Abstract
Human activities are the main sources of antibiotic-resistant genes (ARGs) and mobile genetic elements (MGEs) in the ecosystems of lakes. This research analyzed the abundance of four ARGs (sulI, tetX, cmlA, and aac(6′)-Ib-cr) and one MGE (intI) in sediments from the typical urban and aquacultural polluted areas in Nansi Lake, and further evaluated the risk factors affecting the distribution and occurrence of ARGs. We used 16S rRNA high-throughput sequencing to elucidate the relationship between microbial diversity and ARGs while identifying the possible hosts and sources of ARGs. The results indicated that all five ARGs and MGEs were found in the sampling areas. The abundance of ARGs varied significantly, ranging from 1.29 × 10−6 to 5.59 × 10−4 (copies per 16S rRNA), and the abundance of MGEs was 3.44 × 10−6 to 4.30 × 10−5 (copies per 16S rRNA). The values were relatively higher in the human urban and aquacultural polluted areas than in the pristine environment with minimal nutrient pollution. ARGs exhibited significant correlations with some environmental factors, indicating that environmental factors, such as NH4+-N, total organic carbon (TOC), polystyrene (PS), polyethylene (PE), and polyvinyl chloride (PVC), played crucial roles in the proliferation of ARGs. A network analysis showed that Thermoanaerobaculum, Desulfatiglans, Ignavibacterium, Vibrio, and Spirochaeta were significantly associated with ARGs and MEGs. Meanwhile, these bacterial groups were likely hosts for ARGs and MGEs in the sediments of Nansi Lake. These results underscored the various effects of human activities on the dissemination of ARGs and the composition of microbial communities.
1. Introduction
The presence and evolution of ARGs represent significant challenges for global public health, due to their considerable impact on contemporary medicine [1,2]. ARGs are widely found in diverse surface water bodies [3,4], which were severely polluted due to human activities, such as urban sewage discharge, aquaculture operations, livestock farming, and intensive agriculture [2,5,6]. Given the extensive distribution of ARGs across multiple environments [1,7], ARGs gained recognition as emerging pollutants in 2006. If the current trend of increasing antibiotic resistance continues unchecked, it is projected that, by 2050, antibiotic-resistant bacteria (ARB) could lead to the deaths of 10 million individuals [8,9]. Humans can easily acquire ARGs through interaction with contaminated aquatic environments, which may lead to antibiotic resistance and potentially provoke clinical emergencies [8,9]. In light of this pressing global issue, the World Health Organization (WHO) proposed the One Health approach, which has rapidly gained significant public interest regarding the occurrence of ARGs across different environments over the past few years [8,10]. Gaining insight into the environmental dynamics of ARGs is essential for mitigating the hazards associated with these emerging pollutants to human health.
In contrast to chemical pollutants, ARGs exhibit significant dynamic variability, which can be linked to their intrinsic biological unpredictability [11,12]. It is well established that the transmission of ARGs occurs via both horizontal and vertical gene transfer. Vertical gene transfer pertains to the genetic behavior of ARGs through the reproduction of parent bacteria, while horizontal gene transfer primarily relies on the dissemination of these genetic elements within bacterial communities via MGEs [13,14,15]. The high concentration of bacteria in sediments creates an ideal setting for the horizontal transfer of genes between environmental bacteria and human pathogens [1,3]. This phenomenon is particularly concerning, as it facilitates the spread of ARGs. Research indicates that ARG-associated MGEs are predominantly found in cultured indicator bacteria, such as Enterococcus and coliforms. The positioning of ARGs on various types of MGEs, including plasmids, transposons, and integrons, significantly enhances the likelihood and efficiency of resistance transfer among bacteria, regardless of whether they originate from similar or different sources. This capability underscores the potential risk posed by such genetic exchanges in the context of public health and environmental microbiology [3,13]. Many studies have shown that the abundance of intI, as one of the MGEs, is on the rise due to increased human activities and pollution across various environments, including sewage treatment plants, rivers, and lakes [16,17]. Among these, intI is widely recognized as a crucial MGE facilitating genetic transfer. Given the frequent detection of intI in diverse environmental contexts, its abundance serves as a valuable indirect measure of the proliferation of ARGs [3,18,19].
Lakes frequently function as critical reservoirs for ARGs in aquatic ecosystems due to their relatively confined characteristics, with sediments serving as significant stores for the accumulation of ARGs [20,21]. The variation and dissemination of ARGs are influenced by environmental factors and biotic factors [1,20]. Furthermore, environmental factors, including carbon and nitrogen sources, antibiotic occurrence, and microplastics, impose selection pressures on ARGs, potentially leading to their random appearance or disappearance within the ecosystem [1,10]. When exposed to antibiotics, ARBs exhibit a tendency to acquire additional nutrients or expand their territory, which ultimately leads to an increase in their relative abundance compared to susceptible bacterial strains. This phenomenon can be attributed to the selective pressure exerted by antibiotics, which encourages ARBs to thrive in environments where susceptible bacteria may struggle to survive [1,4]. Consequently, the presence of antibiotics can create an ecological imbalance, favoring the proliferation of ARBs over their more vulnerable counterparts. Additionally, even sublethal doses of antibiotics have been found to exert long-term effects on bacterial physiology. These suboptimal concentrations can enhance both the genetic and phenotypic variability among bacterial populations, fostering a greater diversity of traits within these communities. Notably, low doses of antibiotics can function as signaling molecules, facilitating communication between bacteria. This signaling capability leads to a variety of functional outcomes, including alterations in gene expression, the ability for quorum sensing, the formation of biofilms, and increased virulence. Such responses highlight the complex interactions that can occur in microbial environments under antibiotic stress [12]. While the global distribution and levels of pollution concerning ARGs in sediments have been reported, their dynamic behavior related to different human activities has been explored in only a few studies. Understanding how different human behaviors impact sediment ARGs is important for fully understanding the behavior of ARGs in lake environments.
Nansi Lake serves as a crucial transit point in the eastern route of China’s South-to-North Water Diversion Project, facilitating the transport of water from Jiangsu Province to areas such as Shandong and Tianjin Province [7,22]. This lake serves as a primary source of drinking water for millions of residents in Jining and Zaozhuang, featuring an average depth of 1.5 m. Additionally, it plays an essential role for local communities in terms of work, aquaculture, and recreational activities [7,23]. However, various human activities associated with urbanization and aquaculture have led to the dissemination of different ARGs. The presence of these ARGs within the lake increases the risk of human exposure through contact with the contaminated environment. Despite this, the effects of diverse human activities on the proliferation of ARGs in the lake’s sediment remain poorly understood, which is essential for comprehending the behavior of ARGs in aquatic ecosystems [1,3].
