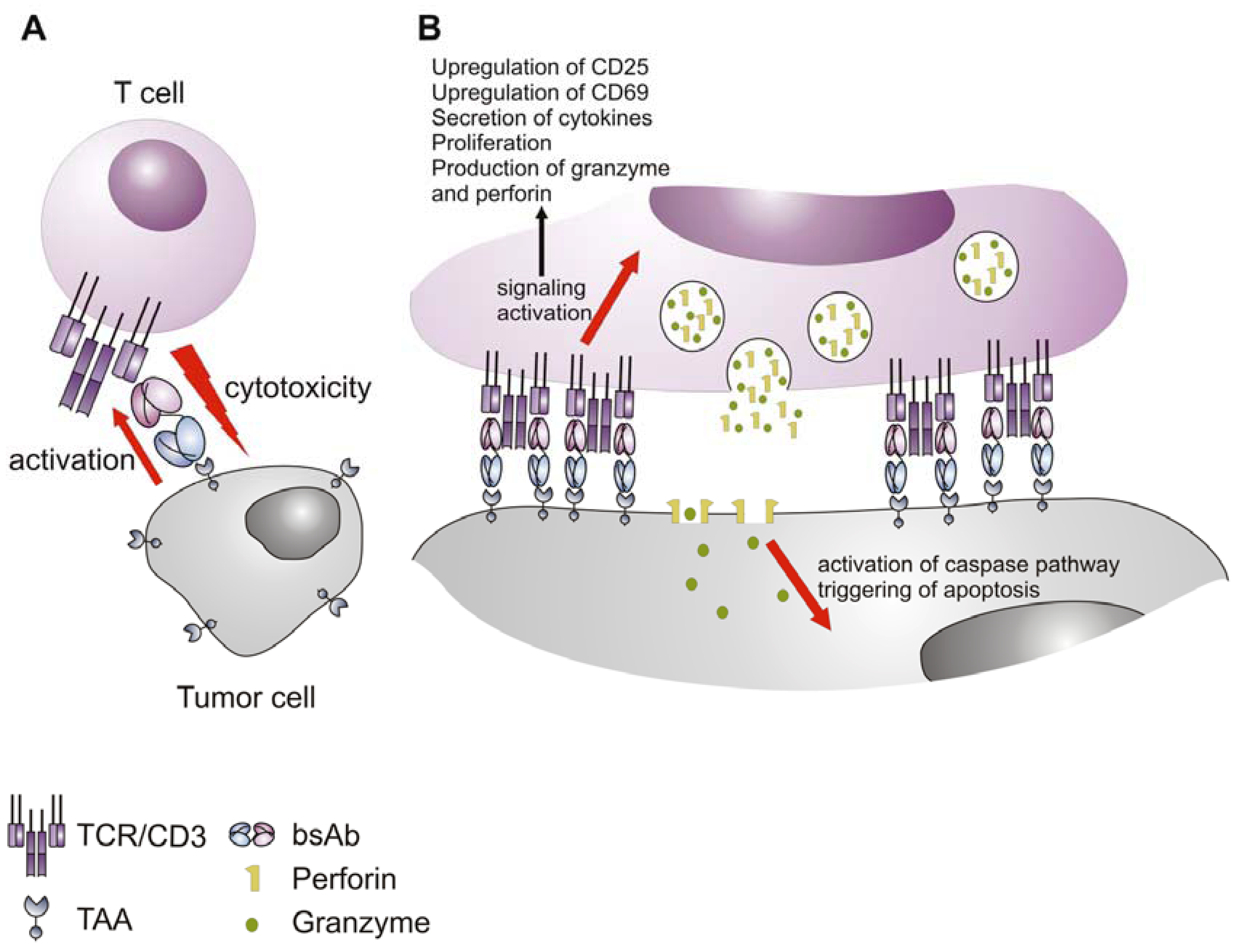
5. Mechanism of Action of the Bispecific Antibodies
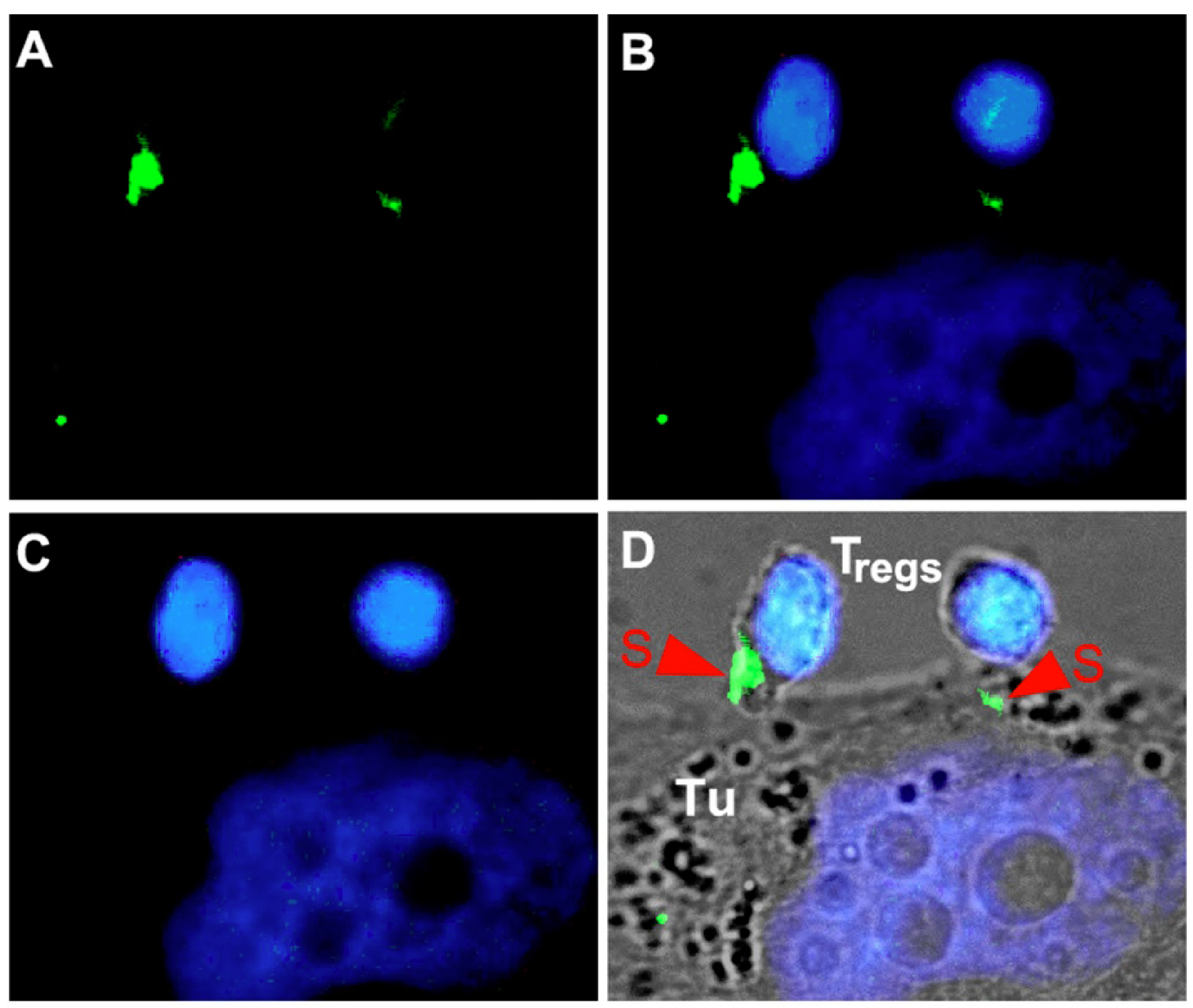

6. Immunoligands
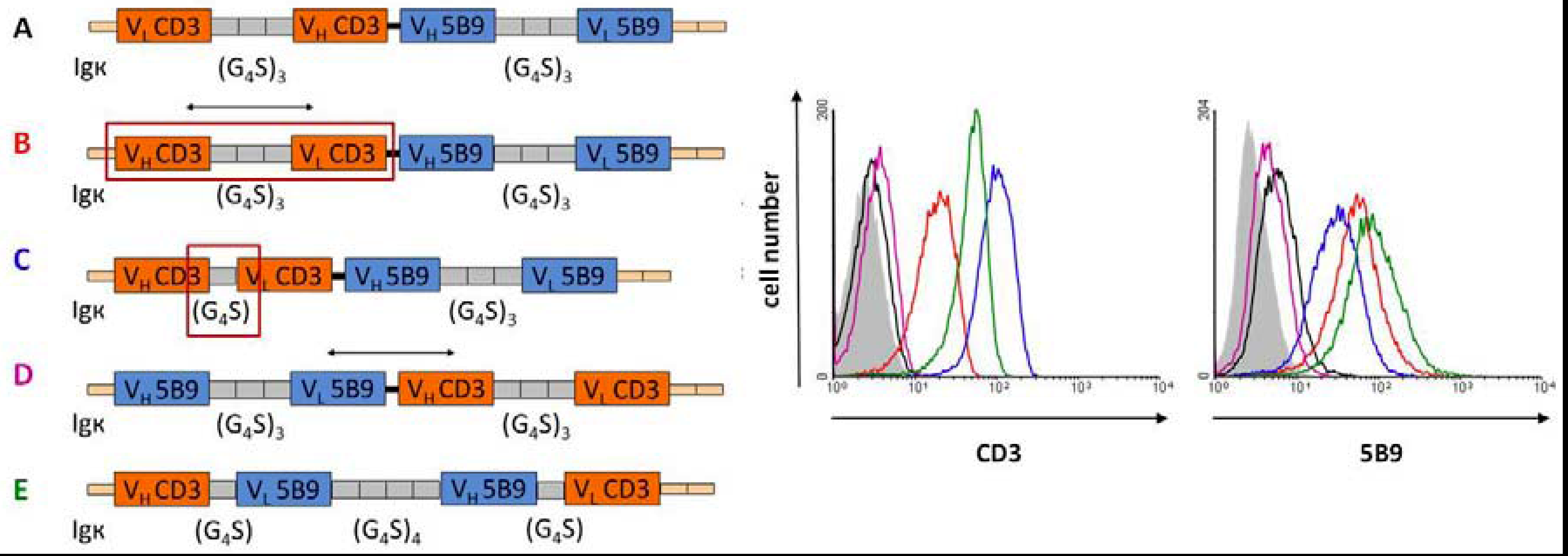
7. Improving the Efficacy and Pharmacokinetics
8. Experimental Section
9. Conclusions
Acknowledgments
References
- Ehrlich, P. Ueber den jetzigen stand der Karzinomforschung. Ned. Tijdschr. Geneeskd. 1909, 273–290. [Google Scholar]
- Burnet, M. Cancer: A biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef]
- Thomas, L. Reactions to homologous tissue antigens in relation to hypersensitivity [Discussion]. In Cellular and Humoral Aspects of the Hypersensitive States; Lawrence, H.S., Ed.; Hoeber-Harper: New York, NY, USA, 1959; pp. 529–533. [Google Scholar]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Parish, C.R. Cancer immunotherapy: The past, the present and the future. Immunol. Cell Biol. 2003, 81, 106–113. [Google Scholar] [CrossRef]
- Dighe, A.S.; Richards, E.; Old, L.J.; Schreiber, R.D. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994, 1, 447–456. [Google Scholar] [CrossRef]
- van den Broek, M.E.; Kagi, D.; Ossendorp, F.; Toes, R.; Vamvakas, S.; Lutz, W.K.; Melief, C.J.; Zinkernagel, R.M.; Hengartner, H. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 1996, 184, 1781–1790. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; Cretney, E.; Trapani, J.A.; Taniguchi, M.; Kawano, T.; Pelikan, S.B.; Crowe, N.Y.; Godfrey, D.I. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 2000, 191, 661–668. [Google Scholar] [CrossRef]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Halliday, G.M.; Patel, A.; Hunt, M.J.; Tefany, F.J.; Barnetson, R.S. Spontaneous regression of human melanoma/nonmelanoma skin cancer: Association with infiltrating CD4+ T cells. World J. Surg. 1995, 19, 352–358. [Google Scholar] [CrossRef]
- Iihara, K.; Yamaguchi, K.; Nishimura, Y.; Iwasaki, T.; Suzuki, K.; Hirabayashi, Y. Spontaneous regression of malignant lymphoma of the breast. Pathol. Int. 2004, 54, 537–542. [Google Scholar] [CrossRef]
- Penn, I. Tumors of the immunocompromised patient. Annu. Rev. Med. 1988, 39, 63–73. [Google Scholar] [CrossRef]
- Buell, J.F.; Gross, T.G.; Woodle, E.S. Malignancy after transplantation. Transplantation 2005, 80, S254–S264. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Scanlan, M.J.; Simpson, A.J.; Old, L.J. The cancer/testis genes: Review, standardization, and commentary. Cancer Immun. 2004, 4, 1. [Google Scholar]
- Haanen, J.B.; Baars, A.; Gomez, R.; Weder, P.; Smits, M.; de Gruijl, T.D.; von Blomberg, B.M.; Bloemena, E.; Scheper, R.J.; van Ham, S.M.; et al. Melanoma-specific tumor-infiltrating lymphocytes but not circulating melanoma-specific T cells may predict survival in resected advanced-stage melanoma patients. Cancer Immunol. Immunother. 2006, 55, 451–458. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Invest. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Smith, P.L.; Lombardi, G.; Foster, G.R. Type I interferons and the innate immune response—More than just antiviral cytokines. Mol. Immunol. 2005, 42, 869–877. [Google Scholar] [CrossRef]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef]
- Smyth, M.J.; Godfrey, D.I.; Trapani, J.A. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2001, 2, 293–299. [Google Scholar] [CrossRef]
- Chan, C.W.; Housseau, F. The 'kiss of death' by dendritic cells to cancer cells. Cell Death Differ. 2008, 15, 58–69. [Google Scholar] [CrossRef]
- Appay, V. The physiological role of cytotoxic CD4(+) T-cells: The holy grail? Clin. Exp. Immunol. 2004, 138, 10–13. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar]
