Abstract
Hydrogeochemistry of a lignite pit lake in Lusatia, Germany, was investigated. Anoxic groundwater from the dump aquifer rich in FeII (average ~5911 µmol/L) and SO4 (average ~14,479 µmol/L) contents enter the lake as subsurface inflow; oxidation and subsequent precipitation of poorly crystallized Fe-oxyhydroxysulfate (schwertmannite) occurs and causes acidification (pH~2.8). However, the removal of dissolved loads as solid phases significantly improves the groundwater quality of the downgradient as an outflow. The rainwater isotopic values (δD ~−8.88‰ and δ18O ~−65.86‰) closely matched with the groundwater showing very little isotopic modification, which points to a short residence time of groundwater. The displacement of δD and δ18O values (slope = 5.3) from the meteoric water line reflected the evaporative enrichment of the lake water. The isotopic signature also revealed longer residence times of epilimnion than the hypolimnion waters which are dominated by groundwater. The lake is dimictic and showed abrupt changes in physicochemical parameters along the interface (~0.30 m thick) when separating the epilimnion (upper 4 m) from the hypolimnion (bottom 1.5 m). Lake sediments were found to be dominated by clay size fraction occurring as laminations (thickness: 1~0.5 mm) that reflect seasonal sedimentation. Higher schwertmannite formation in the south as compared to the north (recharge side) also serves as a scavenger of potentially toxic elements which is probably a natural solution to man-made problems. Schwertmannite transformation to goethite releases sulfate which is reduced and fixed as secondary sulfide minerals over time. Overall, waters are of a Ca–SO4 to Ca–Mg–SO4 type with distinct inflow (FeII/FeIII > 2.5) and outflow (FeII/FeIII < 0.5) of groundwater.
1. Introduction
Mine pits created by lignite (brown coal) mining contain sulfides that oxidize over time and result in acid mine lakes [1]. The acid mine drainage (AMD) associated with mine pit lakes is enriched in SO4, Fe, and acidity [2] and usually contains high concentrations of potentially toxic elements (e.g., As, Pb, Cd) and, therefore, can have a deleterious influence on the surface and groundwater quality [3]. Pit mine lakes are complex geochemical systems fundamentally influenced by geology, climate, groundwater chemistry, mineral precipitation and dissolution, metal adsorption, lake morphology, and biological activity [4,5,6]. Such complexity invites a better understanding of the hydrogeochemical processes occurring in pit lakes [7].
Intensive extraction of lignite in the past has created a network of pit lakes in the mining region of north-eastern Germany. Lignite was obtained by open cast mining which resulted in large pits that were partly filled with overburden and spoil material. Iron sulfides (e.g., pyrite, marcasite) are the most common sulfides in lignite and the mining waste material produced in the course of strip mining generally contained high pyrite content (~1.4 g FeS2/kg [8]). Mining operations lead to the disturbance of overburden and enhance oxidation of sulfide minerals through contact with atmospheric oxygen which serves as an initiation reaction. The general process of sulfide dissolution [9] through a series of intermediate steps involving oxidation, hydrolysis, microbial catalysis, and redox reactions has been described by Nordstrom [10] and Moses et al. [11]. Overall reactions causing acidification are as follows:
FeS2 (s) + 3.5O2 (g) + H2O (l) → Fe2+ (aq) + 2SO42− (aq) + 2H+ (aq)
Fe2+ + 0.25O2 + H+ → Fe3+ + 0.5H2O
FeS2 (s) + 14Fe3+ + 8H2O → 15Fe2+ + 2SO42− + 16H+
Groundwater was lowered by heavy pumping to access the lignite seam during the mining operations. When mining ceased in the area, flooding of the residual pits started which over the years transformed into several post-mining artificial lakes and caused an enormous impact on the landscape. Groundwater rich in dissolved FeII and SO4 concentrations feeds into these lakes. There are about 400 mine lakes [12,13] varying in size and depth and some of these became acidic over time depending primarily upon the groundwater flow direction and the degree of disturbance. Lake 77 is one such acid mine lake, representative of the majority of lakes. This study was aimed at evaluating the hydrogeochemistry of an acid mine lake using hydrogen and oxygen isotopes as investigative tools and determining the baseline situation of metal concentrations. This investigation focused on understanding how the processes occurring produce secondary minerals that may be useful in the management of acidification and the possible use of secondary products.
2. Materials and Methods
2.1. Study Area
The study site, Lake 77 (~0.25 km2, max depth ~7 m), is located in the Lusatia region in the east of Germany, about 100 km south of Berlin (Figure 1). The lake was a post-mining pit turned into a lake when dewatering ceased, and the original groundwater level was established. The lake is fed by the groundwater and is marked by high loads of iron, sulfate, and acidity [14].
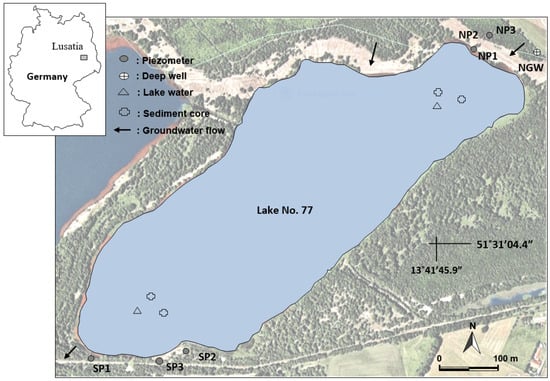
Figure 1.
Location map of the study area.
Lake 77 is thermally stratified (dimictic) and mixes from top to bottom during two periods, spring and fall, in a year. However, in the absence of a winter ice cover, the lake is classified as monomictic, and mixing occurs once a year. During the sampling in October 2010, Lake 77 showed two distinct layers: the epilimnion (upper ~0–4 m) and the hypolimnion (deeper ~4.3–6 m), separated by a thin transition zone called the metalimnion (middle ~4–4.3 m). The average annual temperature is 8.5 °C and the average annual precipitation is 559.8 mm [15]
2.2. Hydrological and Geological Settings
The local groundwater flow direction is from NNE to SSW (Figure 1). Lake 77 receives groundwater influx from the northern aquifer and the subsurface outflow from the lake is into the southern aquifer [16]. The northern bank of the lake is covered with visible waste dump as the overland flow removed the vegetation cover. The local geology consists of tertiary sediments and contains lignite and coal of commercial value [17]. Mineral assemblages in the overburden comprise mostly quartz, kaolinite, feldspar, and mica. Dolomite, calcite, and pyrite are also found in significant amounts.