The objective of this research is to investigate the characteristics of ARGs and the distribution of microbial communities in lake sediments. In this study, quantitative real-time PCR (qPCR) was used to quantify four ARGs from four classes of antibiotics and one MGE gene in the sampling areas. Additionally, microbial diversity in the region was examined through 16S rRNA high-throughput sequencing, which aimed to elucidate the relationship between microbial diversity and ARGs, identify potential sources and hosts for these ARGs, and offer a scientific foundation for the effective utilization and management of antibiotics in lake ecosystems. Furthermore, this study explored the relationship between environmental factors and antibiotic resistance, especially the relationship between microplastic pollution and antibiotic resistance. This research enhances the understanding of pollution levels of ARGs in freshwater settings and provides a scientific guide to the sustainable development of animal husbandry and aquaculture, as well as ecological environment safety management.
2. Materials and Methods
2.1. Study Site and Sampling
Sediment samples were collected from Nansi Lake in Shandong Province, China. The lake is divided into three distinct areas: the upstream area, referred to as AP, which represents a pristine environment with minimal nutrient pollution; the midstream area, known as AA, which is affected by human aquaculture with the use of antibiotics; and the downstream area, designated AU, contaminated by high levels of nitrogen and organic pollutants, situated around the city center and influenced by urban waste (Figure S1, Table S1). Sediment samples were collected in autumn during a single campaign. A total of 27 samples were obtained from the three areas (AP, AA, and AU), with three sampling sites established in each area. At each site, three samples were collected as replicates, consisting of five thoroughly mixed sediment cores taken from a bucket rinsed with sterilized distilled water before collecting different samples. Each sample, weighing roughly 1 kg, was carefully stored in a cooler during the sampling events. Following this, the samples were promptly transported to the laboratory. Then the samples were stored at −80 °C for subsequent DNA extraction and parameter testing.
2.2. Physicochemical Parameters and Microplastic Determination
The sediment samples were air-dried, and 10 g of sediment samples were used for each parameter testing. pH was tested with a pH meter by a soil-to-distilled water ratio of 1:2.5. TOC was assessed by an organic carbon analyzer (Multi N/C2100, Jena, Germany). To extract sulfate, deionized water was utilized at a 1:5 ratio, with subsequent measurement conducted via an ion chromatography system (ICS-1000; Dionex, Sunnyvale, CA, USA). The extraction of nitrate and ammonium from the samples was performed using 2 M KCl (Aladdin, Shanghai, China), followed by analysis with a continuous flow analyzer (Auto Analyzer 3, Seal, Germany) [24]. The parameters of the sediments are presented in Table S1. The variety and amounts of microplastics found in the sediments were identified through Raman spectroscopy, following the methods described previously [25]. Three repetitions were applied for each parameter test. The reagents employed during analysis were all guaranteed reagent grade.
2.3. Quantification of 16S rRNA and ARGs
DNA from each freeze-dried sample was extracted using the MoBio Powersoil DNA Extraction Kit (MoBio, Carlsbad, CA, USA) in accordance with the manufacturer’s guidelines and quantified with the NanoDrop One (Thermo Scientific, Wilmington, DE, USA). The purified DNA was stored at −80 °C for further analysis. qPCR is an exceptionally sensitive technique for measuring microbial abundance. In the present study, 16S rRNA quantification was performed using qPCR with a CFX Connect™ real-time PCR system (Bio-Rad, Hercules, CA, USA). The primers BACT1369F and PROK1492R were utilized for the quantification of 16S rRNA. Additionally, five genes were amplified, comprising four ARGs from distinct categories of antibiotics (tetracycline, sulfonamide, fluoroquinolone, and chloramphenicol resistance genes) and one MGE (primer information is provided in Table S2). Each qPCR reaction was carried out using a reaction mixture of 25 μL that contained SYBR Green I (Roche, Basel, Switzerland). Absolute abundances were calculated by interpolating sample Ct values against the standard curve. Then, they were normalized to 16S rRNA gene copies to calculate the relative abundance.
2.4. High-Throughput Sequencing
The V4–V5 region of 16S rRNA was amplified, employing the primer pair F515/R907. Purified amplicons were pooled in equal molar proportions and subjected to paired-end sequencing (2 × 300 bp) by the Illumina MiSeq platform (Illumina, San Diego, CA, USA) [26,27]. Quality control of the raw fastq files was executed via Trimmomatic, and merging was accomplished with FLASH VI.2.7 [28]. Following this, operational taxonomic units (OTUs) were produced using Uparse (version 7.0.1090; https://drive5.com/uparse/ accessed on 14 May 2025), implementing a similarity threshold of 97%. All the 16S rRNA gene sequences were classified taxonomically against the 16S rRNA database Silva (silva138/16s_bacteria), using a confidence threshold set at 70% [29].
2.5. Statistical Analysis
OTUs for each sample were assessed using the Bray–Curtis distance metric, followed by a principal coordinate analysis (PCoA). Branch diagrams were generated with the linear discriminant analysis effect size (LEfSe) algorithm (http://huttenhower.sph.harvard.edu/galaxy accessed on 14 May 2025). To clarify differences in the microbial taxa, linear discriminant analysis (LDA) effect size was employed [30], with an LDA score of ≥ 2 indicating significant contributions to the model. Network co-occurrence analysis was established using the Maslov–Sneppen method and visualized with Gephi (version 0.9.2) [31]. Figures were generated with Origin software (version 9.1). To evaluate significant differences among groups (p < 0.05), one-way analysis of variance was performed with IBM SPSS Statistics (version 26).
3. Results
3.1. Quantitative Analysis of 16S rRNA and ARGs
The abundance of bacteria determined by 16S rRNA in three areas is summarized in Figure 1A. The relative abundances of four ARGs belonging to four classes of antibiotics and one MGE are depicted in Figure 1B–F. The amplification efficiencies were 0.983, 0.967, 0.973, 0.952, 0.966, and 0.958 for 16S rRNA, sulI, tetX, cmlA, aac(6′)-Ib-cr, and intI, respectively.
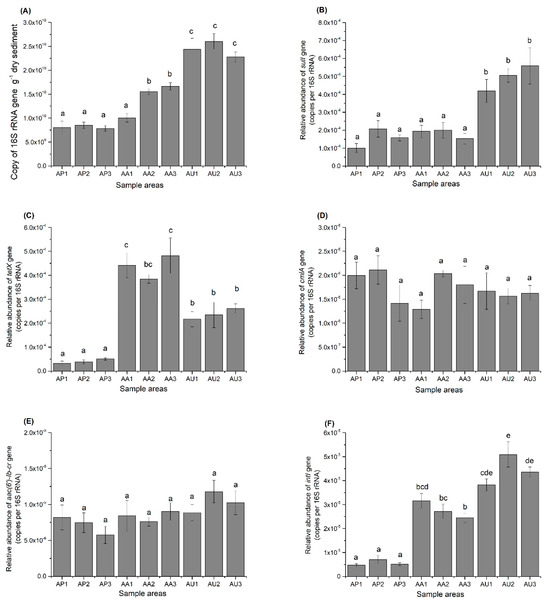
Figure 1.