- Law, T.M.; Motzer, R.J.; Mazumdar, M.; Sell, K.W.; Walther, P.J.; O'Connell, M.; Khan, A.; Vlamis, V.; Vogelzang, N.J.; Bajorin, D.F. Phase III randomized trial of interleukin-2 with or without lymphokine-activated killer cells in the treatment of patients with advanced renal cell carcinoma. Cancer 1995, 76, 824–832. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar]
- Ljunggren, H.G.; Malmberg, K.J. Prospects for the use of NK cells in immunotherapy of human cancer. Nat. Rev. Immunol. 2007, 7, 329–339. [Google Scholar] [CrossRef]
- Cartellieri, M.; Bachmann, M.; Feldmann, A.; Bippes, C.; Stamova, S.; Wehner, R.; Temme, A.; Schmitz, M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Cartellieri, M.; Michalk, I.; von Bonin, M.; Kruger, T.; Stamova, S.; Koristka, S.; Arndt, C.; Feldmann, A.; Schmitz, M.; Wermke, M.; et al. Chimeric Antigen Receptor-Engineered T Cells for Immunotherapy of Acute Myeloid Leukemia. Blood 2011, 118, 1124–1125. [Google Scholar]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar]
- Park, T.S.; Rosenberg, S.A.; Morgan, R.A. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011, 29, 550–557. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar]
- Stroncek, D.F.; Berger, C.; Cheever, M.A.; Childs, R.W.; Dudley, M.E.; Flynn, P.; Gattinoni, L.; Heath, J.R.; Kalos, M.; Marincola, F.M.; et al. New directions in cellular therapy of cancer: A summary of the summit on cellular therapy for cancer. J. Transl. Med. 2012, 10, 48. [Google Scholar] [CrossRef]
- Rivoltini, L.; Canese, P.; Huber, V.; Iero, M.; Pilla, L.; Valenti, R.; Fais, S.; Lozupone, F.; Casati, C.; Castelli, C.; et al. Escape strategies and reasons for failure in the interaction between tumour cells and the immune system: How can we tilt the balance towards immune-mediated cancer control? Expert Opin. Biol. Ther. 2005, 5, 463–476. [Google Scholar] [CrossRef]
- Ferrone, S.; Whiteside, T.L. Tumor microenvironment and immune escape. Surg. Oncol. Clin. N. Am. 2007, 16, 755–774, viii. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef]
- Groth, A.; Kloss, S.; von Strandmann, E.P.; Koehl, U.; Koch, J. Mechanisms of tumor and viral immune escape from natural killer cell-mediated surveillance. J. Innate Immun. 2011, 3, 344–354. [Google Scholar] [CrossRef]
- Weiner, L.M.; Murray, J.C.; Shuptrine, C.W. Antibody-based immunotherapy of cancer. Cell 2012, 148, 1081–1084. [Google Scholar] [CrossRef]
- Raju, T.S. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008, 20, 471–478. [Google Scholar] [CrossRef]
- Chan, A.C.; Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 2010, 10, 301–316. [Google Scholar] [CrossRef]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Neuberger, M.S.; Williams, G.T.; Mitchell, E.B.; Jouhal, S.S.; Flanagan, J.G.; Rabbitts, T.H. A hapten-specific chimaeric IgE antibody with human physiological effector function. Nature 1985, 314, 268–270. [Google Scholar]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Hoogenboom, H.R.; Chames, P. Natural and designer binding sites made by phage display technology. Immunol. Today 2000, 21, 371–378. [Google Scholar] [CrossRef]
- Lonberg, N. Human monoclonal antibodies from transgenic mice. Handb. Exp. Pharmacol. 2008, 69–97. [Google Scholar] [CrossRef]
- Reichert, J.M.; Rosensweig, C.J.; Faden, L.B.; Dewitz, M.C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005, 23, 1073–1078. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Cheson, B.D. Radioimmunotherapy of non-Hodgkin's lymphomas. Curr. Drug Targets 2006, 7, 1293–1300. [Google Scholar] [CrossRef]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A. Targeting T cells with bispecific antibodies for cancer therapy. BioDrugs 2011, 25, 365–379. [Google Scholar] [CrossRef]
