
Understanding the Adsorption Mechanism of Phenol and Para-Chlorophenol onto Sepiolite Clay: A Combined DFT Calculations, Molecular Dynamics Simulations, and Isotherm Analysis

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
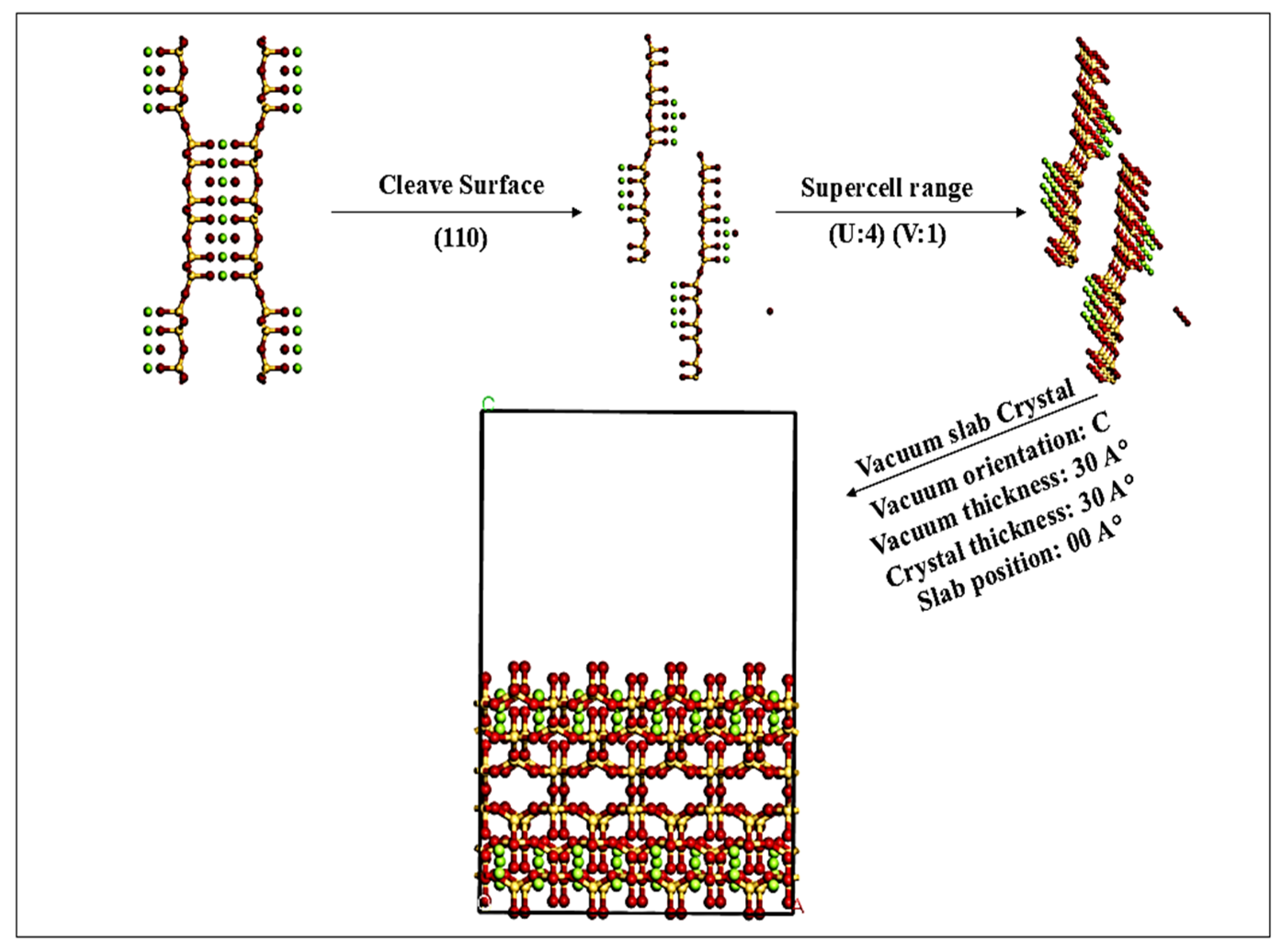
2.1. Adsorbent Model Preparation
2.2. DFT Computational Details
2.3. Adsorption MD Simulation Preparation
3. Results
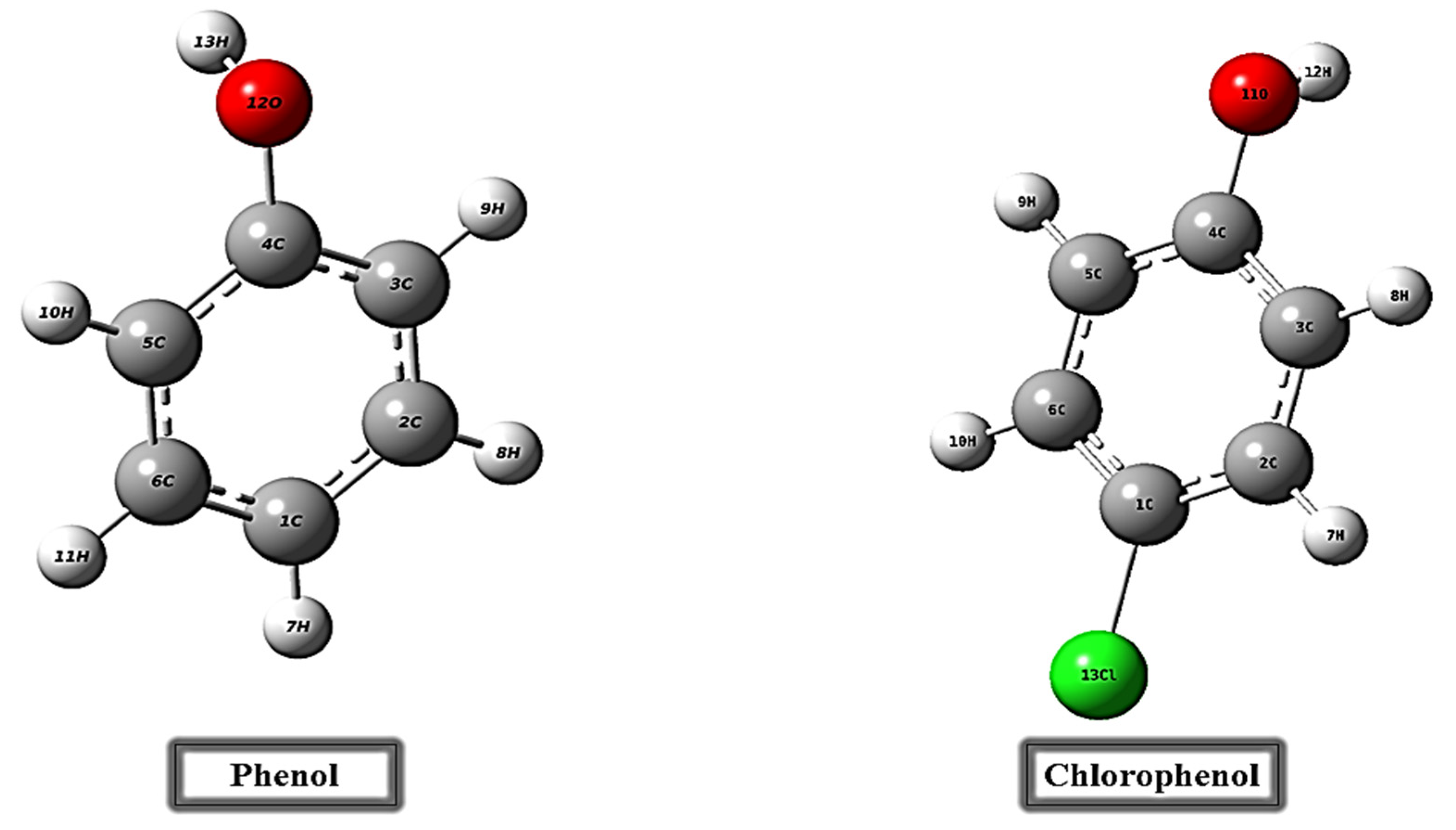
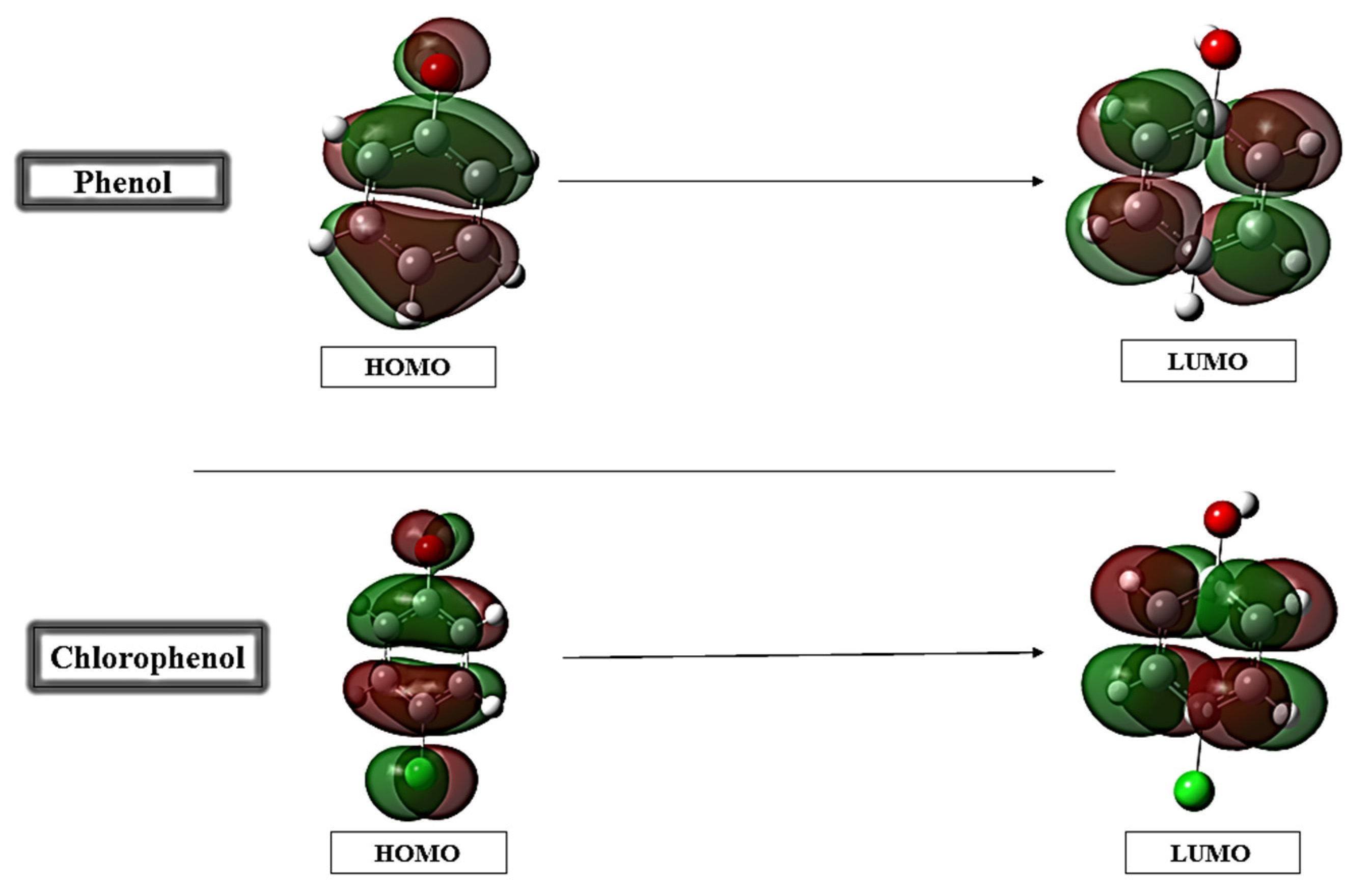
3.1. DFT Results and Analysis
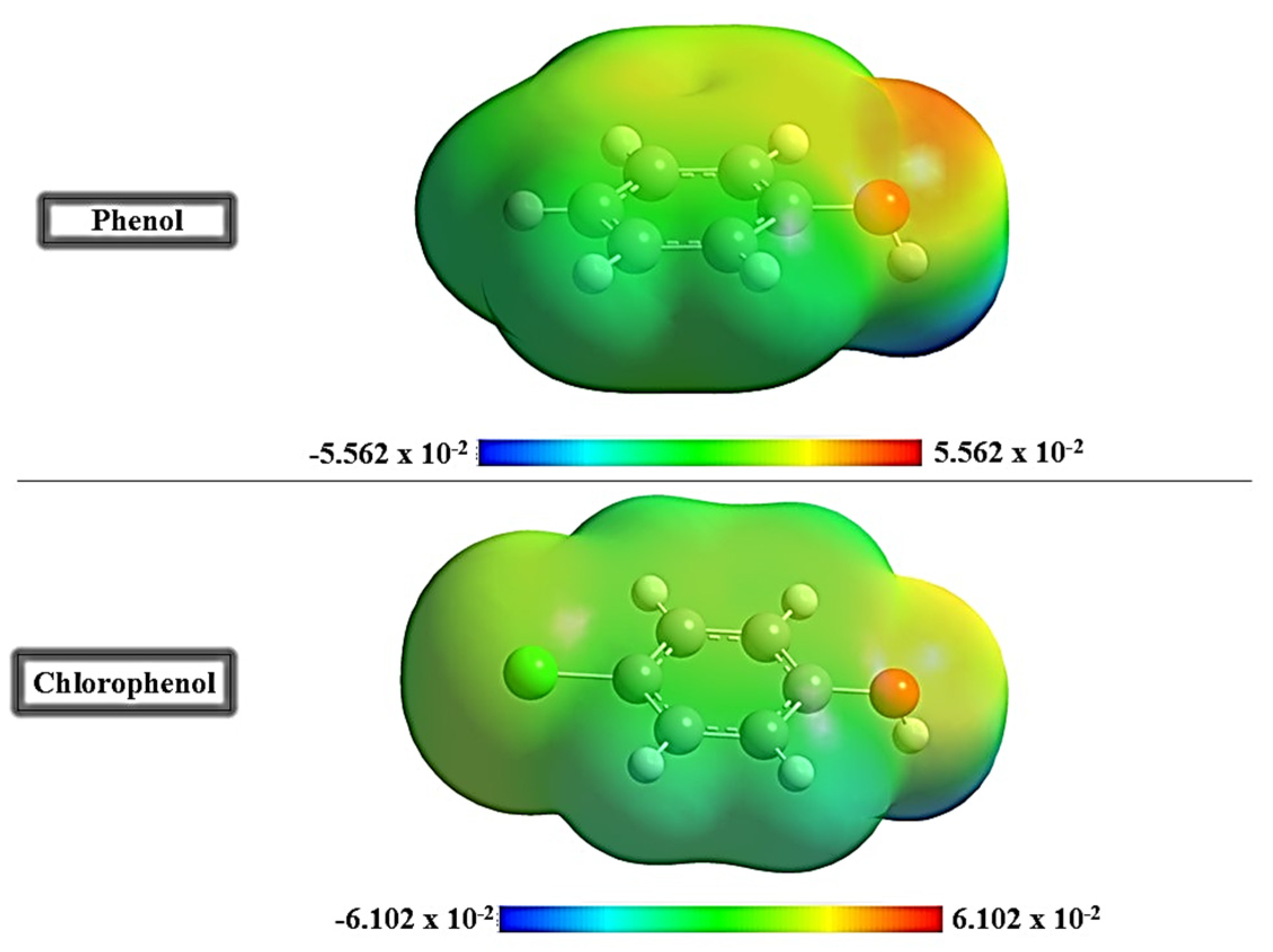
3.2. Molecular Electrostatic Potential (MEP) Maps
3.3. Mulliken Charges and Fukui Indices
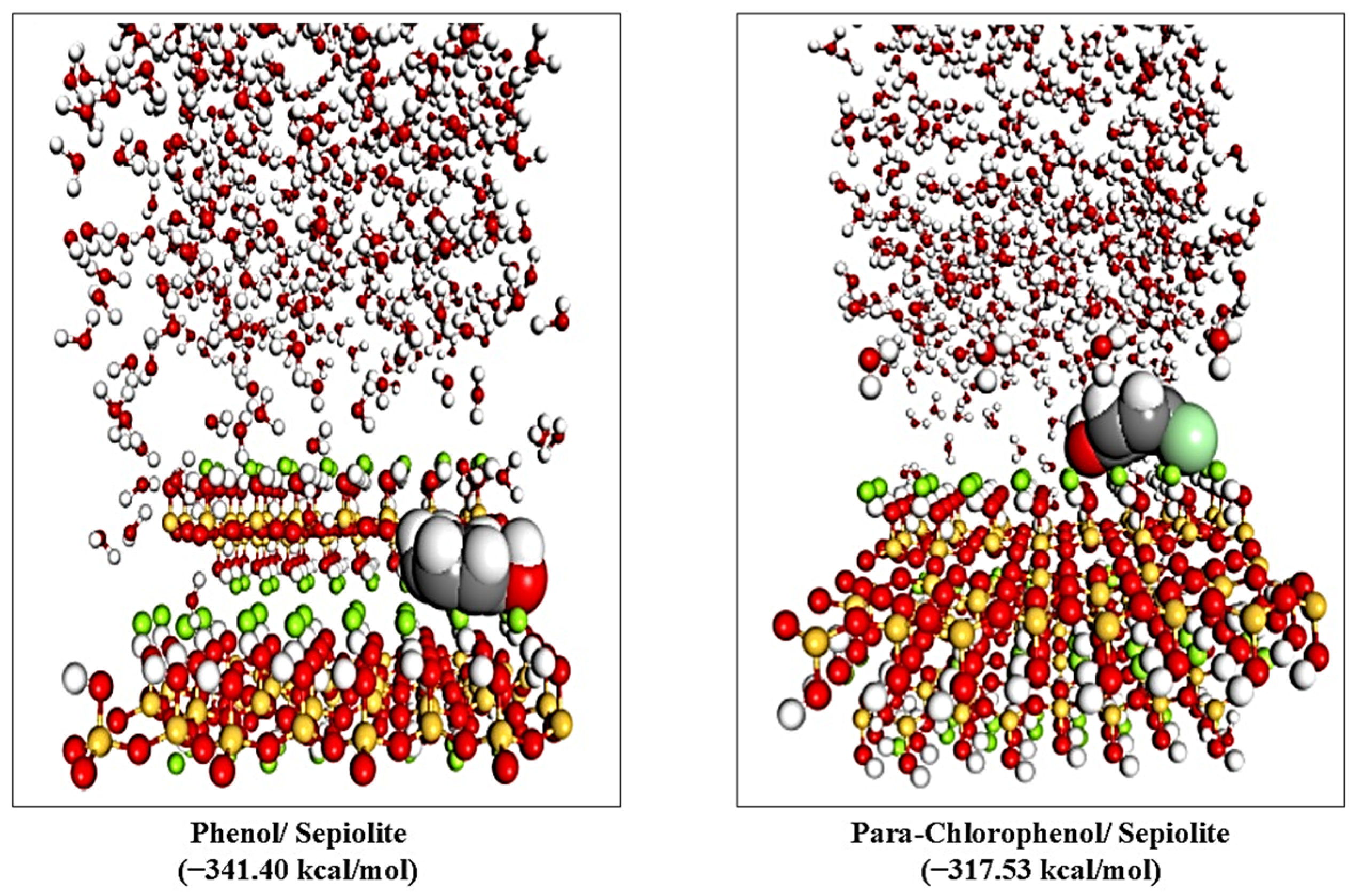
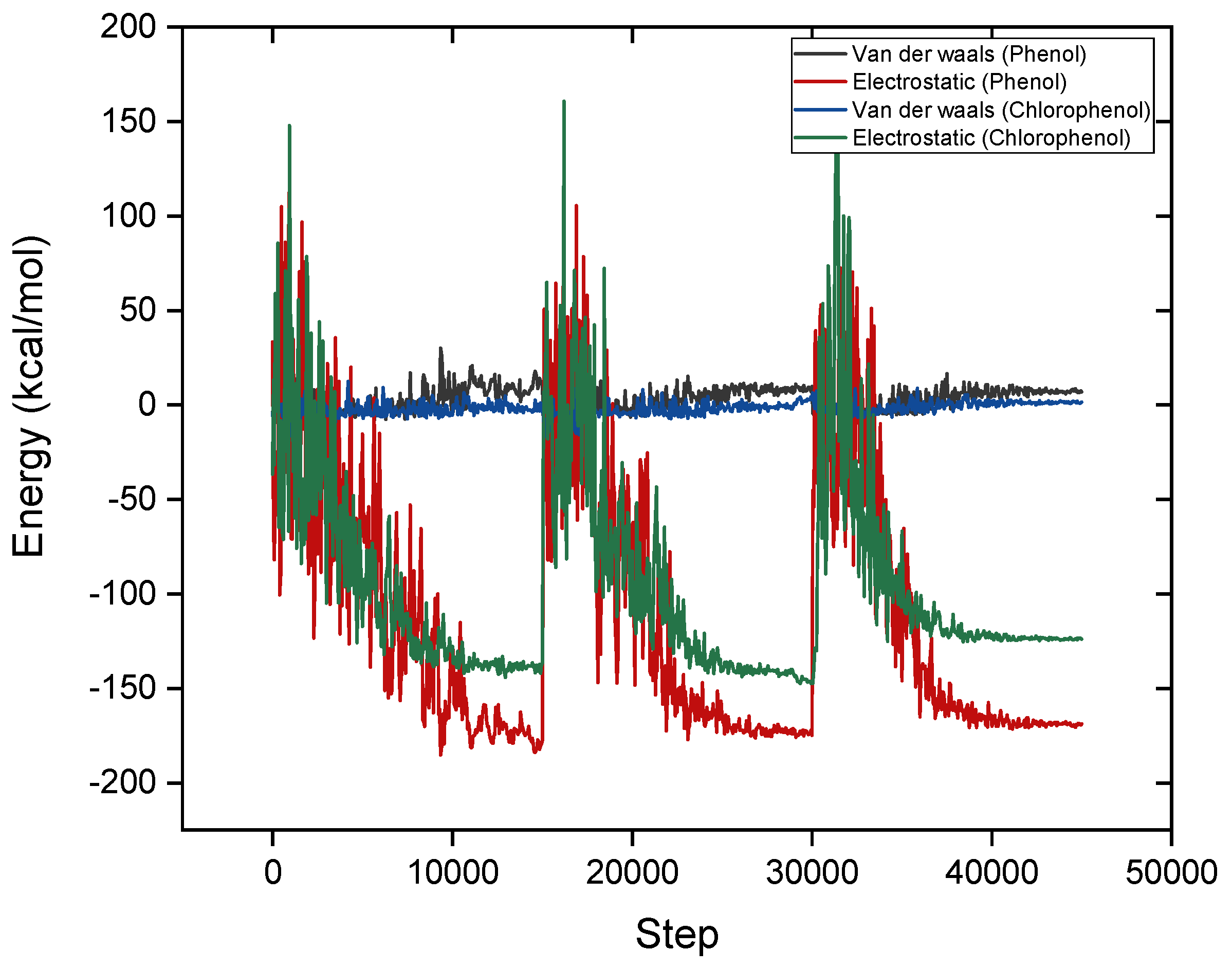
4. Adsorption MD Simulation Results
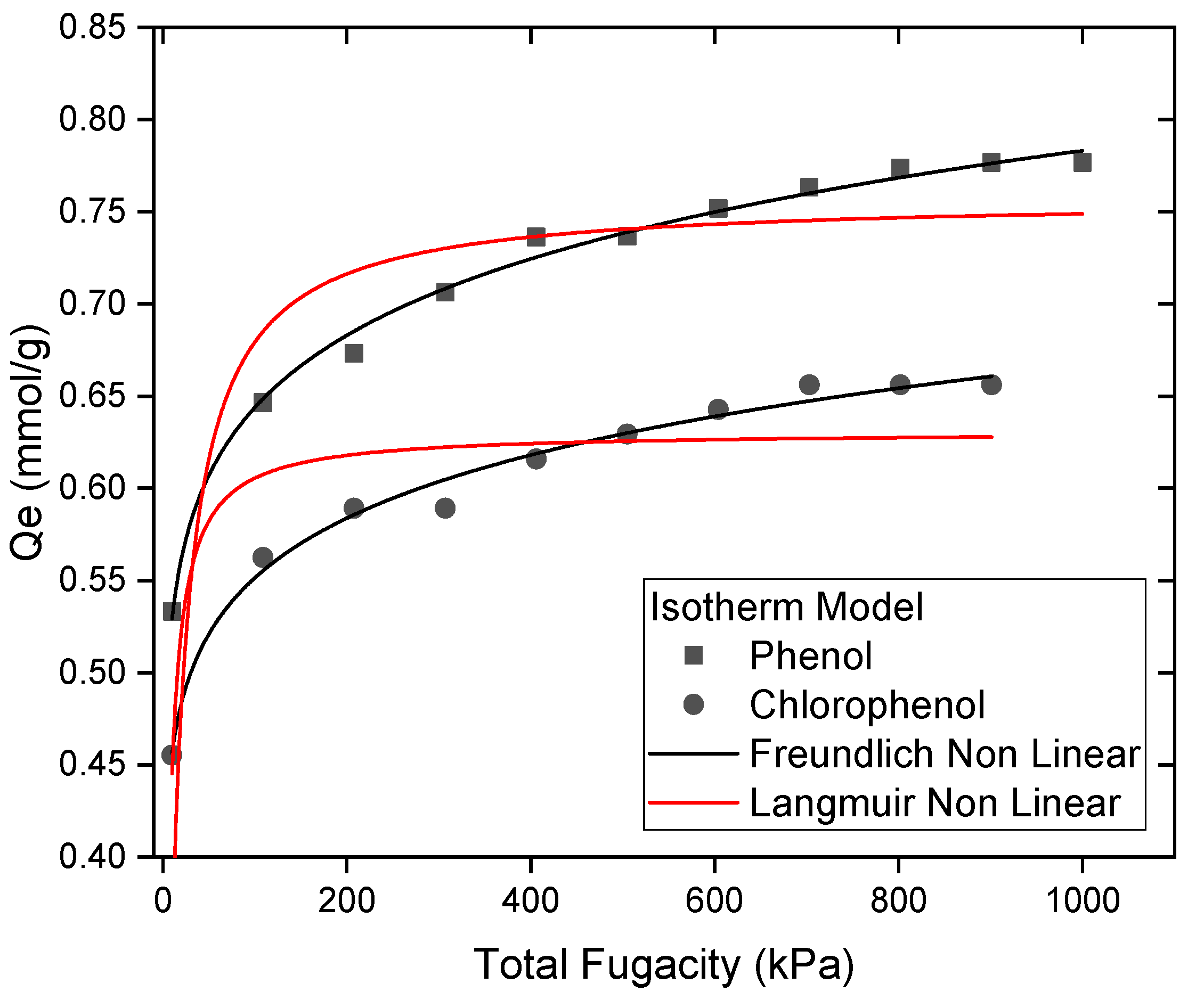
5. Adsorption Isotherm Model
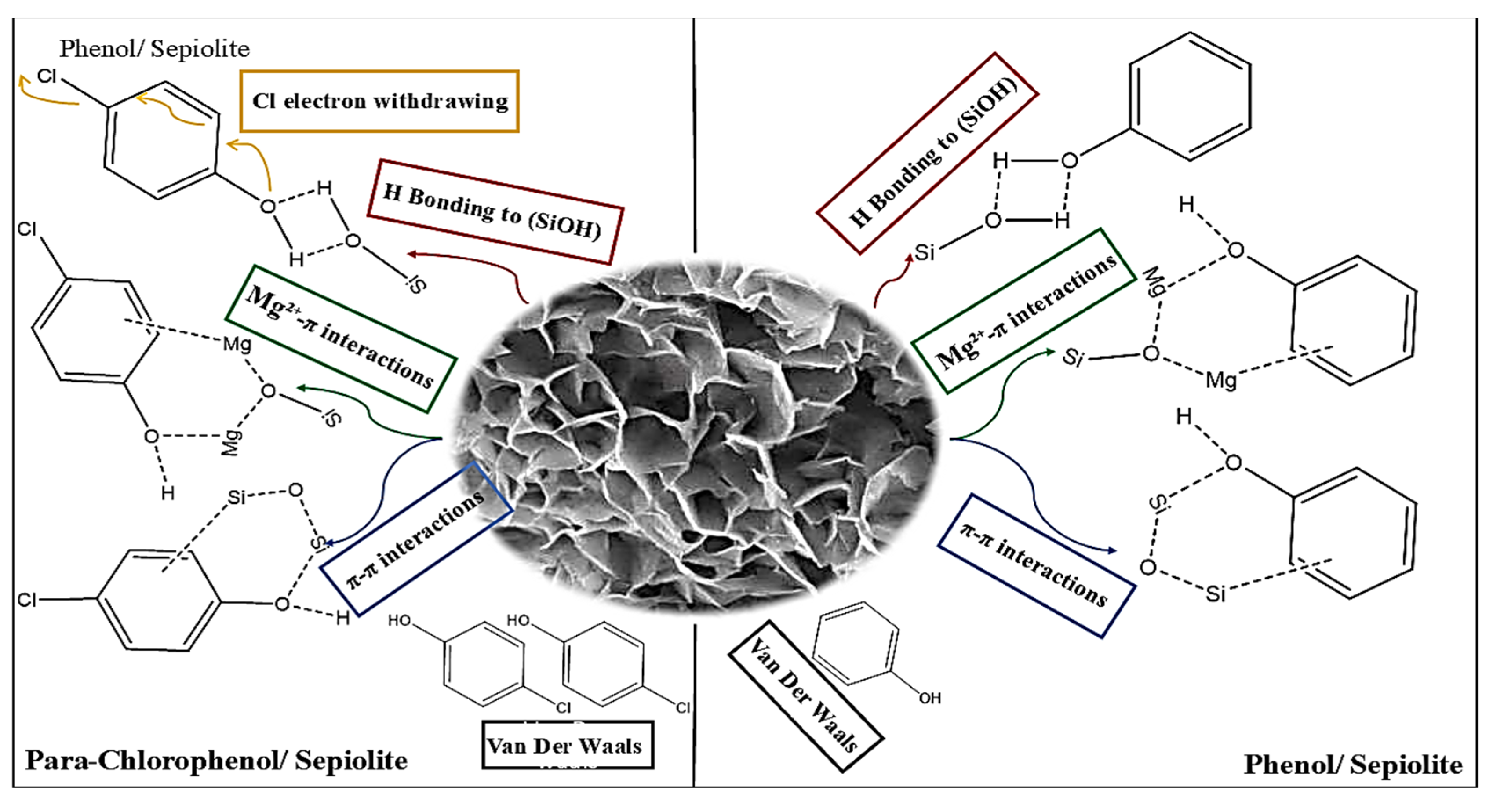
6. Proposed Mechanism
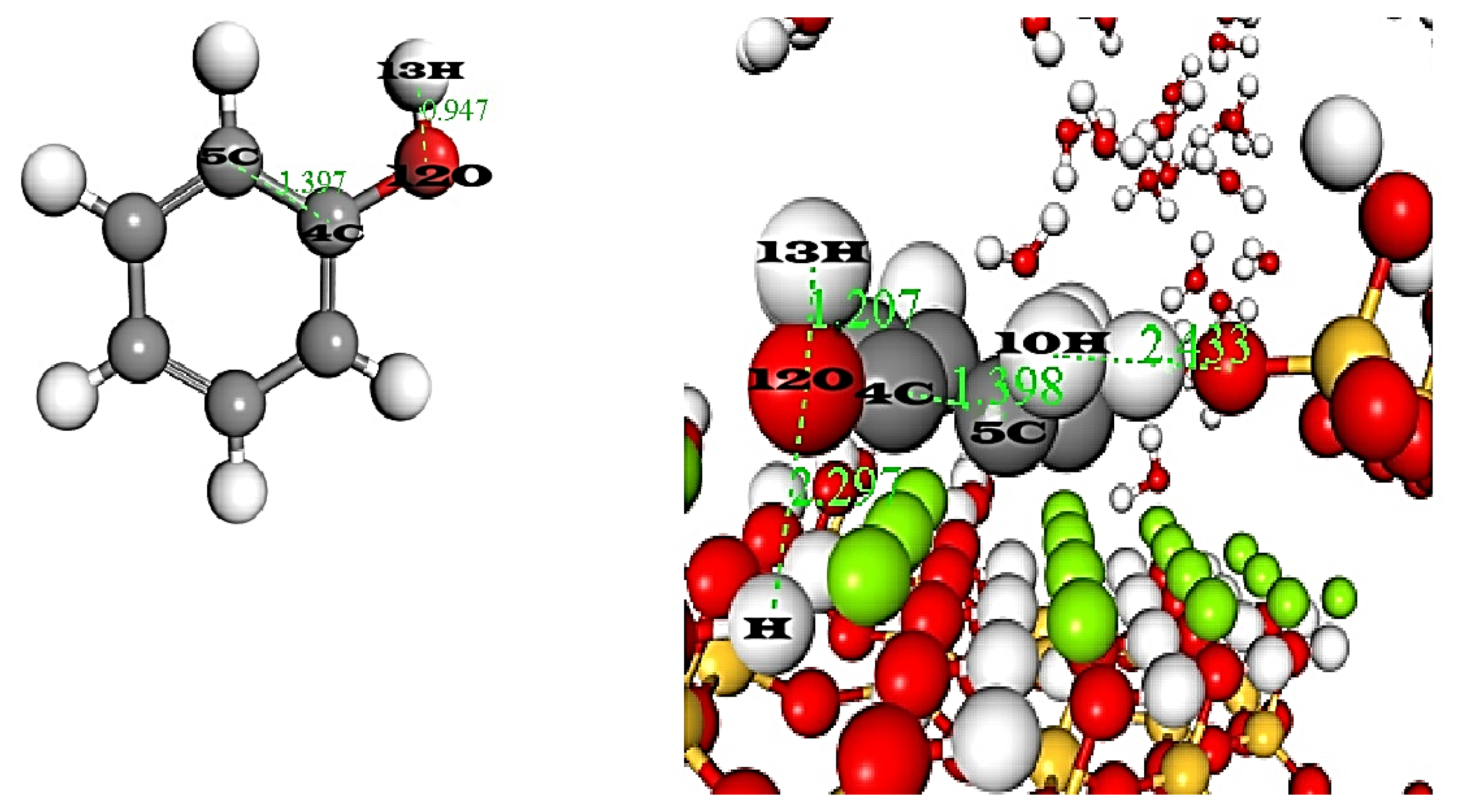
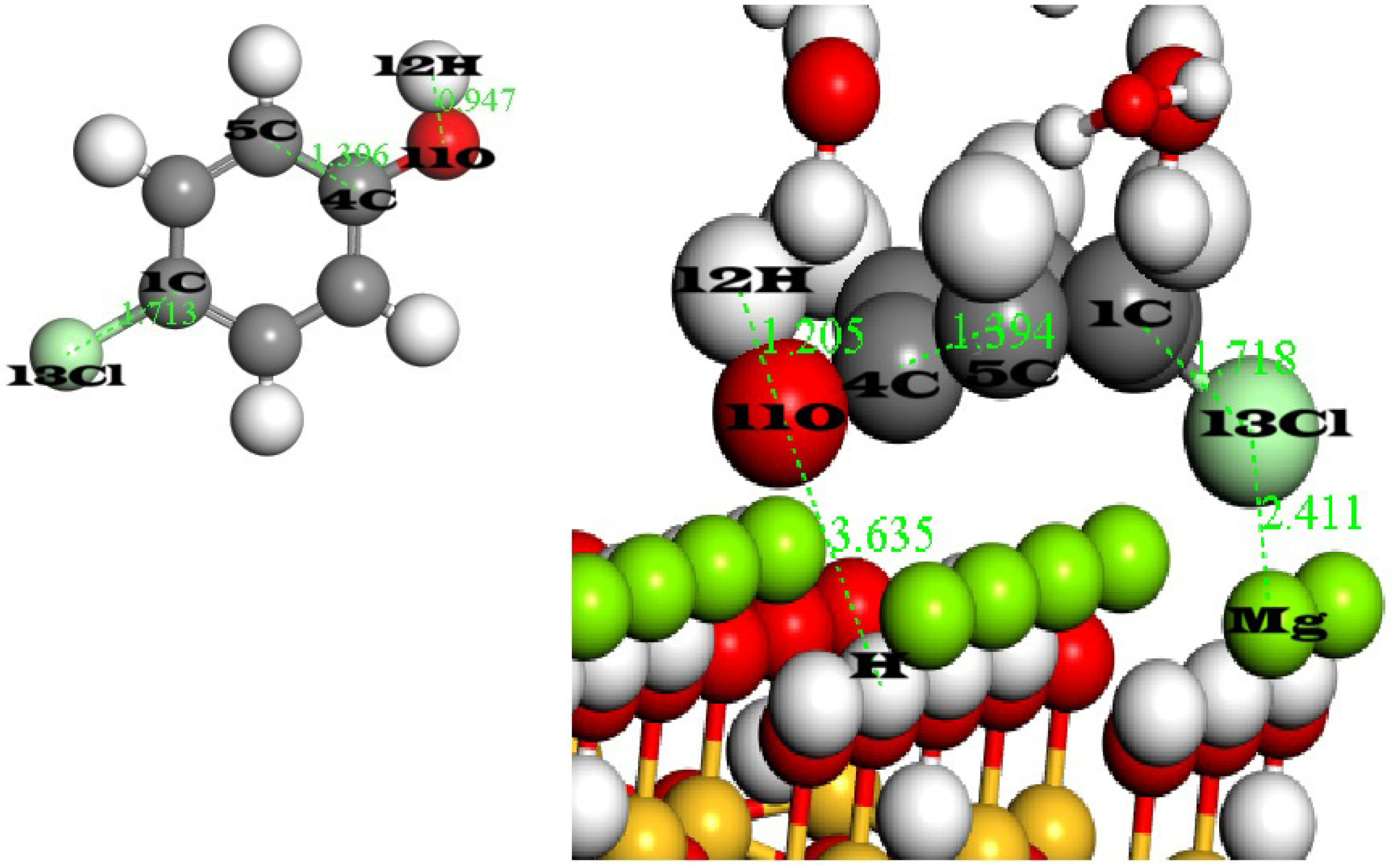
7. Comparative Geometric Analysis
8. Comparison of Phenol/Chlorophenol Adsorption Capacities Across Different Clay
9. Conclusions
- Phenol demonstrates enhanced adsorption efficiency on sepiolite relative to para-chlorophenol, attributed to robust hydrogen bonding and π-cation interactions, as indicated by DFT calculations;
- The hydroxyl group in phenol has a pronounced negative Mulliken charge (−0.428) and notable electrophilic reactivity (fi+ = 0.090), hence augmenting its interaction with silanol (-SiOH) groups;
- The substantial chlorine substituent of para-chlorophenol diminishes its hydrogen bonding capacity and imposes steric hindrance, as evidenced by its less reactive hydroxyl group (fi+ = 0.077) and almost neutral Mulliken charge (−0.020);
- Phenol exhibits reduced deformation energy (+94.18 kcal/mol), facilitating superior structural alignment with sepiolite’s active sites, in contrast to para-chlorophenol (+102.33 kcal/mol);
- Theoretical isotherm findings validate phenol’s superior adsorption capacity (Qmax = 0.78 mmol/g), indicating effective site usage and alignment;