The abundances of 16S rRNA (A), sulI (B), tetX (C), cmlA (D), aac-lb-cr (E), and intI (F) gene copies in sediments from Nansi Lake subjected to various forms of human activities. The values shown represent the means from three replicates along with their standard errors. Distinct letters signify a significant difference (p < 0.05).
In all AU sampling sites and two AA sampling sites, 16S rRNA genes exhibited significantly higher abundance levels compared to all AP sites (p < 0.05). No significant differences were noted between the AP and AA sampling sites. In every location, sulI and tetX were more abundant than cmlA and aac(6′)-Ib-cr. The relative abundance of sulI and tetX varied from 1.01 × 10−4 to 5.59 × 10−4 and from 3.28 × 10−5 to 4.82 × 10−4 (copies per 16S rRNA). Meanwhile, the relative abundances of aac(6′)-Ib-cr and cmlA ranged from 5.75 × 10−6 to 1.17 × 10−5 and from 1.29 × 10−6 to 2.11 × 10−6 (copies per 16S rRNA). In the AU sampling sites, the abundances of both sulI and tetX were significantly higher than those in the AP sampling sites (p < 0.05). Additionally, in all three AA sampling sites, the tetX abundance was significantly elevated than in AP sampling sites. However, no notable variations in sulI relative abundance were detected between the AP and AA areas. A more pronounced difference in the influence of human activities on ARGs emerged when analyzing the relative abundance of intI. The relative abundance of intI was markedly greater in all AA and AU sampling sites compared to the AP sampling sites, with significant differences also observed between the AA and AU sampling sites. However, there were no significant differences for both cmlA and aac(6′)-Ib-cr in all three areas.
3.2. Correlation Analysis Between Sediment Parameters and ARGs
To investigate the connection between sediment parameters and ARGs, a correlation analysis was performed involving six genes and eight sediment parameters, as shown in Table 1. The results indicated significant associations between various sediment parameters and ARGs. Specifically, the 16S rRNA gene exhibited strong positive correlations with NH4+-N and all three kinds of microplastics. Additionally, the relative abundance of sulI displayed significant correlations with NH4+-N, TOC, PE, PVC, but an insignificant correlation with PS. Interestingly, the relative abundance of both tetX and intI showed significantly positive correlations with all three kinds of microplastics and TOC, while intI also exhibited strong positive correlations with NH4+-N compared with tetX. As the least relative abundance of all six genes, cmlA showed nonsignificant positive or negative relations with all eight sediment parameters. However, the relative abundance of aac(6′)-Ib-cr displayed significant correlations with NH4+-N, TOC, PE, and PVC as the less relative abundance of all six genes. These findings highlight the important role that physicochemical parameters and microplastics play in the spread of ARGs. In conclusion, the key parameters that acted as significant mediators were NH4+-N, TOC, PE, PVC, and PS in the study areas.

Table 1.
Importance of environmental parameters related to the abundance of 16S rRNA and ARGs.
3.3. Variations in Microbial Diversity Related to Human Activities
From the three sampling areas, a total of 3,454,952 high-quality sequences of the 16S rRNA gene were acquired through the Illumina MiSeq platform, averaging 127,961 sequences for each sample, with three replicate samples taken at each site across the three areas. Each sample was randomly re-sampled and yielded 60,000 sequences. A total of 8145 OTUs were identified, and the resampled OTU table served for further analysis. An evaluation of the OTUs revealed a high level of diversity among microbial communities. Microbial communities were dominated by Firmicutes (ranging from 12.84% to 36.72% of sequences), Proteobacteria (9.73% to 38.46%), and Chloroflexi (8.65% to 18.45%) across all locations. The α-Diversity indices indicated significant differences in community diversity (p < 0.01) between AP and AU, as well as between AP and AA, such as the Simpson indices and Shannon indices (Table 2). However, no significant differences were observed between samples from AU and AA (p > 0.05). Meanwhile, the ACE and Chao index indicate significant community richness differences (p < 0.01) between AP and RA, while there were no significant differences between AP and AU and between AU and AA. The PCoA analysis of the sequence data illustrated significant clustering based on locations, with the primary principal component (PC) scores registering PC1 = 35.31% and PC2 = 19.76% (Figure 2).
Table 2.
Alpha diversity of bacterial communities accompanied by standard errors.
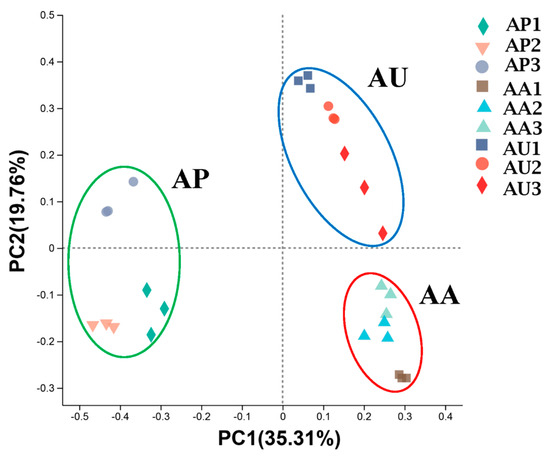
Figure 2.
Principal coordinate analysis of sediment microbiota in Nansi Lake. Each point represents the sediment microbiota of a replicate in AP (green circle), AA (red circle), or AU (blue circle). n = 9 per region.
The primary phyla identified in the three areas included Firmicutes, Proteobacteria, Chloroflexi, Actinobacteriota, and Bacteroidota. In comparison to AP and AU, the relative abundance of Firmicutes was markedly reduced in AA, whereas AA exhibited a notably increased proportion of Chloroflexi. A total of fifteen phyla displayed significant variation between AU and AP, as well as between AA and AP. Differences were also significant among these areas for Proteobacteria, highlighting a distinct microbial community structure within this phylum. We hypothesized that diverse human activities have significantly influenced the microbial community structures observed in AA and AU.
3.4. Differences in Microbial Community Structure Among Areas
We conducted a further investigation to determine whether various human activities influence microbial community structure at diverse taxonomic levels. The microbial taxa that exhibit the most significant differences among areas are illustrated in Figure 3 with LDA scores. At the class level, AU and AA were clearly distinct from AP. AU showcased an elevated presence of Clostridia, Actinobacteria, Thermoleophilia, and Limnochordia, whereas AA exhibited a rise in Anaerolineae, Vicinamibacteria, Acidimicrobiia, and Bacteroidia. Additionally, more pronounced differences among the areas were observed at the order level. AU displayed an increase in Gaiellales, while AA was defined by a prevalence of Anaerolineales, Vicinamibacterales, Ignavibacteriales, Microtrichales, and Pirellulales. Furthermore, AU and AA were also notably different from AP at the family level. AU revealed higher quantities of Clostridiaceae, Bacillaceae, and Hungateiclostridiaceae, in contrast to AA, which was characterized by an increase in Anaerolineaceae and Pirellulaceae.
