
Multi-Heteroatom Doped Fe@CN Activation Peroxomonosulfate for the Removal of Trace Organic Contaminants from Water: Optimizing Fabrication and Performance



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
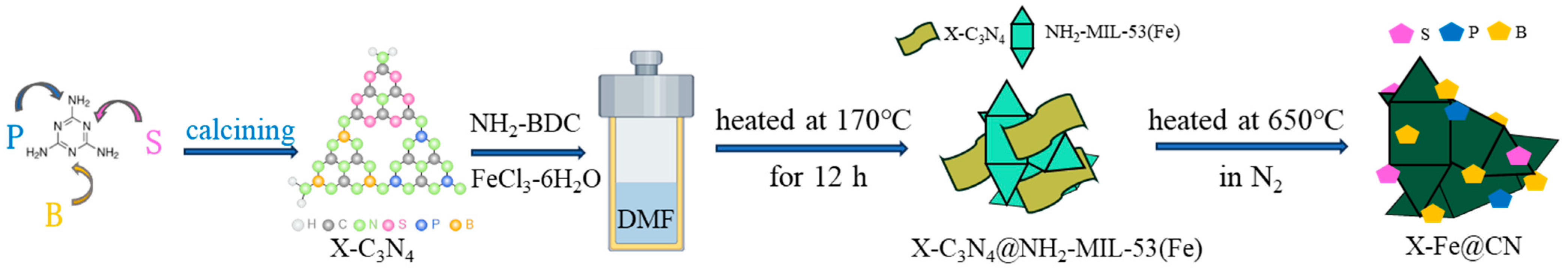