- Nonlinear Freundlich-type behavior for both molecules indicates multilayer adsorption, with phenol establishing a denser and more organized adsorption layer.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmaruzzaman, M.; Mishra, S.R.; Gadore, V.; Yadav, G.; Roy, S.; Bhattacharjee, B.; Bhuyan, A.; Hazarika, B.; Darabdhara, J.; Kumari, K. Phenolic Compounds in Water: From Toxicity and Source to Sustainable Solutions—An Integrated Review of Removal Methods, Advanced Technologies, Cost Analysis, and Future Prospects. J. Environ. Chem. Eng. 2024, 12, 112964. [Google Scholar] [CrossRef]
- Ladeia Ramos, R.; Rezende Moreira, V.; Santos Amaral, M.C. Phenolic Compounds in Water: Review of Occurrence, Risk, and Retention by Membrane Technology. J. Environ. Manag. 2024, 351, 119772. [Google Scholar] [CrossRef] [PubMed]
- Djama, C.; Bouguettoucha, A.; Chebli, D.; Amrane, A.; Tahraoui, H.; Zhang, J.; Mouni, L. Experimental and Theoretical Study of Methylene Blue Adsorption on a New Raw Material, Cynara Scolymus—A Statistical Physics Assessment. Sustainability 2023, 15, 10364. [Google Scholar] [CrossRef]
- Satyam, S.; Patra, S. Innovations and Challenges in Adsorption-Based Wastewater Remediation: A Comprehensive Review. Heliyon 2024, 10, e29573. [Google Scholar] [CrossRef]
- Khader, E.H.; Mohammed, T.J.; Mirghaffari, N.; Salman, A.D.; Juzsakova, T. Removal of Organic Pollutants from Produced Water by Batch Adsorption Treatment|Clean Technologies and Environmental Policy. Clean Technol. Environ. Policy 2022, 24, 713–720. Available online: https://link.springer.com/article/10.1007/s10098-021-02159-z (accessed on 26 March 2025). [CrossRef]
- Benazouz, K.; Bouchelkia, N.; Moussa, H.; Boutheldja, R.; Zamouche, M.; Amrane, A.; Parvathiraja, C.; Al-Lohedan, H.A.; Bollinger, J.-C.; Mouni, L. Efficient Removal of Cu(II) from Wastewater Using Chitosan Derived from Shrimp Shells: A Kinetic, Thermodynamic, Optimization, and Modelling Study. Water 2025, 17, 851. [Google Scholar] [CrossRef]