2.3. Sampling
Lake water (n = 46) was sampled (from the surface to a maximum of the 6 m deep water column) from the sampling locations in the north and the south (Figure 1) in September 2010 (late summer or early autumn). Groundwater (n = 7) from three piezometers from the south and three from the north, and a deep groundwater well was sampled using an automatic pump in precleaned glass vials. A total of four undisturbed lake bottom sediment cores were also collected from the north the south sampling locations using a gravity corer in Plexiglas tubes (⌀~5.9 cm). Sediment cores were kept cool and stored in a specially designed box for transportation to the laboratory. Each sediment core was sectioned into 2 to 4 cm slices (n = 33) based on visual observation of color and texture. Water pH, redox potential (Eh), electrical conductivity (EC), dissolved oxygen (DO), and temperature were measured in situ in lake waters and groundwaters, and in the laboratory for porewaters (n = 33) using standard portable meters. A rainwater sampler was placed close to the study area (<1 km to Lake 77) for the rainwater sample collection for the isotopic analysis of local rain.
2.4. Analytical
All water samples were filtered with 0.45 µm cellulose acetate membrane filters, acidified down to pH < 2 with concentrated HNO3, and refrigerated until the analysis. However, water samples for major anion (SO4, Cl, and NO3) analysis were not acidified. Major trace and rare earth elements (Ca, Mg, Na, K, Fe, Al, Mn, Si, Sr, As, Ba, Cr, Co, Cu, Zn, Ni, Pb, Ce, Dy, Gd, La, Li, Nd, Pr, Rb, Sm, U and V) were analyzed by inductively coupled plasma mass spectrometry (ICP-MS Perkin Elmer ELAN 6100) at the BayCEER laboratory in Germany. The accuracy and precision of the analytical method were verified against certified reference waters (NIST SRM 1643). The ionic balance of measured cations and anions was about 15% or better. Total Fe and FeII were determined spectrophotometrically with phenanthroline [18] after adding a water sample to a pH 4 acetate buffer solution (~0.2 mol/L). IronIII concentration was calculated using the difference between FeII and total Fe. Major anions (Cl, SO4, and NO3) were analyzed by ion chromatography. The total organic carbon (TOC) concentration was measured using the Shimadzu TOC 5050 organic carbon analyzer. The dissolved inorganic carbon (DIC) was determined using the Shimadzu Mini-2 gas chromatograph in the gas phase in 1.8 mL vials closed with Teflon-coated rubber septa after the addition of 4 M HCl to 0.5 mL sample. The DIC concentration was calculated using Henry’s constant (KH = 10−1.5 mol/L. atm). The amount of chromium reducible sulfur (CRS = S° + FeS2) and acid volatile sulfur (AVS = H2S + FeS) in sediments were determined following the method described by Wieder et al. [19]. About 3 mL of ethanol, 5 mL of 5N HCl, and 15 mL of 1M Cr2+ were added to the sample and allowed to boil for 1 h. The reduced sulfur species were released as H2S and were trapped in 0.15 N NaOH. The concentration of reduced sulfur was determined spectrophotometrically.
A mechanical method was used to section sediment cores without the vertical disturbance of the sample. To avoid smearing, a sharp thin slicing plate was used for cutting a series of horizontal slits (thickness~2 cm). Porewater was extracted directly from sediments using Rhizon samplers (artificial root) connected to a standard syringe using the Luer-lock fitting, PVC tubing, and porous membrane filter (~0.2 µm). Porewater from each sectioned sediment core was collected by applying sufficient vacuum and, therefore, no contact with the atmospheric oxygen.
Sediment samples were dried under a vacuum in a freeze drier and grounded to pass through a 2 mm sieve. X-ray powder diffraction (Siemens D5000 goniometer with a Co-Kα1.2-anode; λ = 1.7890 Å) was employed for the mineral identification. Samples were also examined by Fourier Transform Infrared spectroscopy (Vektor 22–FTIR-MIR, Bruker, the spectral resolution of 2 cm−1) in transmission mode as KBr pellets (0.3% sample) from 300 to 4000 cm−1 for mineral characterization. The sediment sample was mounted on the stub and coated with gold for the scanning electron microscope (SEM) and energy dispersive spectrometer (EDS; JEOL-JSM 5310) analysis. Particle size analysis of clay (<2 µm), silt (2–20 µm), and sand (20–2000 µm) size fractions of sediments was performed using a Malvern® Instrument by laser scattering at 5–10% obscuration.
Oxygen and hydrogen isotopes of water and sediment samples were determined at the BayCEER laboratory, University of Bayreuth, Germany. The solid samples were ground (<100 µm) and dried and the high-temperature furnace interfaced with an isotope ratio mass spectrometer (IRMS: Delta V Advantage, Thermo Fisher Scientific, Bremen, Germany; pyrolysis oven: TG pyrolysis oven HTO, HEKAtech, Wegberg, Germany; interface: ConFlo IV, Thermo Fisher Scientific, Bremen, Germany) was used for the simultaneous determination of oxygen (δ18O) and hydrogen (δD) isotope abundances. The isotopic ratios were determined by the analysis of CO and H2 generated by the pyrolysis of the material by employing the technique of temperature conversion elemental analysis. Samples were placed into silver capsules and then dropped into a TC/EA generating the product gases. These gases were separated using a gas chromatograph (GC 5890 series II; Hewlett Packard, Wilmington, NC, USA), sent to an open split interface, and then to the IRMS for analysis. Methods followed in this study have been described in detail by Brand [20] and Dawson et al. [21]. The relative abundances of the H and O isotopes were analyzed by isotope ratio mass spectrometry (IRMS). The isotope ratios are presented in the delta notation as given below:
where δX is the δ value of the heavy isotope X (δ18O or δD) and R is 18O/16O or 2H/H. Certified reference material (Vienna Standard Mean Ocean Water [22]) was used for the calibration. Reproducibility (1 σ) was ±1.2‰ for δD and ±0.4‰ for δ18O. Stable isotope data are expressed in delta (δ) as parts per thousand (‰) relative to V-SMOW. All chemicals used were reagent grade.
δX = [Rsample/Rstandard − 1] × 1000‰
The PHREEQC computer code [23] with the thermodynamic database MINTEQ [24] was used for the chemical speciation of dissolved species, saturation indices (SI) of minerals, and calculation of theoretical pe values based on the redox couple of FeII/FeIII.
3. Results
3.1. Water
The statistical summary of the physical, chemical, and isotopic data of lake surface water, porewater, and groundwater samples are given in Table 1 and Table 2 (also as Supplementary Files in Tables S1–S5). The lake water temperature ranged between 11.8 and 20.6 °C and decreased with depth. The lake was thermally stratified with warmer water at the top (epilimnion) and colder at the bottom. Groundwater temperature varied from 14.2 to 18.6 °C.