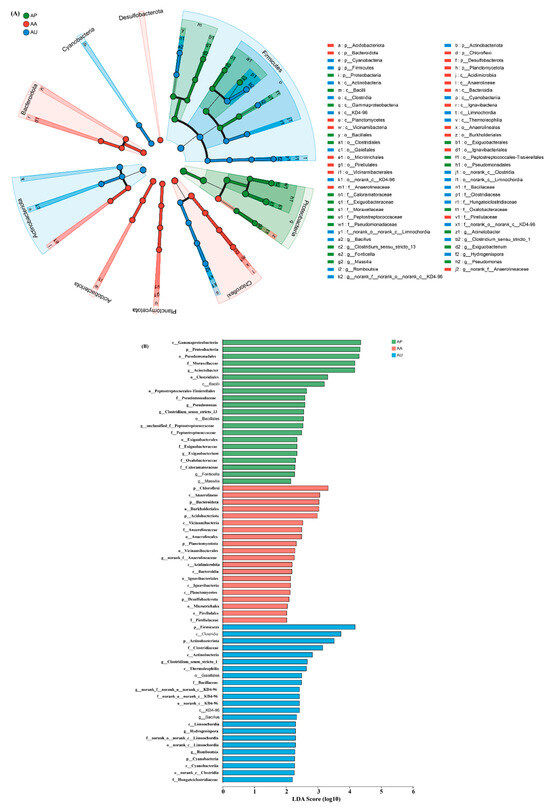
Figure 3.
Taxonomic variations among polluted and non-polluted areas were analyzed using linear discriminant analysis effect size (LEfSe). (A) The cladogram illustrates significant statistical differences across various areas, indicated by the color of the most prevalent genera (green: AP; red: AA; blue: AU). The circles’ diameters reflect the abundance of the taxa. (B) Linear discriminant analysis (LDA) scores were used to evaluate differing genera (green: AP; red: AA; blue: AU). The visualization highlights microbial taxa with elevated LDA scores. n = 9 for each area. p__: phylum; o__: order; f; c__: class; o__: order; f__: family.
To further corroborate the differences in functional populations linked to human intervention, a heat map illustrating the 50 most prominent genera was created (Figure 4). The results showed that the majority of these dominant genera exhibited higher levels in AU and AA in contrast to AP. Certain phylotypes, including Methylocystis, Spirochaeta, Methylocaldum, and Vibrio, were found to be more prevalent in the sediments of both AA and AU, whereas others, like Desulfatiglans, Blastopirellula, and Thiobacillus, were exclusively found enriched in the sediments of AA. Acinetobacter, identified as a major genus, showed the highest abundance in AP, but the abundance significantly decreased in both AA and AU. AA was dominated by Romboutsia, Thiobacillus, and Ignavibacterium, while AU was dominated by Romboutsia, Bacillus, and Hydrogenispora. Clearly, Romboutsia was enriched in both AA and AU compared to AP.
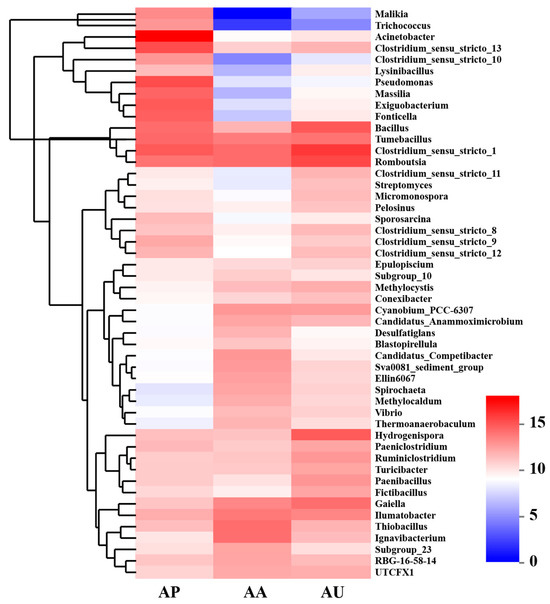
Figure 4.
Heatmap of genera abundance (normalized on a log2 scale) found in lake sediments subjected to varying human interventions.
3.5. Network Analysis Involving Bacterial Taxa and ARGs
This study utilized network analysis to examine the connections of 100 predominant bacterial genera alongside five ARGs and MGEs pertinent to the study area (Figure 5). Overall, the networks based on the 16S rRNA gene comprised 37 nodes and 61 significant (p < 0.05) correlations with a Spearman’s correlation coefficient ρ > 0.7 or < −0.7. Most of the genera belong to Firmicutes and Proteobacteria at the phylum level. Twenty-nine bacterial genera were identified as potential hosts of ARGs, including Spirochaeta, Ignavibacterium, Desulfatiglans, Bacillus, Thiobacillus, Methylocaldum, Trichococcus, Malikia, and Vibrio, which may be associated with tetX and sulI. tetX had a stronger positive correlation with Spirochaeta, Vibrio, and Ignavibacterium than with other genera. sulI had a significant positive correlation with Cyanobium_PCC-6307 and two significant negative correlations with Trichococcus and Malikia, but the correlations were weaker in all correlations. Furthermore, thirty bacterial genera were also identified as potential hosts of MGEs, such as Spirochaeta, Vibrio, Exiguobacterium, Acinetobacter, Pseudomonas, Desulfatiglans, and Methylocystis, which may be associated with intI. intI had strong negative correlations with Fonticella and Malikia, and strong positive correlations with Ignavibacterium and Spirochaeta. Interestingly, both tetX and intI had strong positive correlations with Ignavibacterium and Spirochaeta, indicating that Ignavibacterium and Spirochaeta were hosts of both tetX and intI. cmlA and aac(6′)-Ib-cr did not present identified hosts within the top 100 bacterial genera, indicating that additional hosts may be present in the study areas. These results provide significant understanding regarding the possible hosts of ARGs and MGEs. Nonetheless, further investigations, including the isolation of ARB strains and genome sequencing, are necessary to confirm the identified hosts.