- Zango, Z.U.; Rozaini, M.N.; Bakar, N.H.H.A.; Zango, M.U.; Haruna, M.A.; Dennis, J.O.; Alsadig, A.; Ibnaouf, K.H.; Aldaghri, O.A.; Wadi, I.A. Advancements in Clay Materials for Trace Level Determination and Remediation of Phenols from Wastewater: A Review. Separations 2023, 10, 125. [Google Scholar] [CrossRef]
- Khan, S.; Ajmal, S.; Hussain, T.; Rahman, M.U. Clay-Based Materials for Enhanced Water Treatment: Adsorption Mechanisms, Challenges, and Future Directions. J. Umm Al-Qura Univ. Appl. Sci. 2023. [Google Scholar] [CrossRef]
- Khachay, A.; Cherifi, H.; Yous, R.; Khalladi, R.; Belaid, B. Mechanistic Insights into the Adsorption of Cationic Crystal Violet onto Sepiolite: A Comprehensive Study Combining Experimental and Theoretical Approaches. J. Mol. Struct. 2025, 1325, 140944. [Google Scholar] [CrossRef]
- Junior, H.B.; da Silva, E.; Saltarelli, M.; Crispim, D.; Nassar, E.J.; Trujillano, R.; Rives, V.; Vicente, M.A.; Gil, A.; Korili, S.A.; et al. Inorganic–Organic Hybrids Based on Sepiolite as Efficient Adsorbents of Caffeine and Glyphosate Pollutants. Appl. Surf. Sci. Adv. 2020, 1, 100025. [Google Scholar] [CrossRef]
- Dehmani, Y.; Lgaz, H.; Alrashdi, A.A.; Lamhasni, T.; Abouarnadasse, S.; Chung, I.-M. Phenol Adsorption Mechanism on the Zinc Oxide Surface: Experimental, Cluster DFT Calculations, and Molecular Dynamics Simulations. J. Mol. Liq. 2021, 324, 114993. [Google Scholar] [CrossRef]
- Kuśmierek, K. The Removal of Chlorophenols from Aqueous Solutions Using Activated Carbon Adsorption Integrated with H2O2 Oxidation. React. Kinet. Mech. Catal. 2016, 119, 19–34. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, J.; Li, D.; Ao, Z.; An, T.; Wang, G. Density Functional Theory Study on the Enhanced Adsorption Mechanism of Gaseous Pollutants on Al-Doped Ti2CO2 Monolayer. Sustain. Mater. Technol. 2021, 29, e00294. [Google Scholar] [CrossRef]
- Salahshoori, I.; Mahdavi, S.; Moradi, Z.; Otadi, M.; Zare Kazemabadi, F.; Nobre, M.A.L.; Ali Khonakdar, H.; Baghban, A.; Wang, Q.; Mohammadi, A.H. Advancements in Molecular Simulation for Understanding Pharmaceutical Pollutant Adsorption: A State-of-the-Art Review. J. Mol. Liq. 2024, 410, 125513. [Google Scholar] [CrossRef]