- McLaughlin, P.; Grillo-Lopez, A.J.; Link, B.K.; Levy, R.; Czuczman, M.S.; Williams, M.E.; Heyman, M.R.; Bence-Bruckler, I.; White, C.A.; Cabanillas, F.; et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: Half of patients respond to a four-dose treatment program. J. Clin. Oncol. 1998, 16, 2825–2833. [Google Scholar]
- Witzig, T.E.; White, C.A.; Gordon, L.I.; Wiseman, G.A.; Emmanouilides, C.; Murray, J.L.; Lister, J.; Multani, P.S. Safety of yttrium-90 ibritumomab tiuxetan radioimmunotherapy for relapsed low-grade, follicular, or transformed non-hodgkin's lymphoma. J. Clin. Oncol. 2003, 21, 1263–1270. [Google Scholar]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999, 17, 2639–2648. [Google Scholar]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E., Jr.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef]
- Lundin, J.; Kimby, E.; Bjorkholm, M.; Broliden, P.A.; Celsing, F.; Hjalmar, V.; Mollgard, L.; Rebello, P.; Hale, G.; Waldmann, H.; et al. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002, 100, 768–773. [Google Scholar] [CrossRef]
- Rhee, J.; Hoff, P.M. Angiogenesis inhibitors in the treatment of cancer. Expert Opin. Pharmacother. 2005, 6, 1701–1711. [Google Scholar] [CrossRef]
- Snyder, L.C.; Astsaturov, I.; Weiner, L.M. Overview of monoclonal antibodies and small molecules targeting the epidermal growth factor receptor pathway in colorectal cancer. Clin. Colorectal. Cancer 2005, 5 (Suppl. 2), S71–S80. [Google Scholar] [CrossRef]
- Patel, D.K. Clinical use of anti-epidermal growth factor receptor monoclonal antibodies in metastatic colorectal cancer. Pharmacotherapy 2008, 28, 31S–41S. [Google Scholar] [CrossRef]
- Chames, P.; Baty, D. Bispecific antibodies for cancer therapy: The light at the end of the tunnel? MAbs 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy: Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar]
- Preithner, S.; Elm, S.; Lippold, S.; Locher, M.; Wolf, A.; da Silva, A.J.; Baeuerle, P.A.; Prang, N.S. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol. Immunol. 2006, 43, 1183–1193. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007, 19, 239–245. [Google Scholar] [CrossRef]
- Xie, H.; Blattler, W.A. In vivo behaviour of antibody-drug conjugates for the targeted treatment of cancer. Expert Opin. Biol. Ther. 2006, 6, 281–291. [Google Scholar] [CrossRef]
- Steiner, M.; Neri, D. Antibody-radionuclide conjugates for cancer therapy: Historical considerations and new trends. Clin. Cancer Res. 2011, 17, 6406–6416. [Google Scholar] [CrossRef]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Kontermann, R.E. Recombinant bispecific antibodies for cancer therapy. Acta Pharmacol. Sin. 2005, 26, 1–9. [Google Scholar] [CrossRef]
- Muller, D.; Kontermann, R.E. Bispecific antibodies for cancer immunotherapy: Current perspectives. BioDrugs 2010, 24, 89–98. [Google Scholar] [CrossRef]
- Singer, H.; Kellner, C.; Lanig, H.; Aigner, M.; Stockmeyer, B.; Oduncu, F.; Schwemmlein, M.; Stein, C.; Mentz, K.; Mackensen, A.; et al. Effective elimination of acute myeloid leukemic cells by recombinant bispecific antibody derivatives directed against CD33 and CD16. J. Immunother. 2010, 33, 599–608. [Google Scholar] [CrossRef]
- Silla, L.M.; Chen, J.; Zhong, R.K.; Whiteside, T.L.; Ball, E.D. Potentiation of lysis of leukaemia cells by a bispecific antibody to CD33 and CD16 (Fc gamma RIII) expressed by human natural killer (NK) cells. Br. J. Haematol. 1995, 89, 712–718. [Google Scholar]