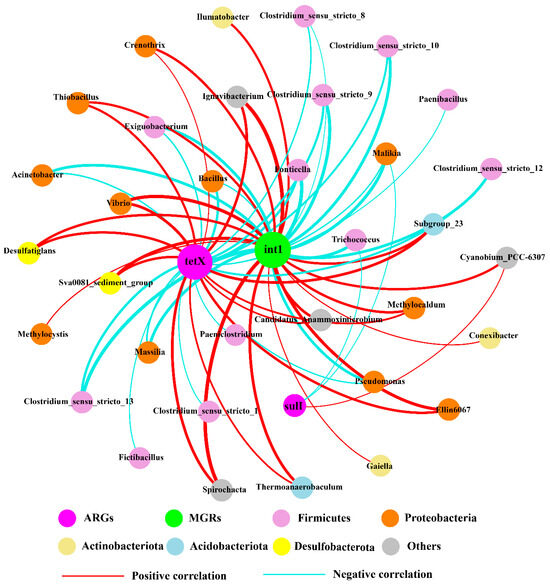
Figure 5.
Co-occurrence network based on bacterial 16S rRNA genera in the sediments. Each node symbolizes a genus, while connections represent strong and significant correlations. The widths of the edges correspond to their weights, and the colors of edges indicate whether the correlation for the interconnected nodes is positive or negative.
4. Discussion
4.1. Distribution of ARGs and MGEs in Nansi Lake
In recent years, the excessive use of antibiotics has led to the emergence of ARGs in microorganisms and animals. The occurrence of ARGs in Nansi Lake aligns with the findings from earlier research, which identified numerous ARGs in the areas affected by urban and aquaculture pollution [7,20]. Furthermore, prior studies have suggested that ARB and ARGs found in lakes could stem from sources including aquaculture establishments, livestock operations, and sewage treatment facilities [8,13,19,27]. For example, the prophylactic use of antibiotics in aquaculture farms has led to a rise in the number of ARBs [5]. The presence of MGEs has also been documented in aquatic ecosystems, including water sources from six urban lakes in Wuhan, Taihu Lake, and sediment collected from lakes along the Yangtze River [20]. Our findings indicated that microbial abundance in the sediments of Nansi Lake was significantly greater across all AU regions compared to those in the AP and AA regions, with the exception of AA1. The concentration of 16S rRNA in sediment samples demonstrated a significant correlation (p < 0.05) with NH4+-N and TOC, with values of 0.827 and 0.701, respectively. Previous research has identified similar associations between microbial abundance and elevated nutrient levels [3,10]. Activities related to urbanization and aquaculture contribute to the dissemination of ARGs in aquatic systems [8,9]. An analysis of samples from various collection sites revealed that the sampling site AU3 exhibited the highest quantity of ARGs (sulI), while the AP1 sampling site displayed the lowest levels. The investigation noted that numerous industrial activities and farms surrounding the AU3 site contributed to high levels of wastewater discharge from the above facilities. Conversely, the AP1 sampling site is located in a region with limited aquaculture activities, suggesting that the complex discharge of urban wastewater might be a factor in the increased presence of ARGs and MGEs observed in this area [4,9]. Comparable results were reported for tetX and intI, indicating that wastewater from urban industries and farms can significantly enrich the concentrations of these genes [26]. Interestingly, increased levels of tetX and intI were also observed at the AA sampling site. Tetracycline ARGs have been widely documented in fish farming and bacteria in aquaculture sites during different sampling years [32]. An analysis of the environment surrounding the AA sampling site revealed a high density of aquaculture farms in the vicinity, coupled with substantial wastewater discharge. This suggests that the release of complex aquaculture wastewater could contribute to the elevated levels of tetX and intI in the region. The application of antibiotics within aquaculture has accelerated the proliferation of ARGs and MGEs [8,20]. Earlier research has revealed a specific relationship between ARGs and the application of antibiotics in aquaculture environments. This suggests that these antibiotics may influence the emergence and spread of ARGs in the aquatic environment [8]. Elevated concentrations of tetX and intI were observed at various sampling locations, potentially linked to the prevalent use of identical antibiotics in both urban and aquaculture activities. Interestingly, an uptick in sulI was exclusively recorded in AU, which may be attributed to the retention of ARGs caused by the localized use of sulfonamides in AU.
In comparison to the overall concentration of ARGs found in Qinghai and Dianchi Lake (0 to 1.5 × 10−4 copies per 16S rRNA), the presence of ARGs in this research area is notably more pronounced [20]. This may be attributed to the more substantial impact of human activities in the eastern region, where extensive urban expansion and aquaculture practices have led to the accumulation of antibiotics, thereby generating and sustaining higher levels of ARGs. For instance, only a small number of individual resistance genes were found in the sediments of Khanka Lake, as well as in various lakes on the Tibetan Plateau. Additionally, strA was observed at a low relative abundance in Namtso Lake and Qinghai Lake [20]. As noted in earlier investigations, these results indicate that the increased abundance of ARGs could be linked to human activities or sources of anthropogenic pollution [8,20]. Among the ARGs examined, sulfonamide ARGs exhibited the highest absolute abundance, compared with tetracycline, fluoroquinolone, and chloramphenicol ARGs, which aligns with results from other lakes in China [20]. Previous research has indicated a certain degree of association between various environmental factors and ARGs, with these parameters indirectly influencing the microbial distribution of ARGs [3,10,19]. The significant ARG pollution identified in this research area may be linked to the region’s sediment parameters. Urban contamination, characterized by elevated TOC and NH4+-N concentrations, creates conditions conducive to microbial reproduction. This extensive growth of bacteria has resulted in increased concentrations of ARGs and MGEs within the region [3,19]. Furthermore, TOC significantly lessens the enzymatic breakdown of nucleic acids due to its strong adsorption capability for ARGs [1]. These findings indicate that increased levels of TOC and NH4+-N in polluted areas create a more conducive environment for the proliferation and longevity of ARGs and MGEs in Nansi Lake. Notably, three types of microplastics (PE, PVC, and PS) identified in this study exhibited a strong positive correlation with ARGs and MGEs, including tetX and intI. Microplastics play a crucial role in the spread and prevalence of ARGs and MGEs in aquatic systems through various mechanisms. They serve as carriers for ARGs and MGEs, facilitating their horizontal transfer among microbial populations. The biofilms that develop on the surfaces of microplastics create an environment conducive to bacteria harboring ARGs and MGEs, including integrons and transposons, thereby increasing the likelihood of genetic exchange [12]. Additionally, microplastics can absorb antibiotics and heavy metals, leading to selective pressures that enhance the concentration of ARGs. For instance, studies indicate that exposure to microplastics made of PE and PVC results in a higher prevalence of antibiotic efflux pump genes, which are directly associated with the enhancement of efflux pump systems [10]. Another consequence of microplastic exposure is the induction of oxidative stress and DNA damage in bacterial cells. The production of reactive oxygen species (ROS) and the resultant ROS-mediated DNA damage activate the SOS response, a cellular mechanism that amplifies genetic variability and facilitates ARG transfer [10]. This selective pressure also alters microbial composition, favoring organisms that harbor ARGs [10,12]. Various forms of microplastics may exert different impacts. For example, PVC microplastics characterized by a higher zeta potential may suppress nitrification and encourage ARG accumulation due to intensified selective pressures. Previous research has also indicated that rainfall and temperature play crucial roles in ARG and MGE transitions in aquatic ecosystems [1,9]. Nonetheless, the sampling events for this investigation took place in the autumn, periods that did not coincide with the rainy season, and showed only minor differences in the temperature of the lake water. The changes in rainfall and water temperature within the lake could be insufficient to contribute to ARG differentiation among the different sampling areas. Furthermore, the flow speeds and rates within the lake at each sampling site were quite low throughout our research campaigns, resulting in minimal disturbances to the associated sedimentary environment, which could have minimal impact on the temporal variation of ARGs and MGEs in Nansi Lake [9].