- Amrhar, O.; Lee, H.-S.; Lgaz, H.; Berisha, A.; Ebenso, E.E.; Cho, Y. Computational Insights into the Adsorption Mechanisms of Anionic Dyes on the Rutile TiO2 (1 1 0) Surface: Combining SCC-DFT Tight Binding with Quantum Chemical and Molecular Dynamics Simulations. J. Mol. Liq. 2023, 377, 121554. [Google Scholar] [CrossRef]
- Khnifira, M.; Mahsoune, A.; Belghiti, M.E.; Khamar, L.; Sadiq, M.; Abdennouri, M.; Barka, N. Combined DFT and MD Simulation Approach for the Study of SO2 and CO2 Adsorption on Graphite (111) Surface in Aqueous Medium. Curr. Res. Green Sustain. Chem. 2021, 4, 100085. [Google Scholar] [CrossRef]
- Zhou, F.; Ye, G.; Gao, Y.; Wang, H.; Zhou, S.; Liu, Y.; Yan, C. Cadmium Adsorption by Thermal-Activated Sepiolite: Application to in-Situ Remediation of Artificially Contaminated Soil. J. Hazard. Mater. 2022, 423, 127104. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. In Proceedings of the Horizons of Quantum Chemistry, Kyoto, Japan, 29 October–3 November 1979; Fukui, K., Pullman, B., Eds.; Springer: Dordrecht, The Netherlands, 1980; pp. 5–15. [Google Scholar]
- Kohn, W.; Sham, L.J. Quantum Density Oscillations in an Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, A1697–A1705. [Google Scholar] [CrossRef]
- Lu, L.; Hu, H.; Hou, H.; Wang, B. An Improved B3LYP Method in the Calculation of Organic Thermochemistry and Reactivity. Comput. Theor. Chem. 2013, 1015, 64–71. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- North, S.C.; Jorgensen, K.R.; Pricetolstoy, J.; Wilson, A.K. Population Analysis and the Effects of Gaussian Basis Set Quality and Quantum Mechanical Approach: Main Group through Heavy Element Species. Front. Chem. 2023, 11, 121554. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Islam, N.; Kaya, S. (Eds.) Conceptual Density Functional Theory and Its Application in the Chemical Domain; Apple Academic Press: New York, NY, USA, 2018; ISBN 978-0-203-71139-2. [Google Scholar]
- Chattaraj, P.K. (Ed.) Chemical Reactivity Theory: A Density Functional View; CRC Press: Boca Raton, FL, USA, 2009; ISBN 978-0-429-13722-8. [Google Scholar]