Table 1.
Lake water physical, chemical, and isotopic data.
Table 2.
Physical, chemical, and isotopic data of porewater and groundwater samples.
The lake water pH ranged between 2.71 and 5.75. While surface water pH in the north increased by about 3 pH units with depth, it slightly decreased (~0.3 pH units) at the south sampling location. The sediment porewater pH ranged from 2.71 to 4.41 and increased with the depth of the sediments at both sampling locations. Most of the pH values were within the range of 2.5 to 4.0 as is normally reported for acid mine drainage. The groundwater pH ranged between 2.97 and 5.61. The lake water redox varied between 173 and 541 mV with an average of 478 mV. At the depth of 4 m, redox potential values reduced abruptly (~three times), especially at the north sampling location.
The lake water EC varied from 2.05 to 2.94 mS/cm and significantly increased at the north sampling location but slightly decreased at the south sampling location with depth. The porewater EC varied between 1.09 and 2.58 mS/cm and decreased with depth at both the north and south sampling locations. The groundwater EC varied from 0.98 to 3.96 mS/cm. The groundwater from the deep groundwater well (NGW; Table 2) registered the highest EC value.
The DIC in the lake water varied from 118.1 to 6933.7 µmol/L and remained nearly unchanged in the upper 4 m water column but increased significantly in the lower water column probably due to the decay of organic matter and remineralization of organic matter to CO2 at the sediment surface. The north sampling location registered four times the DIC concentration in the hypolimnion waters than the south sampling location. All groundwater samples registered higher DIC concentrations ranging from 1251.5 to 5175.9 µmol/L with the highest concentration in the deep groundwater well. The TOC in the lake water ranged between 1.1 to 7.6 mg/L and increased with depth. The porewater TOC concentrations varied between 15.9 and 38.1 mg/L. The groundwater TOC concentrations varied from 10.3 to 19.2 mg/L. The groundwater from the south sampling locations registered higher concentrations than the north sampling location.
The lake water sulfate concentrations varied between 7580.4 and 18,203.2 µmol/L and showed an abrupt increase (~1.5 times) at the depth of 4 m at the north sampling location but did not register any significant change at the south sampling location. The porewater sulfate concentration varied from 4708.6 to 16,637.9 µmol/L and generally decreased with depth; however, no systematic change in concentration was observed. The groundwater SO4 concentrations varied between 2436.3 and 27,203.8 µmol/L by a factor of 11 with higher concentrations from the north as compared to the south. Sulfate was the most dominant anion in each system and showed a strong positive correlation (r ≥ 0.90, p < 0.01) with EC.
The dissolved Cl concentrations varied from 268.2 to 859.7 µmol/L in lake waters, 539.8 to 1254.2 µmol/L in porewaters, and 357.6 to 1593.5 µmol/L in groundwaters. The Cl concentration decreased about three times with depth in the lake water. Groundwater from the south sampling location (lake water outflow) registered two times higher concentrations than the north (lake water inflow) which is mostly due to the enrichment by evaporation in the lake. The NO3 concentration varied from 13.4 to 149.2 µmol/L in lake water, 14.8 to 78.3 µmol/L in porewater, and 8.1 to 112.7 µmol/L in groundwater. The lake water NO3 concentrations increased in the north but decreased slightly in the south with depth. The porewater NO3 concentration did not register a systematic change with depth. The groundwater samples from the north sampling locations measured > three times higher NO3 concentrations as compared to the south.
The lake water FeTotal concentrations varied between 1554.5 and 12,658.1 µmol/L and remained nearly unchanged in the upper water 4 m column. At the depth of 4 m, FeII concentration increased significantly but FeIII concentration decreased at both the north and south sampling locations. The north sampling location showed a significant and abrupt change with depth in FeII concentration (>11,500 µmol/L) along the transition phase (metalimnion). The porewater FeTotal concentrations varied from 2412.6 to 13,574.1 µmol/L and measured higher concentrations in sediment porewaters from the north (average ~9658.8 µmol/L) as compared to the south sampling location (average ~6794.5 µmol/L). The groundwater FeTotal concentrations varied from 737.5 to 15,824.8 µmol/L with higher concentrations in the north (average ~8341.3 µmol/L) than in the south (average ~1250.6 µmol/L). The deep groundwater well registered the highest concentration with a FeII /FeIII molar ratio of 5.9.
Among the major cations, Ca and Mg concentrations increased but Na and K concentrations decreased with depth in lake waters possibly due to the mobilization of the former and fixation of the latter. However, in porewaters, the opposite trend was noted as Na and K concentrations increased slightly but Ca and Mg concentrations decreased with depth. The groundwater from the north registered lower concentrations of Ca, Mg, Na, and K with the exception of deep groundwater wells. Environmental screening of potentially toxic elements in a few selected lake water samples (Figure 2) showed enrichment of As, Ba, Sr, Ce, Cr, Co, Cu, Dy, Gd, La, Li, Nd, Ni, Pb, Pr, Rb, Sm, U, V and Zn as compared to the European geochemical baseline [25] values of surface waters of 1.2, 35.4, 0.3, 0.4, 0.8, 0.3, 1.2, 0.04, 0.05, 0.2, 6.7, 0.2, 2.4, 0.2, 0.05, 2.4, 0.04, 1.7, 0.8 and 6.0 µg/L, respectively (Table S2). Trace metals such as Ag, Bi, Cd, Cs, Nd, Sn, and Zr were below detection limits.
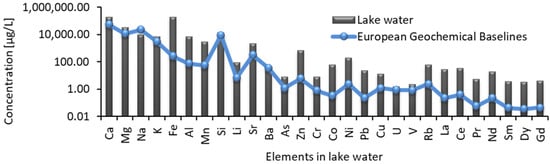
Figure 2.
Dissolved elements in lake water as compared with European geochemical baselines [25] of various elements in stream waters.
3.2. Isotopic Composition
The δ18O values of lake water varied from −18.35 to −4.2‰ and increased slightly with the depth first and then abruptly decreased (up to 3.5 ‰ change) at the depth of 4 m (Table S1). The porewater δ18O values varied from −8.29 to −4.61‰ and decreased with depth (up to 1.4‰ change) at both the north and the south sampling locations. The groundwater δ18O values varied between −8.58 and −7.61‰. The groundwater from the south registered slightly higher δ18O values (average ~−7.83‰) as compared to the north (average ~−8.36‰) sampling location. The local rainwater δ18O value was measured to be −8.88‰. The lake water δD values varied from –62.71 to −40.89‰ and decreased abruptly at the depth of about 4 m (up to 19‰ change). The porewater δD values varied between −60.7 and −44.24‰ and decreased with depth both in the north and the south. The groundwater δD values varied from −65.47 to −59.99‰ with higher δD of groundwater from the south (average ~−60.85‰) as compared to the north (average ~−64.38‰). The local rainwater average δD value was measured to be −65.86‰.