- Cochlovius, B.; Kipriyanov, S.M.; Stassar, M.J.; Christ, O.; Schuhmacher, J.; Strauss, G.; Moldenhauer, G.; Little, M. Treatment of human B cell lymphoma xenografts with a CD3 x CD19 diabody and T cells. J. Immunol. 2000, 165, 888–895. [Google Scholar]
- Blanco, B.; Holliger, P.; Vile, R.G.; Alvarez-Vallina, L. Induction of human T lymphocyte cytotoxicity and inhibition of tumor growth by tumor-specific diabody-based molecules secreted from gene-modified bystander cells. J. Immunol. 2003, 171, 1070–1077. [Google Scholar]
- Schlereth, B.; Fichtner, I.; Lorenczewski, G.; Kleindienst, P.; Brischwein, K.; da Silva, A.; Kufer, P.; Lutterbuese, R.; Junghahn, I.; Kasimir-Bauer, S.; et al. Eradication of tumors from a human colon cancer cell line and from ovarian cancer metastases in immunodeficient mice by a single-chain Ep-CAM-/CD3-bispecific antibody construct. Cancer Res. 2005, 65, 2882–2889. [Google Scholar] [CrossRef]
- Muller, D.; Kontermann, R.E. Recombinant bispecific antibodies for cellular cancer immunotherapy. Curr. Opin. Mol. Ther. 2007, 9, 319–326. [Google Scholar]
- Kiessling, A.; Fussel, S.; Wehner, R.; Bachmann, M.; Wirth, M.P.; Rieber, E.P.; Schmitz, M. Advances in specific immunotherapy for prostate cancer. Eur. Urol. 2008, 53, 694–708. [Google Scholar]
- Arndt, C.; Feldmann, A.; Koristka, S.; Michalk, I.; Cartellieri, M.; Stamova, S.; von Bonin, M.; Bornhauser, M.; Ehninger, G.; Bachmann, M. Redirection of Immune Effector Cells by Bispecific Antibody Systems for the Treatment of Acute Myeloid Leukemia. Blood 2011, 118, 663–664. [Google Scholar]
- Feldmann, A.; Stamova, S.; Bippes, C.C.; Bartsch, H.; Wehner, R.; Schmitz, M.; Temme, A.; Cartellieri, M.; Bachmann, M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: Comparison of different antibody formats. Prostate 2011, 71, 998–1011. [Google Scholar] [CrossRef]
- Fortmuller, K.; Alt, K.; Gierschner, D.; Wolf, P.; Baum, V.; Freudenberg, N.; Wetterauer, U.; Elsasser-Beile, U.; Buhler, P. Effective targeting of prostate cancer by lymphocytes redirected by a PSMA x CD3 bispecific single-chain diabody. Prostate 2011, 71, 588–596. [Google Scholar] [CrossRef]
- Stamova, S.; Cartellieri, M.; Feldmann, A.; Bippes, C.C.; Bartsch, H.; Wehner, R.; Schmitz, M.; von Bonin, M.; Bornhauser, M.; Ehninger, G.; et al. Simultaneous engagement of the activatory receptors NKG2D and CD3 for retargeting of effector cells to CD33-positive malignant cells. Leukemia 2011, 25, 1053–1056. [Google Scholar]
- Milstein, C.; Cuello, A.C. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef]
- Kufer, P.; Lutterbuse, R.; Baeuerle, P.A. A revival of bispecific antibodies. Trends Biotechnol. 2004, 22, 238–244. [Google Scholar] [CrossRef]
- Segal, D.M.; Weiner, G.J.; Weiner, L.M. Bispecific antibodies in cancer therapy. Curr. Opin. Immunol. 1999, 11, 558–562. [Google Scholar]
- Kriangkum, J.; Xu, B.; Nagata, L.P.; Fulton, R.E.; Suresh, M.R. Bispecific and bifunctional single chain recombinant antibodies. Biomol. Eng. 2001, 18, 31–40. [Google Scholar] [CrossRef]
- Holliger, P.; Prospero, T.; Winter, G. "Diabodies": Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar]
- Lawrence, L.J.; Kortt, A.A.; Iliades, P.; Tulloch, P.A.; Hudson, P.J. Orientation of antigen binding sites in dimeric and trimeric single chain Fv antibody fragments. FEBS Lett. 1998, 425, 479–484. [Google Scholar] [CrossRef]
- Volkel, T.; Korn, T.; Bach, M.; Muller, R.; Kontermann, R.E. Optimized linker sequences for the expression of monomeric and dimeric bispecific single-chain diabodies. Protein Eng. 2001, 14, 815–823. [Google Scholar] [CrossRef]