- Kaya, S.; Kaya, C. A New Method for Calculation of Molecular Hardness: A Theoretical Study. Comput. Theor. Chem. 2015, 1060, 66–70. [Google Scholar] [CrossRef]
- Vismara, R.; Terruzzi, S.; Maspero, A.; Grell, T.; Bossola, F.; Sironi, A.; Galli, S.; Navarro, J.A.R.; Colombo, V. CO2 Adsorption in a Robust Iron(III) Pyrazolate-Based MOF: Molecular-Level Details and Frameworks Dynamics From Powder X-Ray Diffraction Adsorption Isotherms. Adv. Mater. 2024, 36, 2209907. [Google Scholar] [CrossRef] [PubMed]
- Sun, H. COMPASS: An Ab Initio Force-Field Optimized for Condensed-Phase ApplicationsOverview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Vlugt, T.J.H.; Krishna, R.; Smit, B. Molecular Simulations of Adsorption Isotherms for Linear and Branched Alkanes and Their Mixtures in Silicalite. J. Phys. Chem. B 1999, 103, 1102–1118. Available online: https://pubs.acs.org/doi/pdf/10.1021/jp982736c (accessed on 26 March 2025). [CrossRef]
- Ouallal, H.; Dehmani, Y.; Moussout, H.; Messaoudi, L.; Azrour, M. Kinetic, Isotherm and Mechanism Investigations of the Removal of Phenols from Water by Raw and Calcined Clays. Heliyon 2019, 5, e01616. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Liu, X.; Zhang, Y.; Zhao, Q.; Ning, P.; Tian, S. Adsorption Behavior of Phenol by Reversible Surfactant-Modified Montmorillonite: Mechanism, Thermodynamics, and Regeneration. Chem. Eng. J. 2018, 334, 1214–1221. [Google Scholar] [CrossRef]
- Becker, H.G.O. Jan Fleming, Frontier Orbitals and Organic Chemical Reactions. 249 S., John Wiley u. Sons LTD., London/New York/Syndney/Toronto 1976. Clothed £ 8,95, Paperb. £ 3,95. J. Für Prakt. Chem. 1978, 320, 879–880. [Google Scholar] [CrossRef]
- Eddy, N.O.; Ita, B.I. QSAR, DFT and Quantum Chemical Studies on the Inhibition Potentials of Some Carbozones for the Corrosion of Mild Steel in HCl. J. Mol. Model. 2011, 17, 359–376. [Google Scholar] [CrossRef]