3.3. Sediment
The physicochemical parameters of sediment samples are shown in Table 3. Contents of total reduced inorganic sulfur (TRIS = AVS + CRS) ranged between 10.3 and 255.3 µmol/g. Both AVS and CRS contents increased with the depth of sediments. The sediment clay size fraction varied between 5.6 and 25.6%, silt size fraction between 9.1 and 78.2%, and sand size fraction between 3.3 and 85.4%. Overall, the silt dominated the clay and sand size fractions (Figure 3 and Table S5). Sediments from the south sampling locations were dominated by fine particles (<10 µm), and sediments from the north sampling locations were enriched in a coarser size fraction (>100 µm). Particle size changes with depth were more abrupt in the north than the south reflecting the burial history and the high input of waste material. One surface soil (0~5 cm depth) sample from the north of the lake measured a high sand size fraction (~86.8%). The sedimentation rate in the lake varies due to in situ processes and the input of mineral material through surface runoff during rainfall events. The south sampling location receives higher sedimentation both as in situ mineralization (autochthonous) and transported (allochthonous) by bank erosion and surface runoff than the north side of the lake. Sediments exhibited different colors from brownish yellow to reddish brown becoming dark gray to black due to the differing mineralogy with depth.
Table 3.
Physicochemical data of sediment cores and a soil sample from north and south sampling locations.
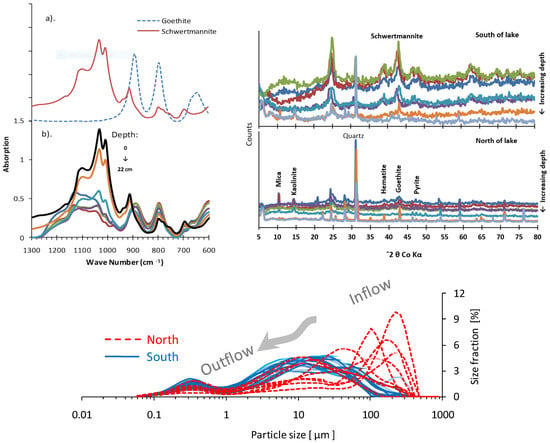
Figure 3.
Particle size distribution of lake sediments. Arrow indicates the water flow direction from the north to the south direction of the lake. X-ray diffractograms of the sediment sample from the north and the south sampling locations of the lake show significant differences in sediment mineralogy. The FTIR spectra of (a) goethite and schwertmannite standards and (b) sediment samples. Note the transformation of schwertmannite to goethite with the depth of lake sediments.
The X-ray diffraction patterns showed the presence of quartz, mica, kaolinite, goethite, and schwertmannite (Figure 3). The sediment samples from the south are dominated by poorly crystallized phase schwertmannite, which differ significantly in X-ray diffractograms from the north sampling location. The broadening of peaks changes with depth as a function of increasing particle size and the degree of crystallinity. Superimposed sharp peaks of detrital quartz and silicate minerals are observed as compared to iron-containing minerals.
The FTIR spectra of sediments are shown in Figure 3. The characteristic FeO-absorption band at 704 cm−1 indicated the presence of schwertmannite. The peak occurring at 610 cm−1 is typical for structural sulfate in schwertmannite and is well developed in upper sediment and gradually disappears with depth. The presence of absorption bands of OH-and O-vibrations at 795 and 892 cm−1 are typical for goethite [26]. All other characteristic bands of both schwertmannite and goethite can be recognized by comparison with standards as shown in Figure 3.
4. Discussion
The lake water is of a Ca–Mg–SO4 to Fe–SO4 type with the order of dominance of cations as Ca2+ > Fe2+ >> Mg2+ > Na+ > K+ and for anions it is SO42− >> Cl− > NO3−. Two hydrochemical facies based on the chemical composition of AMD can be recognized, namely: (1) ferric, oxic, pH~3 and EC~2.1 mS/cm, and (2) ferrous, anoxic, pH~5 and EC~2.5 mS/cm. The former is epilimnion and the latter is hypolimnion separated by an interfacial layer or thermocline (thickness ~30 cm). The chemical composition of the hypolimnion is closer to the groundwater that enters the north side of the lake as subsurface inflow. Almost all of the physicochemical parameters remained nearly unchanged in the epilimnion indicating effective vertical mixing but they increased or decreased abruptly in the hypolimnion. Overall, three types of trends (Figure 4) were observed: (1) DO Eh, Fe3+, K+ and Cl− contents decreased; (2) TOC, DIC, Fe2+, Ca2+ and Mg2+ contents increased; and (3) EC, pH, SO42− and NO3− contents registered an increase in the north but decreased slightly in the south sampling locations. Ferrous (FeII) registered as the dominant iron species (FeII/FeIII > 1) in deeper lake waters, all porewaters, and groundwaters from the north (Figure 4). The FeII/FeIII ratio was the highest (>5) in the deep groundwater well indicating the dissolution of ferrous sulfide followed by mobilization and transport to the lake as subsurface inflow.
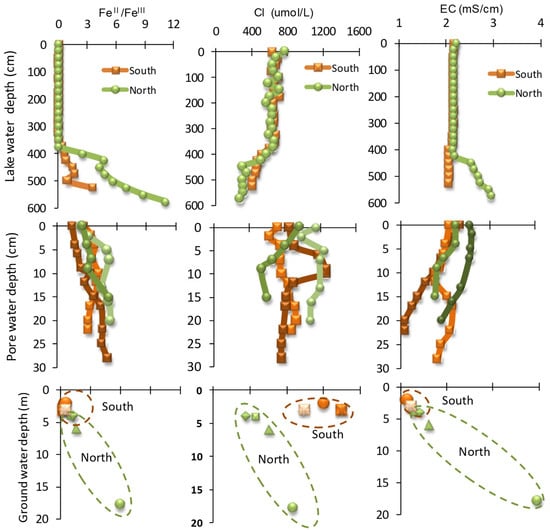
Figure 4.
Vertical changes of Fe II/FeIII, Cl, and EC values in lake water, porewater, and groundwater from the north and south sampling locations.
Geochemical calculations performed with the PHREEQC code revealed the dominant species including Fe+2, FeSO4+, FeOH+2, Fe+3, SO4−2, FeSO4+ and FeSO4 (concentrations > 1 µmol/L) in the AMD. The minerals of interest generally occurring in an acid mine environment include schwertmannite, jarosite, ferrihydrite, goethite, gypsum, and copiapite.