4.2. Microbial Diversity and Community Composition
High-throughput sequencing analysis has shown that various human activities have greatly altered the microbial composition in lake sediments, with significant populations of Firmicutes and Proteobacteria [10,20]. A comprehensive analysis conducted in this research identified that five of the ten most prevalent microorganisms found in Nansi Lake include Firmicutes, Proteobacteria, Chloroflexi, Bacteroidota, and Actinobacteriota. These bacterial groups interact in complex ways within natural ecosystems, aiding in the processes of material cycling, ecological balance, and energy transfer. Moreover, they collectively contribute to the stability and functionality of these ecosystems, though their specific contributions may differ across various environments. It is also crucial to acknowledge that among these bacteria are numerous pathogenic strains, which underscores the necessity of prioritizing management strategies [33,34]. The structure of the microbial community identified in this study at AU and AA is comparable to that observed in regions affected by urban and agricultural pollution in China [24], suggesting a connection to human influences. Despite this, the microbial diversity observed in this area exceeds that recorded in the Hai River, and the dominant microorganisms also show variation, suggesting that varying latitudes may exert an influence on microbial distribution. Studies have indicated that numerous microorganisms are related to environmental parameters [35]. A more pronounced presence of Acidobacteria was noted in AU, suggesting that environmental factors may affect their abundance at these sampling sites. Acidobacteria predominantly prefer acidic conditions [8]. Thus, the greater percentage of Acidobacteria at these locations likely results from their lower pH.
This research identified the 50 dominant bacterial genera at the genus level, which together accounted for 59.34% of the total genera, a figure significantly greater than what was observed in western lakes [24]. The elevated nutrient levels in Nansi Lake may render it less conducive for microbial survival, resulting in a more diverse range of microorganisms in this water body. The commonly found microbial genera in sediment samples impacted by diverse human activities in Nansi Lake differ considerably, likely due to changes in water quality [36,37]. Microorganisms that thrive in aquaculture environments may not be suited for urban water bodies. Notably, Vibrio was not among the top 10 dominant genera in the current study, which may be attributed to human-induced alterations in the water conditions of Nansi Lake, potentially leading to a decline in Vibrio populations. Despite not being the predominant bacterium in this region, some Vibrio species may still pose health hazards to humans and animals. As an example, Vibrio parahaemolyticus exhibited resistance to several antibiotics in shrimp, shellfish, and sea cucumbers for the use of antibiotics in aquaculture farms [12]. Consequently, in order to ensure water quality and safety, it is essential to implement strategies aimed at preventing and managing Vibrio species in both aquaculture and urban environments [38].
4.3. Network Analysis at the Genus Level
Aquatic ecosystems rely heavily on microorganisms, and the spread of ARGs can disturb their microbial composition. Previous studies have shown that pathogenic bacteria present in wastewater demonstrate antibiotic resistance, underscoring the significance of microorganisms in the dispersal of ARGs in aquatic settings [27,39]. An analysis of network co-occurrence revealed that Thermoanaerobaculum, Desulfatiglans, Ignavibacterium, Vibrio, and Spirochaeta might serve as primary potential reservoirs for ARGs in Nansi Lake, indicating that their presence could enhance the dissemination of these genes in the environment [40]. Additionally, Vibrio has been shown to exhibit resistance to erythromycin, tetracyclines, chloramphenicol, and sulfonamides [41,42]. The results of this investigation support earlier findings, reaffirming that Vibrio has developed mechanisms to resist various antibiotics. ARGs and pathogens in aquatic environments pose potential health hazards to several groups, including workers in processing, fishermen, and consumers [43]. Moreover, some researches have confirmed that the rise and dissemination of ARGs are tightly connected to environmental parameters and the structure of microbial communities. Additionally, studies reveal a notable relationship between ARGs and heavy metals, along with nutrients such as TOC and NH4+-N, illustrating a co-effect on the spread of ARGs throughout aquatic systems [25,35]. In this research, some ARGs exhibited no association with the 100 most prevalent bacterial genera, suggesting that these prevalent genera in the examined region do not act as potential reservoirs for all ARGs. At the same time, a noteworthy relationship was identified between ARGs and environmental factors such as TOC, NH4+-N, PE, PVC, and PS [3,10,44], highlighting the significant impact of environmental parameters on ARGs across various areas. The investigation also uncovered a complicated interaction between ARGs and microorganisms that warrants further focus. To more effectively determine the hosts of ARGs, earlier studies predominantly utilized bacterial isolation methods. Therefore, gene sequencing or experiments aimed at isolating bacteria are recommended for additional confirmation [45]. Furthermore, the environmental conditions associated with urban and aquaculture pollution in the area are multifaceted. Enhancing monitoring and regulation during the release of wastewater from urban and aquacultural activities is essential. It is crucial to implement filtration or ultraviolet disinfection techniques to substantially mitigate the discharge of antibiotics and ARGs, ensuring effective management [46].
Nonetheless, for various aquatic systems characterized by unique ecological conditions, such as deepwater, saline, alkaline, and lagoon lakes, it is crucial to grasp their inherent characteristics, and a thorough comparative assessment is necessary before referencing the findings of this research. Moreover, other potential influencing elements, including antibiotic residues, climatic factors, and water mass dynamics, should also be taken into account in ongoing studies to investigate their impacts on the transition of AGRs and MGEs in Nansi Lake.
5. Conclusions
The research indicated that sulI and tetX are more prevalent than other ARGs in the urban and aquacultural polluted regions of Nansi Lake. Concurrently, intI functions as a primary MGE, serving as a vector that facilitates the dissemination of ARGs in marine environments, thereby posing a significant threat to aquatic ecosystems. Meanwhile, some environmental factors influenced by different human activities, such as NH4+-N, TOC, PS, PE, and PVC, played important roles in the diffusion of ARGs. The predominant microbial species identified in the research areas include Firmicutes, Proteobacteria, Chloroflexi, Bacteroidetes, and Actinobacteria. Notably, the potential carriers of ARGs in these areas consist of Thermoanaerobaculum, Desulfatiglans, Ignavibacterium, Vibrio, and Spirochaeta. To protect the environment, it is essential to manage the release of urban and aquaculture wastewater and mitigate the negative effects of pollutants.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/w17101523/s1: Figure S1: Sampling areas in Nansi Lake. Latitude is shown on the right side, while longitude is displayed along the bottom; Figure S2: Phylotypes that show significant differences among the sampling areas at the phylum level; Table S1: Environmental parameters of lake sediment at each sampling area along with standard errors; Table S2: Primer sequences used for the quantification of 16S rRNA and ARGs.