- Bahrani-Pour, M.; Beheshti, A.; Sedaghat, T.; Samiee, S.; Hosen, M.A.; Kawsar, S.M.A. A Comparison between the Anti-Thyroid Properties of Methimazole and Its Derivatives with Two S Donor Atoms Endorsed by Iodine Adsorption Capacity, Docking and DFT Calculations. J. Mol. Struct. 2024, 1296, 136819. [Google Scholar] [CrossRef]
- Chen, M.; Gu, Q.; Shao, H.; Liu, H.; Luan, J.; Yan, Z.; Liu, W.; Ke, X. How PPY/CMC Aerogels Possess Selective Adsorption Capacity for Norfloxacin: Coupling Molecular Scale Interpretation with Experiments. Chem. Eng. J. 2023, 464, 142485. [Google Scholar] [CrossRef]
- Ourhzif, E.-M.; Ketatni, E.M.; Akssira, M.; Troin, Y.; Khouili, M. Crystal Structure, Hirshfeld Surface Analysis and DFT Studies of Euphorbioside Monohydrate a Major Bisnorsesquiterpene Isolated from Euphorbia Resinifera Latex. J. Mol. Struct. 2021, 1241, 130511. [Google Scholar] [CrossRef]
- Hasanov, R.; Sadıkoğlu, M.; Bilgiç, S. Electrochemical and Quantum Chemical Studies of Some Schiff Bases on the Corrosion of Steel in H2SO4 Solution. Appl. Surf. Sci. 2007, 253, 3913–3921. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Obot, I.B.; Onyeachu, I.B.; Wazzan, N.; Al-Amri, A.H. Theoretical and Experimental Investigation of Two Alkyl Carboxylates as Corrosion Inhibitors for Steel in Acidic Medium. J. Mol. Liq. 2019, 279, 190–207. [Google Scholar] [CrossRef]
- Bahamon, D.; Khalil, M.; Belabbes, A.; Alwahedi, Y.; Vega, L.F.; Polychronopoulou, K. A DFT Study of the Adsorption Energy and Electronic Interactions of the SO2 Molecule on a CoP Hydrotreating Catalyst. RSC Adv. 2021, 11, 2947–2957. [Google Scholar] [CrossRef]
- Boumya, W.; Khnifira, M.; Machrouhi, A.; Abdennouri, M.; Sadiq, M.; Achak, M.; Serdaroğlu, G.; Kaya, S.; Şimşek, S.; Barka, N.; et al. Adsorption of Eriochrome Black T on the Chitin Surface: Experimental Study, DFT Calculations and Molecular Dynamics Simulation. J. Mol. Liq. 2021, 331, 115706. [Google Scholar] [CrossRef]
- Bourzi, H.; Oukhrib, R.; El Ibrahimi, B.; Oualid, H.A.; Abdellaoui, Y.; Balkard, B.; El Issami, S.; Hilali, M.; Bazzi, L.; Len, C. Furfural Analogs as Sustainable Corrosion Inhibitors—Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media. Sustainability 2020, 12, 3304. Available online: https://www.mdpi.com/2071-1050/12/8/3304 (accessed on 26 March 2025). [CrossRef]