The groundwater from the south contains a less dissolved load than the groundwater from the north as shown by lower SO4 and Fetotal contents. There is a systematic decrease along the water flow direction from the north (water inflow) and the south (water outflow) sampling locations. The reduction in the total dissolved load is due to the precipitation of oxyhydroxysulfate phases.
High Fe and SO4 concentrations in the AMD indicate significant oxidation and subsequent dissolution of pyrite and other sulphides (e.g., chalcopyrite, sphalerite). A strong correlation of EC with SO4 (r = 0.95) and Fe (r = 0.87) concentrations confirms conductivity as a useful indicator of the degree of contamination. Groundwater with high concentrations of solutes enters the lake and remains in the bottom layer and is not subject to vertical mixing until the season changes, which is when the lake seems to undergo a complete turnover and become holomictic. Such chemical and thermal stratification is more pronounced in the north as compared to the south of the lake. During the turnover event, the oxic epilimnion (DO ~ 8 mg/L) homogenizes with the hypolimnion (DO ≤ 1 mg/L) resulting in oxidation of FeII to FeIII and subsequent formation of large poorly crystallized FeIII-(hydr)oxides. The prevailing pH-redox conditions and availability of SO4 and FeIII in the lake leads to schwertmannite (Fe8O8(OH)6SO4; Equation (4)) formation over other iron oxyhydroxysulfate solid phases. The higher affinity of FeIII over FeII with SO4 favors schwertmannite formation which rains out of the water column and forms a reddish-brown gel at the bottom of the lake. The acidity generation, solubility, and speciation of various components including Fe and SO4 are, therefore, controlled by schwertmannite precipitation.
8Fe3+ + SO42− + 14 H2O → Fe8O8(OH)6SO4 ↓ + 22 H+
Fe8O8(OH)6SO4 (S) + 2 H2O → FeOOH(S) + H2SO4
The initially formed poorly crystallized schwertmannite changes over time to the more stable endmember mineral goethite (α-FeOOH; Equation (5)). This transformation involves changes from the unstable tunnel to a more stable structural arrangement [27] by the dissolution of schwertmannite first and then formation as goethite [28]. Refait et al. [29] presented a detailed account of the chemical composition and Gibbs standard free energy of the formation of Fe(II)-Fe(III) hydroxysulphates. The south sampling location recorded thicker layers of reddish-brown amorphous material than the north.
The lake water attains thermal and chemical stratification following the turnover events causing semi-oxic to anoxic conditions along the sediment–water interface. The anaerobic conditions in lake sediments promote sulfate reduction depending upon the availability of SO4 and the degradable organic matter which is usually the limiting factor. A gradual increase in porewater pH with depth is most likely due to the partial neutralization by the bacterial reduction of sulfate and ferric (Equations (6) and (7)) with organic matter (electron source) as given below:
2CH2O + SO42− + 2 H+ → 2 CO2 + H2S + 2 H2O
¼ CH2O + Fe(OH)3 + 2 H+ → Fe2+ + ¼ CO2 + 11/4 H2O
There are two types of organic matter present in sediments: fresh plant material and lignite inherent to the parent substrate. The latter is not easily biodegradable by the organisms and, therefore, the organic matter could be a limiting factor in the alkalinity generation processes. The high specific surface area of amorphous Fe-(hydr)oxides can be used by bacteria at a high rate and provide high energy gains as compared to the crystallized phases. Roden [30] showed a positive correlation between bacterial iron reduction and the surface area of various iron hydroxides in batch experiments. Sulfate reduction to H2S and the availability of FeII lead to the formation of the amorphous phase mackinawite (FeS; Equation (8)) which is unstable and forms a more stable endmember mineral pyrite (FeS2; Equation (9)). The simplified chemical reactions can be shown as:
Fe2+ + H2S → FeS (am) + 2 H+
FeS + S˚ → FeS2
The gradual increase of CRS with a depth of lake sediments indicated the secondary pyrite formation. The color of sediments became darker with depth.
Open-air lake water results in a systematic isotopic enrichment by evaporation, which depends upon the radiation, humidity, wind, and lake morphometry [31]. A plot of δD against δ18O of waters showed a possible subsequent modification of lake water composition by evaporation (Figure 5). The surface water evaporation has left an isotopic signature shown by the gradual movement away from the meteoric water line (slope = 8). This shift of the line determines the degree of evaporation and shows a unique signature of Lake 77. The δD and δ18O values of local rain were plotted slightly lower than the global meteoric water. Porewaters showed a wide isotopic range (δD = 16.5‰ and δ18O = 3.7‰) and were plotted close to the lake and groundwaters. The isotopic composition of groundwater from the north sampling locations (especially the deep water well) were plotted near the local rain revealing that the waters are likely derived from the rain without significant modification during transport. Shorter residence time in the unsaturated zone permitted fast recharge with limited to no isotopic enrichment of the groundwater. However, groundwater from the south (lake water outflow) is slightly enriched in δ18O (~1‰) due to the isotopic fractionation resulting from oxidation/reduction processes. Habicht et al. [32] showed that dissimilatory sulfate reduction causes considerable oxygen isotopic fractionation. The measured values for δ18O of the deep lake water showed a much lighter isotope composition (−8.35‰) as compared to the water taken near the surface of the lake (−5.78‰). The deeper water (~5 m depth) was found to be reflecting the groundwater input. A similar observation has been reported by Seebach et al. [33] in Lake Waldsee which is a deeper lake (~9 m) than Lake 77 and is also located in the lignite mining district of Germany. The lake water evaporation causes isotopic fractionation, which enriches the broth δ18O and δD in surface waters [34]. The lake water residence time is important and requires more isotopic data to be determined accurately. Knöller and Strauch [35] used isotopes to determine the water residence of 5 years in an acid mine lake (ML 111) in the area.