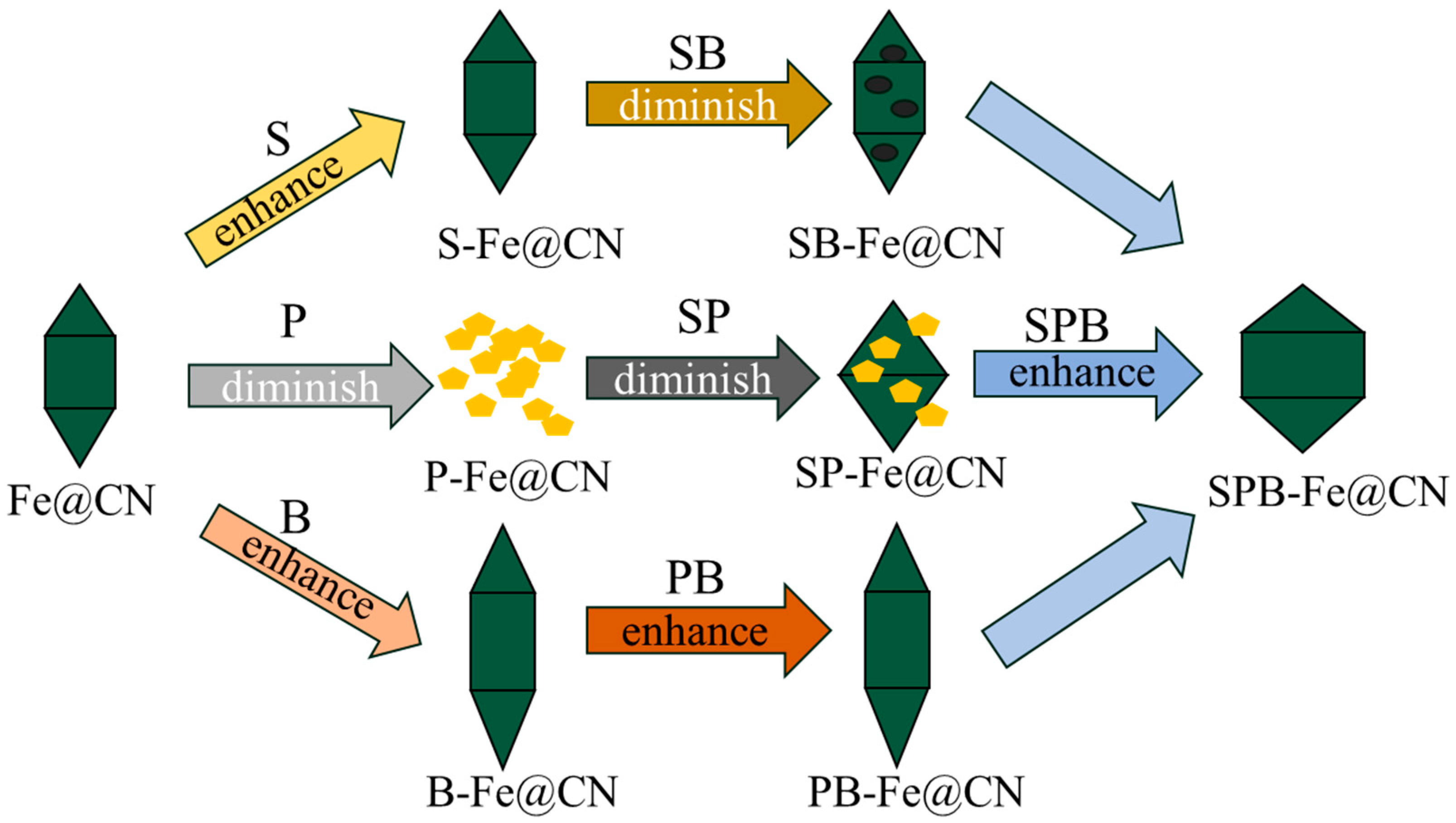
2.2. Synthesis of Heteroatom-Doped Catalysts
2.2.1. Synthesis of X-C3N4
2.2.2. Synthesis of X-C3N4@NH2-MIL-53(Fe) and X-Fe@CN
2.3. Experimental Procedures
2.4. Characterization and Analysis Methods