- Zhao, Z.; Fu, D.; Ma, Q. Adsorption Characteristics of Bisphenol A from Aqueous Solution onto HDTMAB-Modified Palygorskite. Sep. Sci. Technol. 2014, 49, 81–89. Available online: https://www.tandfonline.com/doi/full/10.1080/01496395.2013.818693 (accessed on 25 March 2025). [CrossRef]
- Zheng, S.; Sun, Z.; Park, Y.; Ayoko, G.A.; Frost, R.L. Removal of Bisphenol A from Wastewater by Ca-Montmorillonite Modified with Selected Surfactants. Chem. Eng. J. 2013, 234, 416–422. [Google Scholar] [CrossRef]
- Bhattacharyya, K.G.; Dey, D.G. Adsorption of 2-Chlorophenol on Kaolin. SSRN. Available online: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2979013 (accessed on 25 March 2025).
- Moghazy, M.A. High-Efficiency Adsorptive Removal of Phenol from Aqueous Solution Using Natural Red Clay and ZnO Nanoparticles. ChemistrySelect 2022, 7, e202104074. [Google Scholar] [CrossRef]
- Messaid, B.; Djemai, I.; Boudouh, I.; Robustillo, M.D. Assessment of the Performance of an Acid-Activated Composite Made of Na-Montmorillonite Algerian Clay to Remove Phenol and 4-Chlorophenol from Water. Desalination Water Treat. 2025, 322, 101088. [Google Scholar] [CrossRef]
- Ge, M.; Ge, M.; Du, M.; Liang, G.; Hu, G.; Jahangir Alam, S.M. Adsorption Analyses of Phenol from Aqueous Solutions Using Magadiite Modified with Organo-Functional Groups: Kinetic and Equilibrium Studies. Materials 2019, 12, 96. Available online: https://www.mdpi.com/1996-1944/12/1/96 (accessed on 25 March 2025). [CrossRef] [PubMed]
- Ghogomu, J.N.; Noufame, D.T.; Tamungang, E.B.N.; Ajifack, D.L.; Ndi, J.N.; Ketcha, J.M. Adsorption of Phenol from Aqueous Solutions onto Natural and Thermallymodified Kaolinitic Materials. Int. J. Biol. Chem. Sci. 2015, 8, 2325. Available online: https://www.researchgate.net/publication/281689674_Adsorption_of_phenol_from_aqueous_solutions_onto_natural_and_thermallymodified_kaolinitic_materials (accessed on 25 March 2025).