Author Contributions
Conceptualization, R.W.; methodology, R.W. and C.D.; software, H.L.; validation, H.L.; formal analysis, R.W. and M.L.; investigation, R.W., M.L., and X.Y.; resources, Q.C.; data curation, R.W., M.L., and Q.C.; writing—original draft preparation, R.W.; writing—review and editing, M.L. and Q.C.; visualization, H.L. and X.Y.; supervision, Q.C.; project administration, H.Z. and H.W.; funding acquisition, Q.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Shandong Provincial Natural Science Foundation of China (ZR2023QC324 and ZR2021MC115), the Shandong Provincial University Youth Innovation and Technology Program (2020KJE008 and 2024KJG077), and the Research Start-Up Funds of Zaozhuang University (1020749 and 1020733).
Data Availability Statement
Raw sequences, metadata, and programs in this study will be made available by the authors upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, Y.Y.; Chu, K.J.; Hua, Z.L.; Li, Q.M.; Lu, Y.; Ye, F.Z.; Dong, Y.Y.; Li, X.Q. Dynamics of antibiotic resistance genes in the sediments of a water-diversion lake and its human exposure risk behaviour. Sci. Total Environ. 2024, 929, 172563. [Google Scholar] [CrossRef]
- Truong, T.; Hoang, T.L.; Tran, L.T.; Pham, T.P.T.; Le, T.H. Prevalence of antibiotic resistance genes in the Saigon River impacted by anthropogenic activities. Water 2021, 13, 2234. [Google Scholar] [CrossRef]
- Zhang, L.L.; Chen, H.D.; Gao, S.; Song, Y.M.; Zhao, Y.; Tang, W.Z.; Cui, J.S. Antibiotic resistance genes and mobile genetic elements in different rivers: The link with antibiotics, microbial communities, and human activities. Sci. Total Environ. 2024, 919, 170788. [Google Scholar] [CrossRef]
- Zhang, X.X.; Zhang, T.; Fang, H. Antibiotic resistance genes in water environment. Appl. Microbiol. Biotechnol. 2009, 82, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Jia, L.; Liu, H.; Zhao, N.; Deng, Q.X.; Zhu, C.H.; Zhang, B. Distribution and transfer of antibiotic resistance genes in coastal aquatic ecosystems of Bohai Bay. Water 2022, 14, 938. [Google Scholar] [CrossRef]
- Ding, C.; Gong, Z.; Zhang, K.; Jiang, W.; Kang, M.; Tian, Z.; Zhang, Y.; Li, Y.; Ma, J.; Yang, Y.; et al. Distribution and model prediction of antibiotic resistance genes in Weishan Lake based on the indication of Chironomidae larvae. Water Res. 2022, 222, 118862. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Q.; Ren, X.Y.; Zhang, Y.K.; Ju, H.Y.; Liu, J.; Xie, J.; Altaf, M.M.; Diao, X.P. Distribution characteristics of antibiotic resistance genes and microbial diversity in the inshore aquaculture area of Wenchang, Hainan, China. Sci. Total Environ. 2024, 914, 169695. [Google Scholar] [CrossRef]
- Xu, Z.X.; Jia, Y.; Huang, B.; Zhao, D.M.; Long, X.; Hu, S.Y.; Li, C.Q.; Dao, G.; Chen, B.; Pan, X.J. Spatial distribution, pollution characteristics, and health risks of antibiotic resistance genes in China: A review. Environ. Chem. Lett. 2023, 21, 2285–2309. [Google Scholar] [CrossRef]
- Zeng, Q.Z.; Xiang, J.X.; Yang, C.Y.; Wu, J.X.; Li, Y.X.; Sun, Y.A.; Liu, Q.W.; Shi, S.N.; Gong, Z. Microplastics affect nitrogen cycling and antibiotic resistance genes transfer of sediment. Chem. Eng. J. 2023, 454, 140193. [Google Scholar] [CrossRef]
- Ellabaan, M.M.H.; Munck, C.; Porse, A.; Imamovic, L.; Sommer, M.O.A. Forecasting the dissemination of antibiotic resistance genes across bacterial genomes. Nat. Commun. 2021, 12, 2435. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.S.; Yin, G.Y.; Liu, M.; Chen, C.; Jiang, Y.H.; Hou, L.J.; Zheng, Y.L. A systematic review of antibiotics and antibiotic resistance genes in estuarine and coastal environments. Sci. Total Environ. 2021, 777, 146009. [Google Scholar] [CrossRef] [PubMed]
- Karkman, A.; Do, T.T.; Walsh, F.; Virta, M.P.J. Antibiotic-resistance genes in waste water. Trends Microbiol. 2018, 26, 220–228. [Google Scholar] [CrossRef]
- Li, S.N.; Zhang, C.F.; Li, F.X.; Hua, T.; Zhou, Q.X.; Ho, S.H. Technologies towards antibiotic resistance genes (ARGs) removal from aquatic environment: A critical review. J. Hazard. Mater. 2021, 411, 125148. [Google Scholar] [CrossRef]
- Gomes, R.P.; Oliveira, T.R.; Rodrigues, A.B.; Ferreira, L.M.; Vieira, J.D.G.; Carneiro, L.C. Occurrence of antibiotic resistance genes, antibiotics-resistant and multi-resistant bacteria and their correlations in one river in Central-Western Brazil. Water 2023, 15, 747. [Google Scholar] [CrossRef]
- Xie, K.P.; Zeng, Q.Z.; Yu, S.H.; Luo, H.J.; Zhang, Y.S.; Ma, C.W.; Hu, H.Y.; Shi, S.N.; Gong, Z. Contrasting distribution of microbial communities, functional genes, and antibiotic resistance genes in produced water treatment plants with different treatment technologies. Water 2024, 16, 195. [Google Scholar] [CrossRef]
- Shen, M.N.; Hu, X.W.; Li, M.; Lyu, C.; Hu, Y.; Bu, X.D.; Chen, T.; Cai, H.; Li, C.Y.; Liu, J.H.; et al. Distribution of antibiotic resistance genes and their association with microbes in wastewater treatment plants: A metagenomics analysis. Water 2023, 15, 1587. [Google Scholar] [CrossRef]
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. Fems Microbiol. Rev. 2011, 35, 790–819. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Song, W.J.; Lin, H.; Wang, W.B.; Du, L.N.; Xing, W. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 2018, 116, 60–73. [Google Scholar] [CrossRef]
- Shi, X.M.; Shen, Z.Q.; Shao, B.; Shen, J.Z.; Wu, Y.N.; Wang, S.L. Antibiotic resistance genes profile in the surface sediments of typical aquaculture areas across 15 major lakes in China. Environ. Pollut. 2024, 347, 123709. [Google Scholar] [CrossRef]