- Bippes, C.C.; Feldmann, A.; Stamova, S.; Cartellieri, M.; Schwarzer, A.; Wehner, R.; Schmitz, M.; Rieber, E.P.; Zhao, S.; Schakel, K.; et al. A novel modular antigen delivery system for immuno targeting of human 6-sulfo LacNAc-positive blood dendritic cells (SlanDCs). PLoS One 2011, 6, e16315. [Google Scholar]
- Kontermann, R.E.; Korn, T.; Jerome, V. Recombinant adenoviruses for in vivo expression of antibody fragments. Methods Mol. Biol. 2003, 207, 421–433. [Google Scholar]
- Ren-Heidenreich, L.; Davol, P.A.; Kouttab, N.M.; Elfenbein, G.J.; Lum, L.G. Redirected T-cell cytotoxicity to epithelial cell adhesion molecule-overexpressing adenocarcinomas by a novel recombinant antibody, E3Bi, in vitro and in an animal model. Cancer 2004, 100, 1095–1103. [Google Scholar] [CrossRef]
- Stamova, S.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Koristka, S.; Bartsch, H.; Bippes, C.C.; Wehner, R.; Schmitz, M.; von Bonin, M.; et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol. Immunol. 2011, 49, 474–482. [Google Scholar] [CrossRef]
- Stamova, S.; Feldmann, A.; Cartellieri, M.; Arndt, C.; Koristka, S.; Apel, F.; Wehner, R.; Schmitz, M.; Bornhauser, M.; von Bonin, M.; et al. Generation of single-chain bispecific green fluorescent protein fusion antibodies for imaging of antibody-induced T cell synapses. Anal. Biochem. 2012, 423, 261–268. [Google Scholar]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; McBride, W.J.; Sharkey, R.M.; Chang, C.H. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting. Proc. Natl. Acad. Sci. USA 2006, 103, 6841–6846. [Google Scholar]
- Saerens, D.; Ghassabeh, G.H.; Muyldermans, S. Single-domain antibodies as building blocks for novel therapeutics. Curr. Opin. Pharmacol. 2008, 8, 600–608. [Google Scholar] [CrossRef]
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989, 341, 544–546. [Google Scholar]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- Gill, D.S.; Damle, N.K. Biopharmaceutical drug discovery using novel protein scaffolds. Curr. Opin. Biotechnol. 2006, 17, 653–658. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef]
- Friedman, M.; Lindstrom, S.; Ekerljung, L.; Andersson-Svahn, H.; Carlsson, J.; Brismar, H.; Gedda, L.; Frejd, F.Y.; Stahl, S. Engineering and characterization of a bispecific HER2 × EGFR-binding affibody molecule. Biotechnol. Appl. Biochem. 2009, 54, 121–131. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Cochlovius, B.; Schafer, H.J.; Moldenhauer, G.; Bahre, A.; Le Gall, F.; Knackmuss, S.; Little, M. Synergistic antitumor effect of bispecific CD19 × CD3 and CD19 x CD16 diabodies in a preclinical model of non-Hodgkin's lymphoma. J. Immunol. 2002, 169, 137–144. [Google Scholar]
- Asano, R.; Sone, Y.; Makabe, K.; Tsumoto, K.; Hayashi, H.; Katayose, Y.; Unno, M.; Kudo, T.; Kumagai, I. Humanization of the bispecific epidermal growth factor receptor × CD3 diabody and its efficacy as a potential clinical reagent. Clin. Cancer Res. 2006, 12, 4036–4042. [Google Scholar] [CrossRef]
- Buhler, P.; Wolf, P.; Gierschner, D.; Schaber, I.; Katzenwadel, A.; Schultze-Seemann, W.; Wetterauer, U.; Tacke, M.; Swamy, M.; Schamel, W.W.; et al. A bispecific diabody directed against prostate-specific membrane antigen and CD3 induces T-cell mediated lysis of prostate cancer cells. Cancer Immunol. Immunother. 2008, 57, 43–52. [Google Scholar] [CrossRef]
- Grosse-Hovest, L.; Hartlapp, I.; Marwan, W.; Brem, G.; Rammensee, H.G.; Jung, G. A recombinant bispecific single-chain antibody induces targeted, supra-agonistic CD28-stimulation and tumor cell killing. Eur. J. Immunol. 2003, 33, 1334–1340. [Google Scholar] [CrossRef]