- Richards, S.; Bouazza, A. Phenol Adsorption in Organo-Modified Basaltic Clay and Bentonite. Appl. Clay Sci. 2007, 37, 133–142. [Google Scholar] [CrossRef]
- Yildiz, A.; Gür, A. Adsorption of Phenol and Chlorophenols on Pure and Modified Sepiolite. J. Serbian Chem. Soc. 2024, 72, 467–474. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ionization energy (I) | |
| Electron affinity (A) | |
| Chemical potential (μ) | χ = |
| Hardness (η) | |
| Softness (S) | |
| Global electrophilicity index (ω) | |
| Nucleophilicity (ε) | |
| Back-donation energy (ΔE back-donation) |
| Parameters | Phenol | Para-Chlorophenol |
|---|---|---|
| EHOMO | −6.783 | −6.856 |
| ELUMO | −0.431 | −0.5091 |
| ΔEGAP | 6.352 | 6.077 |
| I | 6.783 | 6.586 |
| A | 0.431 | 0.509 |
| χ | 3.607 | 3.547 |
| μ | −3.607 | −3.547 |
| η | 3.176 | 3.038 |
| S | 0.31 | 0.33 |
| ω | 2.04 | 2.07 |
| ε | 0.49 | 0.48 |
| ΔE back-donation | −0.794 | −0.759 |
| Phenol | Para-Chlorophenol | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atom | Charge | fi+ | fi− | fi∘ | Atom | N | fi+ | fi− | fi° |
| 1C | −0.09858 | 0.119488 | 0.001053 | 0.120541 | 1C | −0.08987 | 0.023476 | −0.01381 | 0.009662 |
| 2C | −0.09028 | 0.045774 | 0.097578 | 0.143352 | 2C | −0.08196 | 0.034383 | 0.07010 | 0.104487 |
| 3C | −0.07859 | 0.070632 | 0.109257 | 0.179889 | 3C | −0.08360 | 0.041943 | 0.07469 | 0.116637 |
| 4C | 0.113792 | 0.079907 | 0.000894 | 0.080801 | 4C | 0.283182 | 0.067005 | 0.01401 | 0.081022 |
| 5C | −0.07859 | 0.070632 | 0.109257 | 0.179889 | 5C | −0.08360 | 0.041942 | 0.07469 | 0.116637 |
| 6C | −0.09028 | 0.045774 | 0.097578 | 0.143352 | 6C | −0.08196 | 0.034383 | 0.07010 | 0.104487 |
| 7H | 0.096526 | 0.093138 | 0.091068 | 0.184206 | 7H | 0.111408 | 0.084951 | 0.11736 | 0.202313 |
| 8H | 0.099092 | 0.08087 | 0.103984 | 0.184854 | 8H | 0.103076 | 0.090871 | 0.12457 | 0.215447 |
| 9H | 0.101857 | 0.082546 | 0.102757 | 0.185303 | 9H | 0.103076 | 0.09087 | 0.12457 | 0.215446 |
| 10H | 0.101857 | 0.082546 | 0.102757 | 0.185303 | 10H | 0.111408 | 0.084951 | 0.11736 | 0.202313 |
| 11H | 0.099092 | 0.08087 | 0.103984 | 0.184854 | 11O | −0.59202 | 0.077178 | 0.04015 | 0.117329 |
| 12O | −0.42810 | 0.090302 | 0.041483 | 0.131785 | 12H | 0.321034 | 0.049966 | 0.037224 | 0.08719 |
| 13H | 0.252234 | 0.057519 | 0.038353 | 0.095872 | 13Cl | −0.02014 | 0.278081 | 0.148948 | 0.427029 |
| Compound | Total Energy (kcal/mol) | Adsorption Energy (kcal/mol) | Rigid Adsorption Energy (kcal/mol) | Deformation Energy (kcal/mol) |
|---|---|---|---|---|
| Phenol | −349.26 | −341.40 | −435.58 | 94.18 |
| Para-chlorophenol | −327.39 | −317.53 | −419.86 | 102.33 |
| Interaction Type | Trend (Phenol vs. Para-Chlorophenol) | Molecular Basis | Effect on Mechanism | Impact on Adsorption |
|---|---|---|---|---|
| Hydrogen Bonding | Phenol: Strong stabilization (−170 kcal/mol). | Phenol -OH group forms strong hydrogen bonds with -SiOH groups. Chlorine in para-chlorophenol withdraws electron density, reducing -OH polarity. | Phenol aligns optimally for hydrogen bonding, enhancing interaction strength. Para-chlorophenol -OH is less nucleophilic, weakening its H-bonding ability. | Phenol: Stronger adsorption. Para-chlorophenol: Weaker adsorption. |
| π-Cation Interactions | Phenol: Significant contribution. Para-chlorophenol: Reduced contribution. | Phenol electron-rich aromatic ring interacts strongly with Mg2⁺ ions in sepiolite. Chlorine in para-chlorophenol reduces electron density on the aromatic ring. | Phenol π-cloud engages Mg2⁺ effectively, stabilizing adsorption. Para-chlorophenol depleted π-cloud weakens π-cation attraction. | Phenol: Enhanced adsorption. Para-chlorophenol: Reduced adsorption. |
| Van der Waals | Phenol: Moderate contributions. Para-chlorophenol: Stronger contributions. | Phenol -OH group is compact and less polarizable. Chlorine in para-chlorophenol is larger and highly polarizable, enhancing dispersive forces. | Para-chlorophenol bulky -Cl group increases the surface contact area, enhancing van der Waals forces. Phenol relies more on electrostatic interactions. | Phenol: Minor role. Para-chlorophenol: Compensates for weaker electrostatics. |
| Steric Effects | Phenol: Minimal hindrance. Para-chlorophenol: Significant hindrance. | Phenol -OH group is compact and does not impede surface alignment. Para-chlorophenol bulky -Cl group disrupts molecular orientation near the surface. | Para-chlorophenol struggles to align optimally for hydrogen bonding and π-interactions. Phenol maintains close surface contact without steric clashes. | Phenol: Favors adsorption. Para-chlorophenol: Hinders adsorption. |
| Aromatic Interactions | Phenol: Strong stacking with the silicate framework. Para-chlorophenol: Weaker stacking. | Phenol aromatic ring retains full electron density for edge-to-face stacking with sepiolite silicate framework. Chlorine distorts ring symmetry and electron density. | Phenol stabilizes through effective aromatic stacking. Para-chlorophenol-distorted ring symmetry reduces stacking efficiency. | Phenol: Strengthens adsorption. Para-chlorophenol: Weakens adsorption. |
| Adsorbent | Phenol Qe (mmol/g) | Chlorophenol Qe (mmol/g) | References |
| Palygorskite | 1.49 | / | [43] |
| Ca-Montmorillonite | 0.37 | / | [44] |
| Kaolin | / | 0.65 | [45] |
| Red clay | 0.89 | / | [46] |
| Na-Montmorillonite | / | 0.23 | [47] |
| Magadiite | 0.55 | / | [48] |
| Meta kaolinite | 0.32 | / | [49] |
| Na-Bentonite | 1.09 | / | [50] |
| Sepiolite | / | 0.3 | [51] |
| HDTMA-Sepiolite | / | 0.7 | [51] |
| Sepiolite | 0.78 | 0.66 | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khachay, A.; Yous, R.; Khalladi, R.; Cherifi, H.; Belaid, B.; Alharthi, M.N.; Salvestrini, S.; Mouni, L. Understanding the Adsorption Mechanism of Phenol and Para-Chlorophenol onto Sepiolite Clay: A Combined DFT Calculations, Molecular Dynamics Simulations, and Isotherm Analysis. Water 2025, 17, 1335. https://doi.org/10.3390/w17091335
Khachay A, Yous R, Khalladi R, Cherifi H, Belaid B, Alharthi MN, Salvestrini S, Mouni L. Understanding the Adsorption Mechanism of Phenol and Para-Chlorophenol onto Sepiolite Clay: A Combined DFT Calculations, Molecular Dynamics Simulations, and Isotherm Analysis. Water. 2025; 17(9):1335. https://doi.org/10.3390/w17091335
Chicago/Turabian StyleKhachay, Abdelhak, Radia Yous, Razika Khalladi, Hakima Cherifi, Bouthaina Belaid, Maymounah N. Alharthi, Stefano Salvestrini, and Lotfi Mouni. 2025. "Understanding the Adsorption Mechanism of Phenol and Para-Chlorophenol onto Sepiolite Clay: A Combined DFT Calculations, Molecular Dynamics Simulations, and Isotherm Analysis" Water 17, no. 9: 1335. https://doi.org/10.3390/w17091335
APA StyleKhachay, A., Yous, R., Khalladi, R., Cherifi, H., Belaid, B., Alharthi, M. N., Salvestrini, S., & Mouni, L. (2025). Understanding the Adsorption Mechanism of Phenol and Para-Chlorophenol onto Sepiolite Clay: A Combined DFT Calculations, Molecular Dynamics Simulations, and Isotherm Analysis. Water, 17(9), 1335. https://doi.org/10.3390/w17091335