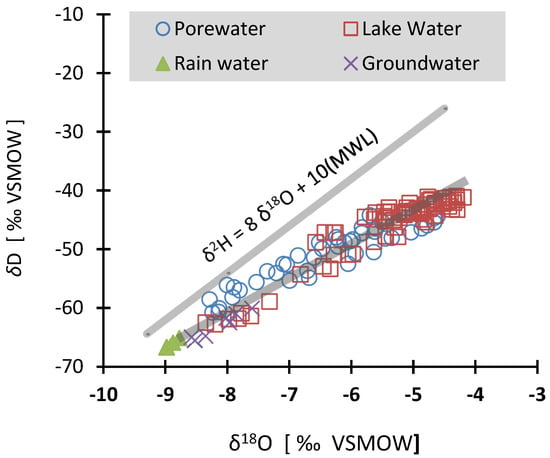
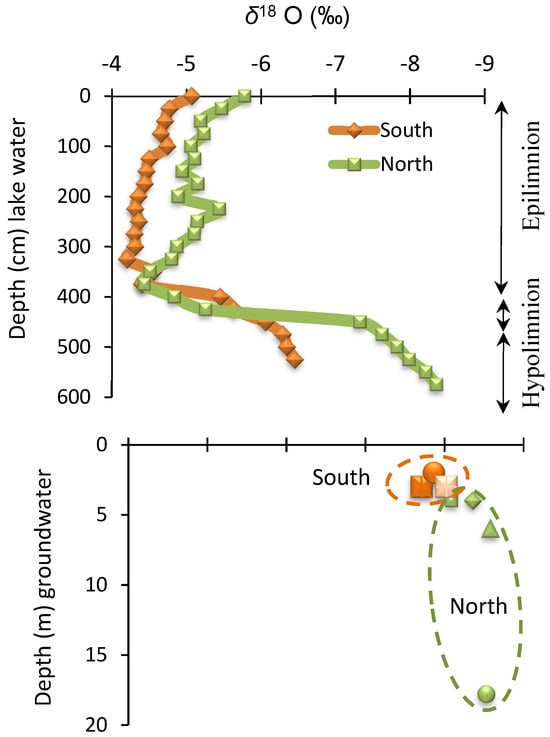
Figure 5.
Plot of δD vs. δ18O of lake surface water, porewater, groundwater, and local rainwater relative to the global meteoric water line (MWL; Craig [36]). Changes in δ18O values with the depth of lake water and groundwater. Note the δ18O values have a shift of about 1‰ in the epilimnion and about 2‰ in the hypolimnion from north to south (~800 m apart).
The epilimnion waters were found to be considerably enriched (>3‰) than the hypolimnion waters, groundwaters, and the local meteoric water (Figure 5). The recording of evaporative losses as isotopic enrichment depends on the water residence time and was reflected in the unique slope (s = 5.3) of δD versus δ18O plot. Lake water from the south showed more enrichment than the lake water from the north due to longer residence times and higher accumulative evaporative losses as the two sampling stations are about 800 m apart. However, the top 100 cm of lake water registered a slight δ18O depletion (~1‰) which is most probably due to the mixing of rainwater with lake surface water. The top lake surface water also showed δD depletion of ~5‰. The epilimnion waters seem to have much longer residence times than hypolimnion waters. The hypolimnion waters are marked by lateral flow and epilimnion waters by vertical mixing and even the wind-induced mixing between the hypolimnion and epilimnion seems to be impeded until the turnover event.
A simplified conceptual geochemical model Is depicted in Figure 6. In the northern part of the recharge area, overburden mixed with excavated waste material, which was transported and moved around during the mining of lignite beds in the past, now forms a ‘dump aquifer’. Over time pyrite weathering products are stored in porewater or as secondary mineral phases. The rain and snowmelt water infiltrating the dump aquifer carry oxidation products (e.g., FeII, Ca2+, H+, SO42−) to the groundwater which enters the lake as subsurface inflow. The groundwater from the northern aquifer showed 1.9 times higher EC values as compared to the groundwater from the south (lake outflow). Dissolved FeTotal and SO4 showed a significant reduction in concentrations, 6.7 and 3.4 times, respectively, from the north to the south groundwater along the flow path (<1 km apart). The difference in concentration of 7030 umol/L of FeTotal and 10,225 umol/L of SO4 is retained in the lake as solid products. High surface area and reactivity of amorphous phases also act as a scavenger of dissolved elements further decreasing EC values. Schwertmannite is a great sorbent for trace metals in AMD systems but metal sorption is highly pH dependent for many metals including Pb and As around 4 to 5 pH values, and Cu, Zn, and Cd around circumneutral conditions [6]. Reduction in a dissolved load indicated that the lake naturally works as a mini treatment plant that improves water quality downstream. Since the conditions are favorable for schwertmannite formation, the solid phase can be used as a product of commercial value. The thickness of lake sediment cores and the percentage of fine particles (size < 50 µm) in samples from the south sampling location were measured to be higher than the north, pointing to a higher sedimentation rate. It is, therefore, likely that the lake’s depth will reduce faster in the south as compared to the north over time. The higher fine particle size fraction and XRD analysis also supported a high percentage of fine-size amorphous materials.
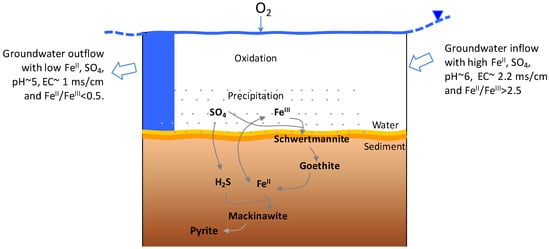
Figure 6.
A simplified conceptual geochemical model showing the formation of minerals.
Acid waters are environments with extreme levels of pH, sulfate, and iron and, hence, support selective biological communities. At the sediment depth of about 5 cm, abundance of diatoms was observed that gradually disappeared in the bottom sediments exhibiting most likely a boom-and-bust lifestyle. However, only one species of diatom (genus Eunotia) of rectangular shape (up to 50 µm long) was observed (Figure 7a) which can be used as an indicator species. The diversity of diatoms is associated with conductivity, alkalinity, and silicon contents. Zalack et al. [37] developed a diatom index of AMD severity which allows assessment of impairment, sensitivity, and recovery of diatom communities.
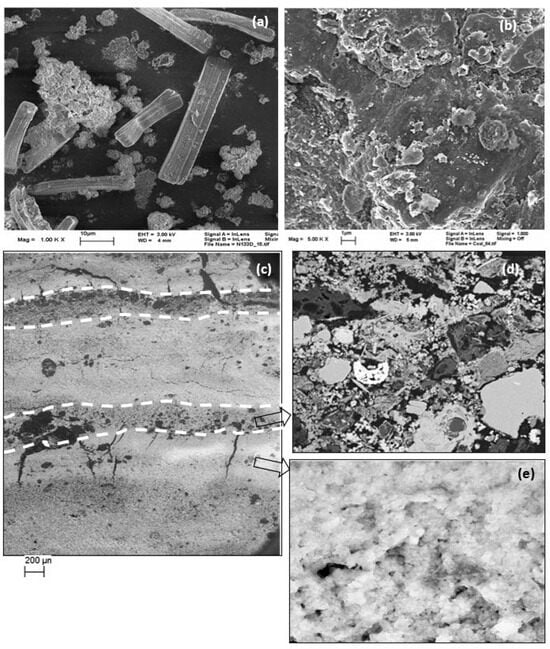
Figure 7.