- Otz, T.; Grosse-Hovest, L.; Hofmann, M.; Rammensee, H.G.; Jung, G. A bispecific single-chain antibody that mediates target cell-restricted, supra-agonistic CD28 stimulation and killing of lymphoma cells. Leukemia 2009, 23, 71–77. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Grosse-Hovest, L.; Wick, W.; Minoia, R.; Weller, M.; Rammensee, H.G.; Brem, G.; Jung, G. Supraagonistic, bispecific single-chain antibody purified from the serum of cloned, transgenic cows induces T-cell-mediated killing of glioblastoma cells in vitro and in vivo. Int. J. Cancer 2005, 117, 1060–1064. [Google Scholar] [CrossRef]
- Mack, M.; Riethmuller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar]
- Topp, M.S.; Kufer, P.; Gokbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar]
- Molhoj, M.; Crommer, S.; Brischwein, K.; Rau, D.; Sriskandarajah, M.; Hoffmann, P.; Kufer, P.; Hofmeister, R.; Baeuerle, P.A. CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis. Mol. Immunol. 2007, 44, 1935–1943. [Google Scholar]
- Feldmann, A.; Arndt, C.; Töpfer, K.; Stamova, S.; Krone, C.M.; Koristka, S.; Michalk, I.; Lindemann, D.; Schmitz, M.; Temme, A.; et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T Cells. J. Immunol. 2012, in press.. [Google Scholar]
- Haagen, I.A.; de Lau, W.B.; Bast, B.J.; Geerars, A.J.; Clark, M.R.; de Gast, B.C. Unprimed CD4+ and CD8+ T cells can be rapidly activated by a CD3 × CD19 bispecific antibody to proliferate and become cytotoxic. Cancer Immunol. Immunother. 1994, 39, 391–396. [Google Scholar] [CrossRef]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef]
- Buhler, P.; Molnar, E.; Dopfer, E.P.; Wolf, P.; Gierschner, D.; Wetterauer, U.; Schamel, W.W.; Elsasser-Beile, U. Target-dependent T-cell activation by coligation with a PSMA × CD3 diabody induces lysis of prostate cancer cells. J. Immunother. 2009, 32, 565–573. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar]
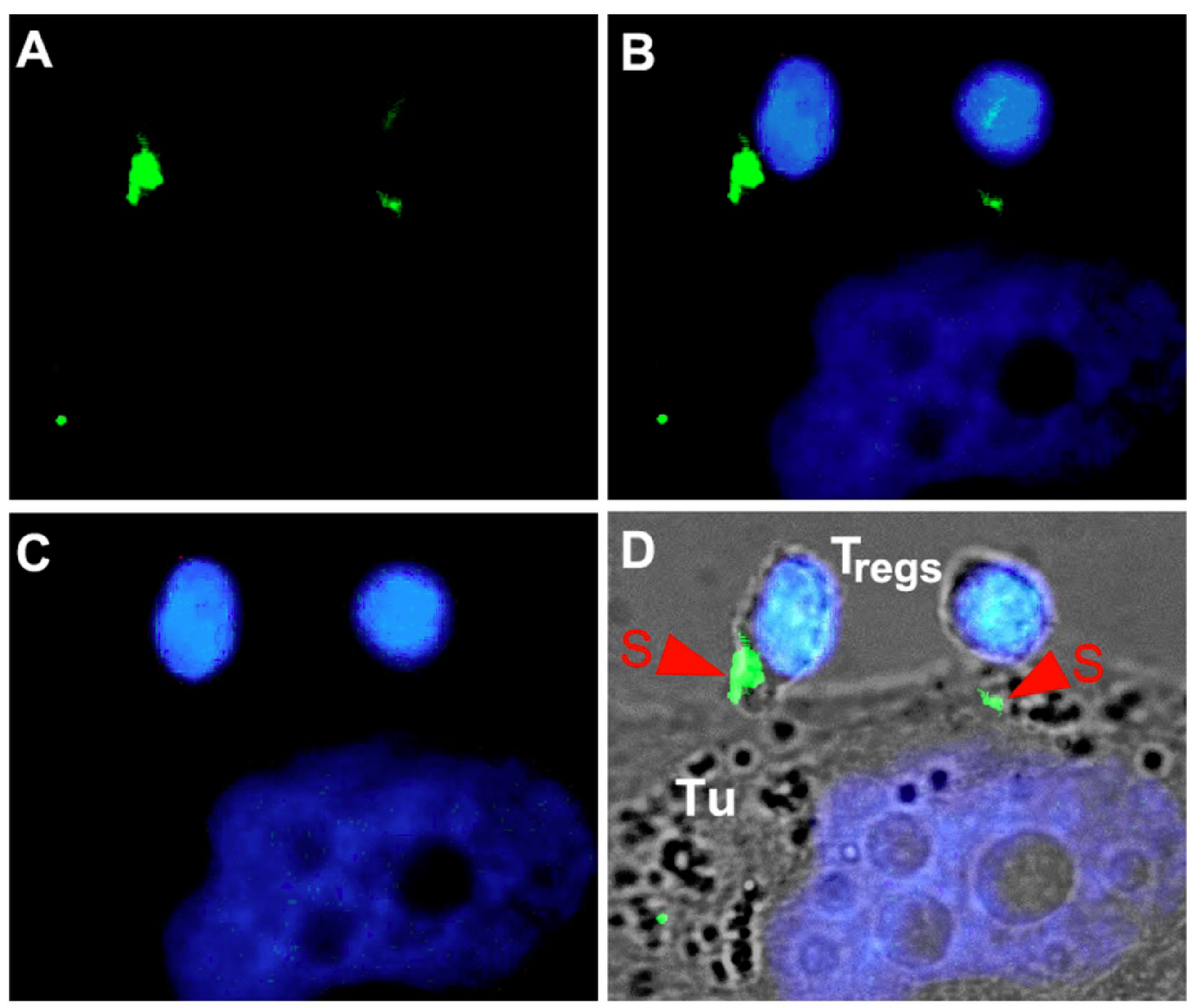
- Koristka, S.; Cartellieri, M.; Theil, A.; Feldmann, A.; Arndt, C.; Stamova, S.; Michalk, I.; Topfer, K.; Temme, A.; Kretschmer, K.; et al. Retargeting of human regulatory T cells by single-chain bispecific antibodies. J. Immunol. 2012, 188, 1551–1558. [Google Scholar]
- Hoffmann, P.; Hofmeister, R.; Brischwein, K.; Brandl, C.; Crommer, S.; Bargou, R.; Itin, C.; Prang, N.; Baeuerle, P.A. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 2005, 115, 98–104. [Google Scholar] [CrossRef]
- Boesteanu, A.C.; Katsikis, P.D. Memory T cells need CD28 costimulation to remember. Semin. Immunol. 2009, 21, 69–77. [Google Scholar] [CrossRef]
- Koristka, S.; Cartellieri, M.; Theil, A.; Arndt, C.; Feldmann, A.; Michalk, I.; Schmitz, M.; Kretschmer, K.; Bornhauser, M.; Ehninger, G.; et al. Antigen-Specific Redirection of Human Regulatory T Cells by Bispecific Antibodies. Blood 2011, 118, 1725–1726. [Google Scholar]
- Hombach, A.A.; Kofler, D.; Rappl, G.; Abken, H. Redirecting human CD4+CD25+ regulatory T cells from the peripheral blood with pre-defined target specificity. Gene Ther. 2009, 16, 1088–1096. [Google Scholar] [CrossRef]
- Lanier, L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 2003, 15, 308–314. [Google Scholar]
- Moretta, L.; Moretta, A. Unravelling natural killer cell function: Triggering and inhibitory human NK receptors. EMBO J. 2004, 23, 255–259. [Google Scholar] [CrossRef]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar]
- Fuertes, M.B.; Girart, M.V.; Molinero, L.L.; Domaica, C.I.; Rossi, L.E.; Barrio, M.M.; Mordoh, J.; Rabinovich, G.A.; Zwirner, N.W. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J. Immunol. 2008, 180, 4606–4614. [Google Scholar]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar]