- Curran, J.F.; Zaggia, L.; Quero, G.M. Metagenomic characterization of microbial pollutants and antibiotic- and metal-resistance genes in sediments from the canals of Venice. Water 2022, 14, 1161. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, X.; Lu, S.; Zhang, J.; Wang, W. Occurrence of typical antibiotics in Nansi Lake’s inflowing rivers and antibiotic source contribution to Nansi Lake based on principal component analysis-multiple linear regression model. Chemosphere 2020, 242, 125269. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Shen, X.; Shen, D.; Wang, K.; Jiang, X.; Qadeer, A. Regional differences in lead (Pb) and tetracycline (TC) binding behavior of sediment dissolved organic matter (SDOM): Effects of DOM heterogeneity and microbial degradation. J. Hazard. Mater. 2024, 474, 134785. [Google Scholar] [CrossRef]
- Wang, R.; Xu, S.; Jiang, C.; Zhang, Y.; Bai, N.; Zhuang, G.; Bai, Z.; Zhuang, X. Impacts of human activities on the composition and abundance of sulfate-reducing and sulfur-oxidizing microorganisms in polluted river sediments. Front. Microbiol. 2019, 10, 231. [Google Scholar] [CrossRef]
- Ballent, A.; Corcoran, P.L.; Madden, O.; Helm, P.A.; Longstaffe, F.J. Sources and sinks of microplastics in Canadian Lake Ontario nearshore, tributary and beach sediments. Mar. Pollut. Bull. 2016, 110, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Pazda, M.; Kumirska, J.; Stepnowski, P.; Mulkiewicz, E. Antibiotic resistance genes identified in wastewater treatment plant systems—A review. Sci. Total Environ. 2019, 697, 134023. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.Q.; Vu, H.P.; Nguyen, L.N.; Wang, Q.L.; Djordjevic, S.P.; Donner, E.; Yin, H.B.; Nghiem, L.D. Monitoring antibiotic resistance genes in wastewater treatment: Current strategies and future challenges. Sci. Total Environ. 2021, 783, 146964. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Maslov, S.; Sneppen, K. Specificity and stability in topology of protein networks. Science 2002, 296, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Nonaka, L.; Suzuki, S. Occurrence of tetracycline resistance genes tet(M) and tet(S) in bacteria from marine aquaculture sites. FEMS Microbiol. Lett. 2004, 237, 147–156. [Google Scholar] [CrossRef]
- Laverock, B.; Gilbert, J.A.; Tait, K.; Osborn, A.M.; Widdicombe, S. Bioturbation: Impact on the marine nitrogen cycle. Biochem. Soc. Trans. 2011, 39, 315–320. [Google Scholar] [CrossRef]
- Li, J.; Dong, C.; Lai, Q.; Wang, G.; Shao, Z. Frequent occurrence and metabolic versatility of Marinifilaceae bacteria as key players in organic matter mineralization in global deep seas. mSystems 2022, 7, e00864-22. [Google Scholar] [CrossRef]
- Zhang, L.; Ju, Z.; Su, Z.; Fu, Y.; Zhao, B.; Song, Y.; Wen, D.; Zhao, Y.; Cui, J. The antibiotic resistance and risk heterogeneity between urban and rural rivers in a pharmaceutical industry dominated city in China: The importance of social-economic factors. Sci. Total Environ. 2022, 852, 158530. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Iqbal, M.; Zeng, Z.; Lian, Y.; Zheng, A.; Zhao, M.; Li, Z.; Wang, G.; Li, Z.; Xie, J. Comparative analysis of microbial community structure in the ponds with different aquaculture model and fish by high-throughput sequencing. Microb. Pathog. 2020, 142, 104101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, J.; Wang, N.; Wang, H.; Yu, L. Metagenomic analysis reveals microbiome and resistome in the seawater and sediments of Kongsfjorden (Svalbard, High Arctic). Sci. Total Environ. 2022, 809, 151937. [Google Scholar] [CrossRef]
- Park, J.; Lee, C.S. Vibrio vulnificus infection. N. Engl. J. Med. 2018, 379, 375. [Google Scholar] [CrossRef]
- He, L.X.; He, L.Y.; Gao, F.Z.; Wu, D.L.; Ye, P.; Cheng, Y.X.; Chen, Z.Y.; Hu, L.X.; Liu, Y.S.; Chen, J.; et al. Antibiotics, antibiotic resistance genes and microbial community in grouper mariculture. Sci. Total Environ. 2022, 808, 152042. [Google Scholar] [CrossRef]
- Watkins, R.R.; Bonomo, R.A. Overview: Global and local impact of antibiotic resistance. Infect. Dis. Clin. North Am. 2016, 30, 313–322. [Google Scholar] [CrossRef]
- Labella, A.; Gennari, M.; Ghidini, V.; Trento, I.; Manfrin, A.; Borrego, J.J.; Lleo, M.M. High incidence of antibiotic multi-resistant bacteria in coastal areas dedicated to fish farming. Mar. Pollut. Bull. 2013, 70, 197–203. [Google Scholar] [CrossRef] [PubMed]
- He, L.X.; He, L.Y.; Gao, F.Z.; Zhang, M.; Chen, J.; Jia, W.L.; Ye, P.; Jia, Y.W.; Hong, B.; Liu, S.S.; et al. Mariculture affects antibiotic resistome and microbiome in the coastal environment. J. Hazard. Mater. 2023, 452, 131208. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lan, B.; Fei, H.; Wang, S.; Zhu, G. Heavy metal could drive co-selection of antibiotic resistance in terrestrial subsurface soils. J. Hazard. Mater. 2021, 411, 124848. [Google Scholar] [CrossRef]
- Wang, J.H.; Lu, J.; Zhang, Y.X.; Wu, J.; Luo, Y.; Liu, H. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 2018, 253, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Haldar, A.; Bhattacharyya, M.; Ghosh, A. Anthropogenic influence shapes the distribution of antibiotic resistant bacteria (ARB) in the sediment of Sundarban estuary in India. Sci. Total Environ. 2019, 647, 1626–1639. [Google Scholar] [CrossRef]
- Su, H.C.; Liu, Y.S.; Pan, C.G.; Chen, J.; He, L.Y.; Ying, G.G. Persistence of antibiotic resistance genes and bacterial community changes in drinking water treatment system: From drinking water source to tap water. Sci. Total Environ. 2018, 616–617, 453–461. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).