SEM images of diatoms (a), weathered lignite (b), and layers (c) with a mixture of minerals as a dark band (d) and Fe-(hydr)oxide (e).
Lignite surface morphology revealed etched pits, layers, hills, and valley-like structural features which are randomly distributed throughout the micrograph (Figure 7b). The mass percent composition of major elements was observed to be in the order of abundance C (69%) > O > Fe > S > Ca > Al (0.2%).
Sediment cores showed laminations of alternating dark and brown layers (Figure 7c) of about 1 to 0.5 mm thickness. These layers were well developed in the middle of the core and became dominantly brownish in the upper part and darker at the bottom. Each layer most likely represents the seasonal lamination characterized by contrasting conditions [38] such as an ice cover and summer storms resulting in rhythmic cycling. Sedimentation may also partially present the burial history when the lake was flooded after mining stopped in 1965. Undisturbed sediment core samples comprising of layers were analyzed using scanning electron microscopy (Figure 7c–e) which revealed a dark layer of mixtures of minerals (e.g., quartz, clay minerals) mostly originating from lignite and a bright layer consisting of FeIII-(hydr)oxides (e.g., goethite, schwertmannite). The dark layers probably represent a high-energy environment of deposition in which water flow with a random mixture of transported material settles to the lake bottom to form a blanket over the sediments. The light-colored homogenous layers on the other hand represent a calm period during which precipitation of Fe-oxides takes place. Such calm periods could be the winter season snow when there is no overland flow.
5. Conclusions
The measured physicochemical parameters of the AMD showed stratification with a steep concentration gradient along the thermocline (thickness ~30 cm). The thermal and chemical stratification is more pronounced in the groundwater inflow side of the lake as compared to the lake outflow. Evaporative enrichment of δD and δ18O revealed longer residence times of epilimnion waters (FeII << FeIII; vertical mixing) than the hypolimnion waters (FeII >> FeIII; lateral flow) which are dominated by groundwater. Rainwater isotopic signatures of δD and δ18O closely match with the groundwater showing very little or no isotopic modification pointing to the relatively short residence times of the groundwater.
Under the prevailing pH-redox conditions, the dissolved load entering the lake precipitates as solid phases, dominantly as schwertmannite, converts to goethite over time. Anoxic conditions in deeper sediments promote sulfate and iron reduction that result in secondary pyrite formation and also slightly raise the porewater pH. Seasonal changes are recorded as fine laminations of alternating dark and brown layers. Both groundwater and overland flow remove clay and silt-size particles from the north (recharge side) and deposit them to the south of the lake. Lake turnover events enhance oxidation and Fe-oxyhydroxysulfate precipitation and, therefore, improve water quality during lake water outflow. Lake 77, therefore, naturally works as a mini-treatment plant. Low levels of potentially toxic elements in sediments and a high rate of schwertmannite formation, carry the potential of small-scale commercial mining. However, slight enrichment of dissolved traces and REEs is observed in the lake water as compared to the European geochemical baselines of stream waters. The most likely fate of REEs is the fixation (i.e., adsorption, absorption) as secondary minerals in the lake sediments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/w15244273/s1, Table S1: Lake water physical, chemical and isotopic data; Table S2: Trace and rare earth elements in lake water; Table S3: Physical, chemical and isotopic data of pore waters; Table S4: Groundwater physical, chemical and isotopic data from the north and south sampling locations of the Lake 77; Table S5: Physicochemical data of sediment cores and a soil sample from north and south sampling locations.
Author Contributions
Conceptualization, K.S.; methodology, K.S.; software, T.K.F. and Q.u.Z.; formal analysis, K.S.; investigation, K.S.; resources, K.S.; data curation, K.S.; writing—original draft preparation, K.S.; writing—review and editing, Q.u.Z. and T.K.F.; visualization, K.S.; project administration, K.S.; funding acquisition, T.K.F. All authors have read and agreed to the published version of the manuscript.
Funding
We extend our appreciation to the Researchers Supporting Project at King Saud University, Riyadh, Saudi Arabia, for funding this research project, (Fund no. RSP2023R487).
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
Thanks to the Alexander von Humboldt Foundation (AvH), Germany, for the financial support in the completion of this research work and the postdoc fellowship that helped carry out this research. Thanks also faculty members and staff for the support regarding the sampling and chemical analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Acharya, B.S.; Kharel, G. Acid mine drainage from coal mining in the United States–An overview. J. Hydrol. 2020, 588, 125061. [Google Scholar] [CrossRef]
- Eary, L.E. Geochemical and equilibrium trends in mine pit lakes. Appl. Geochem. 1999, 14, 963–987. [Google Scholar] [CrossRef]
- Cortecci, G.; Boschetti, T.; Dinelli, E.; Cabella, R. Sulphur isotopes, trace elements and mineral stability diagrams of waters from the abandoned Fe–Cu mines of Libiola and Vigonzano (Northern Apennines, Italy). Water Air Soil Pollut. 2008, 192, 85–103. [Google Scholar] [CrossRef]
- Castendyk, D.N.; Webster-Brown, J.G. Sensitivity analyses in pit lake prediction, Martha mine, New Zealand 2: Geochemistry, water–rock reactions, and surface adsorption. Chem. Geol. 2007, 244, 56–73. [Google Scholar] [CrossRef]
- Castro, J.; Moore, J. Pit lakes: Their characteristics and the potential for their remediation. Environ. Geol. 2000, 39, 1254–1260. [Google Scholar] [CrossRef]
- Espana, J.S.; Pamo, E.L.; Santofimia, E.; Aduvire, O.; Reyes, J.; Barettino, D. Acid mine drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, mineralogy and environmental implications. Appl. Geochem. 2005, 20, 1320–1356. [Google Scholar] [CrossRef]