- Germain, C.; Larbouret, C.; Cesson, V.; Donda, A.; Held, W.; Mach, J.P.; Pelegrin, A.; Robert, B. MHC class I-related chain A conjugated to antitumor antibodies can sensitize tumor cells to specific lysis by natural killer cells. Clin. Cancer Res. 2005, 11, 7516–7522. [Google Scholar]
- von Strandmann, E.P.; Hansen, H.P.; Reiners, K.S.; Schnell, R.; Borchmann, P.; Merkert, S.; Simhadri, V.R.; Draube, A.; Reiser, M.; Purr, I.; et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 2006, 107, 1955–1962. [Google Scholar]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar]
- Groh, V.; Rhinehart, R.; Randolph-Habecker, J.; Topp, M.S.; Riddell, S.R.; Spies, T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2001, 2, 255–260. [Google Scholar] [CrossRef]
- Kim, Y.J.; Han, M.K.; Broxmeyer, H.E. 4-1BB regulates NKG2D costimulation in human cord blood CD8+ T cells. Blood 2008, 111, 1378–1386. [Google Scholar]
- Doubrovina, E.S.; Doubrovin, M.M.; Vider, E.; Sisson, R.B.; O'Reilly, R.J.; Dupont, B.; Vyas, Y.M. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J. Immunol. 2003, 171, 6891–6899. [Google Scholar]
- Kim, Y.J.; Stringfield, T.M.; Chen, Y.; Broxmeyer, H.E. Modulation of cord blood CD8+ T-cell effector differentiation by TGF-beta1 and 4-1BB costimulation. Blood 2005, 105, 274–281. [Google Scholar] [CrossRef]
- Parmiani, G.; Rivoltini, L.; Andreola, G.; Carrabba, M. Cytokines in cancer therapy. Immunol. Lett. 2000, 74, 41–44. [Google Scholar] [CrossRef]
- Masztalerz, A.; Van Rooijen, N.; Den Otter, W.; Everse, L.A. Mechanisms of macrophage cytotoxicity in IL-2 and IL-12 mediated tumour regression. Cancer Immunol. Immunother. 2003, 52, 235–242. [Google Scholar]
- Emminger, W.; Emminger-Schmidmeier, W.; Peters, C.; Susani, M.; Hawliczek, R.; Hocker, P.; Gadner, H. Capillary leak syndrome during low dose granulocyte-macrophage colony-stimulating factor (rh GM-CSF) treatment of a patient in a continuous febrile state. Blut 1990, 61, 219–221. [Google Scholar] [CrossRef]
- Stern, A.C.; Jones, T.C. The side-effect profile of GM-CSF. Infection 1992, 20 (Suppl. 2), S124–S127. [Google Scholar] [CrossRef]
- Schwartz, R.N.; Stover, L.; Dutcher, J. Managing toxicities of high-dose interleukin-2. Oncology (Williston Park) 2002, 16, 11–20. [Google Scholar]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar]
- Neri, D.; Bicknell, R. Tumour vascular targeting. Nat. Rev. Cancer 2005, 5, 436–446. [Google Scholar] [CrossRef]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef]
- Ortiz-Sanchez, E.; Helguera, G.; Daniels, T.R.; Penichet, M.L. Antibody-cytokine fusion proteins: Applications in cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 609–632. [Google Scholar] [CrossRef]
- Ott, M.G.; Marme, F.; Moldenhauer, G.; Lindhofer, H.; Hennig, M.; Spannagl, R.; Essing, M.M.; Linke, R.; Seimetz, D. Humoral response to catumaxomab correlates with clinical outcome: Results of the pivotal phase II/III study in patients with malignant ascites. Int. J. Cancer 2012, 130, 2195–2203. [Google Scholar]
- Meredith, R.F.; Khazaeli, M.B.; Plott, W.E.; Saleh, M.N.; Liu, T.; Allen, L.F.; Russell, C.D.; Orr, R.A.; Colcher, D.; Schlom, J.; et al. Phase I trial of iodine-131-chimeric B72.3 (human IgG4) in metastatic colorectal cancer. J. Nucl. Med. 1992, 33, 23–29. [Google Scholar]
- Steffens, M.G.; Boerman, O.C.; Oosterwijk-Wakka, J.C.; Oosterhof, G.O.; Witjes, J.A.; Koenders, E.B.; Oyen, W.J.; Buijs, W.C.; Debruyne, F.M.; Corstens, F.H.; et al. Targeting of renal cell carcinoma with iodine-131-labeled chimeric monoclonal antibody G250. J. Clin. Oncol. 1997, 15, 1529–1537. [Google Scholar]
- Pavlinkova, G.; Colcher, D.; Booth, B.J.; Goel, A.; Wittel, U.A.; Batra, S.K. Effects of humanization and gene shuffling on immunogenicity and antigen binding of anti-TAG-72 single-chain Fvs. Int. J. Cancer 2001, 94, 717–726. [Google Scholar] [CrossRef]