- Skousen, J.G.; Ziemkiewicz, P.F.; McDonald, L.M. Acid mine drainage formation, control and treatment: Approaches and strategies. Extr. Ind. Soc. 2019, 6, 241–249. [Google Scholar] [CrossRef]
- LUA. Wasserbeschaffenheit in Tagebaurestseen. Studien und Tagungsberichte Band 6; Landesamt für Umwelt: Brandenburg, Germany, 1995. [Google Scholar]
- Sienkiewicz, E.; Gąsiorowski, M. The evolution of a mining lake-From acidity to natural neutralization. Sci. Total Environ. 2016, 557, 343–354. [Google Scholar] [CrossRef]
- Kirk Nordstrom, D. Aqueous pyrite oxidation and the consequent formation of secondary iron minerals. Acid Sulfate Weather 1982, 10, 37–56. [Google Scholar]
- Moses, C.O.; Nordstrom, D.K.; Herman, J.S.; Mills, A.L. Aqueous pyrite oxidation by dissolved oxygen and by ferric iron. Geochim. Cosmochim. Acta 1987, 51, 1561–1571. [Google Scholar] [CrossRef]
- Fyson, A.; Nixdorf, B.; Kalin, M.; Steinberg, C. Mesocosm studies to assess acidity removal from acidic mine lakes through controlled eutrophication. Ecol. Eng. 1998, 10, 229–245. [Google Scholar] [CrossRef]
- Nixdorf, B.; Lessmann, D.; Deneke, R. Mining lakes in a disturbed landscape: Application of the EC Water Framework Directive and future management strategies. Ecol. Eng. 2005, 24, 67–73. [Google Scholar] [CrossRef]
- Blodau, C.; Peine, A.; Hoffmann, S.; Peiffer, S. Organic matter diagenesis in acidic mine lakes. Acta Hydrochim. Hydrobiol. 2000, 28, 123–135. [Google Scholar] [CrossRef]
- BMVBS. Federal Ministry of Transport, Building, and Urban Development; Bundesministerium für Verkehr, Bau und Stadtentwicklung: Berlin, Germany, 2011. [Google Scholar]
- Knoll, D.; Schafer, W.; Weber, L. Grundwasseranbindung von alten Tagebaurestseen im iederlausitzer Braunkohletagebaugebiet. Grundwasser 1999, 2, 55–61. [Google Scholar] [CrossRef]
- Blodau, C. A review of acidity generation and consumption in acidic coal mine lakes and their watersheds. Sci. Total Environ. 2006, 369, 307–332. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Goto, K.; Yotsuyanagi, T.; Nagayama, M. Spectrophotometric determination of iron (II) with 1, 10-phenanthroline in the presence of large amounts of iron (III). Talanta 1974, 21, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Wieder, R.K.; Lang, G.E.; Granus, V.A. An evaluation of wet chemical methods for quantifying sulfur fractions in fresh-water wetland peat. Limnol. Oceanogr. 1985, 30, 1109–1115. [Google Scholar] [CrossRef]
- Brand, W. PreCon: A fully automated interface for the Pre-Gc concentration of trace gases on air for isotopic analysis. Isot. Environ. Health Stud. 1995, 31, 277–284. [Google Scholar] [CrossRef]
- Dawson, T.E.; Mambelli, S.; Plamboeck, A.H.; Templer, P.H.; Tu, K.P. Stable isotopes in plant ecology. Annu. Rev. Ecol. Syst. 2002, 33, 507–559. [Google Scholar] [CrossRef]
- Gonfiantini, R. Standards for stable isotope measurements in natural compounds. Nature 1978, 271, 534–536. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C. User’s guide to PHREEQC (Version 2): A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. Water-Resour. Investig. Rep. 1999, 99, 312. [Google Scholar]
- Allison, J.D.; Brown, D.S.; Novo-Gradac, K.J. MINTEQA2/PRODEFA2, A Geochemical Assessment Model for Environmental Systems: Version 3.0 User’s Manual; EPA Report 600/3-91/021; Environmental Research Laboratory. U.S. Environmental Protection Agency: Athens, GA, USA, 1991; p. 93. [Google Scholar]
- Salminen, R.; Tarvainen, T.; Demetriades, A.; Duris, M.; Fordyce, F.; Gregorauskiene, V.; Kahelin, H.; Kivisilla, J.; Klaver, G.; Klein, H. FOREGS Geochemical Mapping Field Manual. 1998. Available online: https://nora.nerc.ac.uk/id/eprint/19017/1/FOREGSmanual.pdf (accessed on 16 April 2019).
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrence and Uses, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1996; p. 143. [Google Scholar]
- Blgham, J.; Schwertmann, U.; Carlson, L.; Murad, E. A poorly crystallized oxyhydroxysulfate of iron formed by bacterial oxidation of Fe (II) in acid mine waters. Geochim. Cosmochim. Acta 1990, 54, 2743–2758. [Google Scholar] [CrossRef]
- Regenspurg, S.; Brand, A.; Peiffer, S. Formation and stability of schwertmannite in acidic mining lakes. Geochim. Cosmochim. Acta 2004, 68, 1185–1197. [Google Scholar] [CrossRef]
- Refait, P.; Bon, C.; Simon, L.; Bourrie, G.; Trolard, F.; Bessière, J.; Génin, J.-M. Chemical composition and Gibbs standard free energy of formation of Fe (II)-Fe (III) hydroxysulphate green rust and Fe (II) hydroxide. Clay Miner. 1999, 34, 499–510. [Google Scholar] [CrossRef]
- Roden, E.E. Fe (III) oxide reactivity toward biological versus chemical reduction. Environ. Sci. Technol. 2003, 37, 1319–1324. [Google Scholar] [CrossRef]
- Henderson, A.K.; Shuman, B.N. Hydrogen and oxygen isotopic compositions of lake water in the western United States. Geol. Soc. Am. Bull. 2009, 121, 1179–1189. [Google Scholar] [CrossRef]
- Habicht, K.S.; Canfield, D.E. Sulfur isotope fractionation during bacterial sulfate reduction in organic-rich sediments. Geochim. Cosmochim. Acta 1997, 61, 5351–5361. [Google Scholar] [CrossRef]
- Seebach, A.; Dietz, S.; Lessmann, D.; Knoeller, K. Estimation of lake water—Groundwater interactions in meromictic mining lakes by modelling isotope signatures of lake water. Isot. Environ. Health Stud. 2008, 44, 99–110. [Google Scholar] [CrossRef]
- Clark, I.D.; Fritz, P. Environmental Isotopes in Hydrogeology; CRC Press: New York, NY, USA, 2013. [Google Scholar]
- Knöller, K.; Strauch, G. The application of stable isotopes for assessing the hydrological, sulfur, and iron balances of acidic mining lake ML 111 (Lusatia, Germany) as a basis for biotechnological remediation. Water Air Soil Pollut. Focus 2002, 2, 3–14. [Google Scholar] [CrossRef]
- Craig, H. Isotopic variations in meteoric waters. Science 1961, 133, 1702–1703. [Google Scholar] [CrossRef]
- Zalack, J.T.; Smucker, N.J.; Vis, M.L. Development of a diatom index of biotic integrity for acid mine drainage impacted streams. Ecol. Indic. 2010, 10, 287–295. [Google Scholar] [CrossRef]
- Xin, R.; Banda, J.F.; Hao, C.; Dong, H.; Pei, L.; Guo, D.; Wei, P.; Du, Z.; Zhang, Y.; Dong, H. Contrasting seasonal variations of geochemistry and microbial community in two adjacent acid mine drainage lakes in Anhui Province, China. Environ. Pollut. 2021, 268, 115826. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).