- Verhoeyen, M.; Milstein, C.; Winter, G. Reshaping human antibodies: Grafting an antilysozyme activity. Science 1988, 239, 1534–1536. [Google Scholar]
- Jolliffe, L.K. Humanized antibodies: Enhancing therapeutic utility through antibody engineering. Int. Rev. Immunol. 1993, 10, 241–250. [Google Scholar] [CrossRef]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Vaughan, T.J.; Williams, A.J.; Pritchard, K.; Osbourn, J.K.; Pope, A.R.; Earnshaw, J.C.; McCafferty, J.; Hodits, R.A.; Wilton, J.; Johnson, K.S. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 1996, 14, 309–314. [Google Scholar] [CrossRef]
- Weiner, L.M. Fully human therapeutic monoclonal antibodies. J. Immunother. 2006, 29, 1–9. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs 2009, 23, 93–109. [Google Scholar] [CrossRef]
- Muller, D.; Karle, A.; Meissburger, B.; Hofig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar]
- Stork, R.; Muller, D.; Kontermann, R.E. A novel tri-functional antibody fusion protein with improved pharmacokinetic properties generated by fusing a bispecific single-chain diabody with an albumin-binding domain from streptococcal protein G. Protein Eng. Des. Sel. 2007, 20, 569–576. [Google Scholar] [CrossRef]
- Stork, R.; Campigna, E.; Robert, B.; Muller, D.; Kontermann, R.E. Biodistribution of a bispecific single-chain diabody and its half-life extended derivatives. J. Biol. Chem. 2009, 284, 25612–25619. [Google Scholar]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef]
- Tijink, B.M.; Laeremans, T.; Budde, M.; Stigter-van Walsum, M.; Dreier, T.; de Haard, H.J.; Leemans, C.R.; van Dongen, G.A. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: Taking advantage of modular Nanobody technology. Mol. Cancer Ther. 2008, 7, 2288–2297. [Google Scholar] [CrossRef]
- Marvin, J.S.; Zhu, Z. Recombinant approaches to IgG-like bispecific antibodies. Acta Pharmacol. Sin. 2005, 26, 649–658. [Google Scholar] [CrossRef]
- Stork, R.; Zettlitz, K.A.; Muller, D.; Rether, M.; Hanisch, F.G.; Kontermann, R.E. N-Glycosylation as novel strategy to improve pharmacokinetic properties of bispecific single-chain diabodies. J. Biol. Chem. 2008, 283, 7804–7812. [Google Scholar]
- Chapman, A.P. PEGylated antibodies and antibody fragments for improved therapy: A review. Adv. Drug Deliv. Rev. 2002, 54, 531–545. [Google Scholar] [CrossRef]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM × anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stamova, S.; Koristka, S.; Keil, J.; Arndt, C.; Feldmann, A.; Michalk, I.; Bartsch, H.; Bippes, C.C.; Schmitz, M.; Cartellieri, M.; et al. Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs. Antibodies 2012, 1, 172-198. https://doi.org/10.3390/antib1020172
Stamova S, Koristka S, Keil J, Arndt C, Feldmann A, Michalk I, Bartsch H, Bippes CC, Schmitz M, Cartellieri M, et al. Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs. Antibodies. 2012; 1(2):172-198. https://doi.org/10.3390/antib1020172
Chicago/Turabian StyleStamova, Slava, Stefanie Koristka, Juliane Keil, Claudia Arndt, Anja Feldmann, Irene Michalk, Holger Bartsch, Claudia C. Bippes, Marc Schmitz, Marc Cartellieri, and et al. 2012. "Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs" Antibodies 1, no. 2: 172-198. https://doi.org/10.3390/antib1020172
APA StyleStamova, S., Koristka, S., Keil, J., Arndt, C., Feldmann, A., Michalk, I., Bartsch, H., Bippes, C. C., Schmitz, M., Cartellieri, M., & Bachmann, M. (2012). Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs. Antibodies, 1(2), 172-198. https://doi.org/10.3390/antib1020172