3. Results and Discussion
3.1. Characterization of X-Fe@CNs and Their Precursors
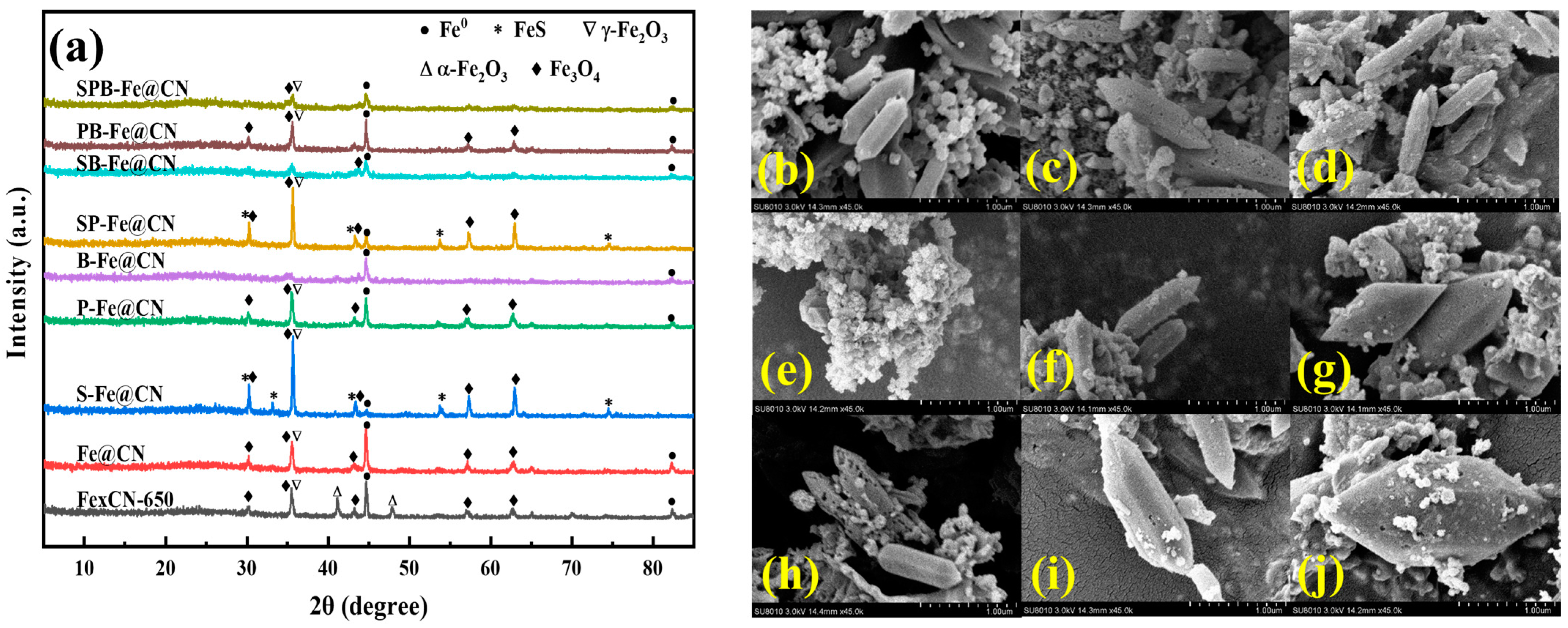
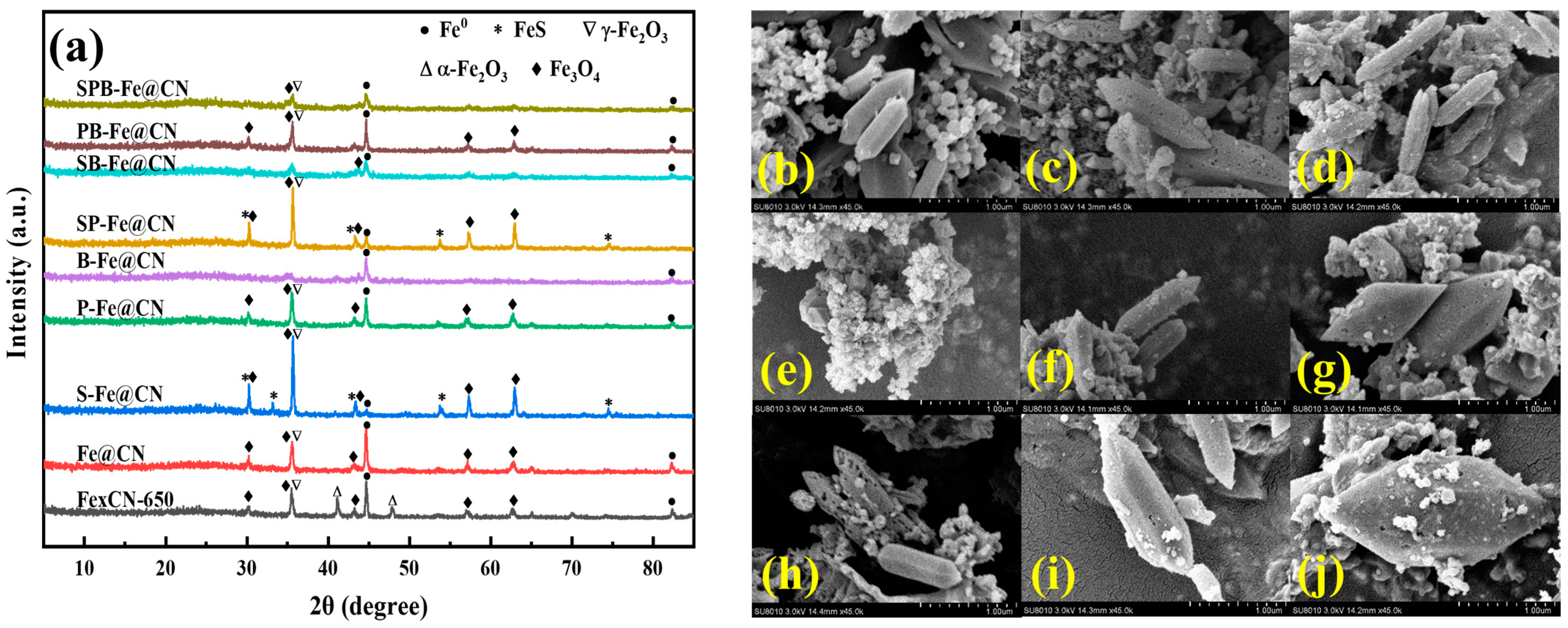
3.1.1. XRD and SEM of X-C3N4
3.1.2. XRD and SEM of X-C3N4@NH2-MIL-53(Fe)
3.1.3. XRD and SEM of X-Fe@CN
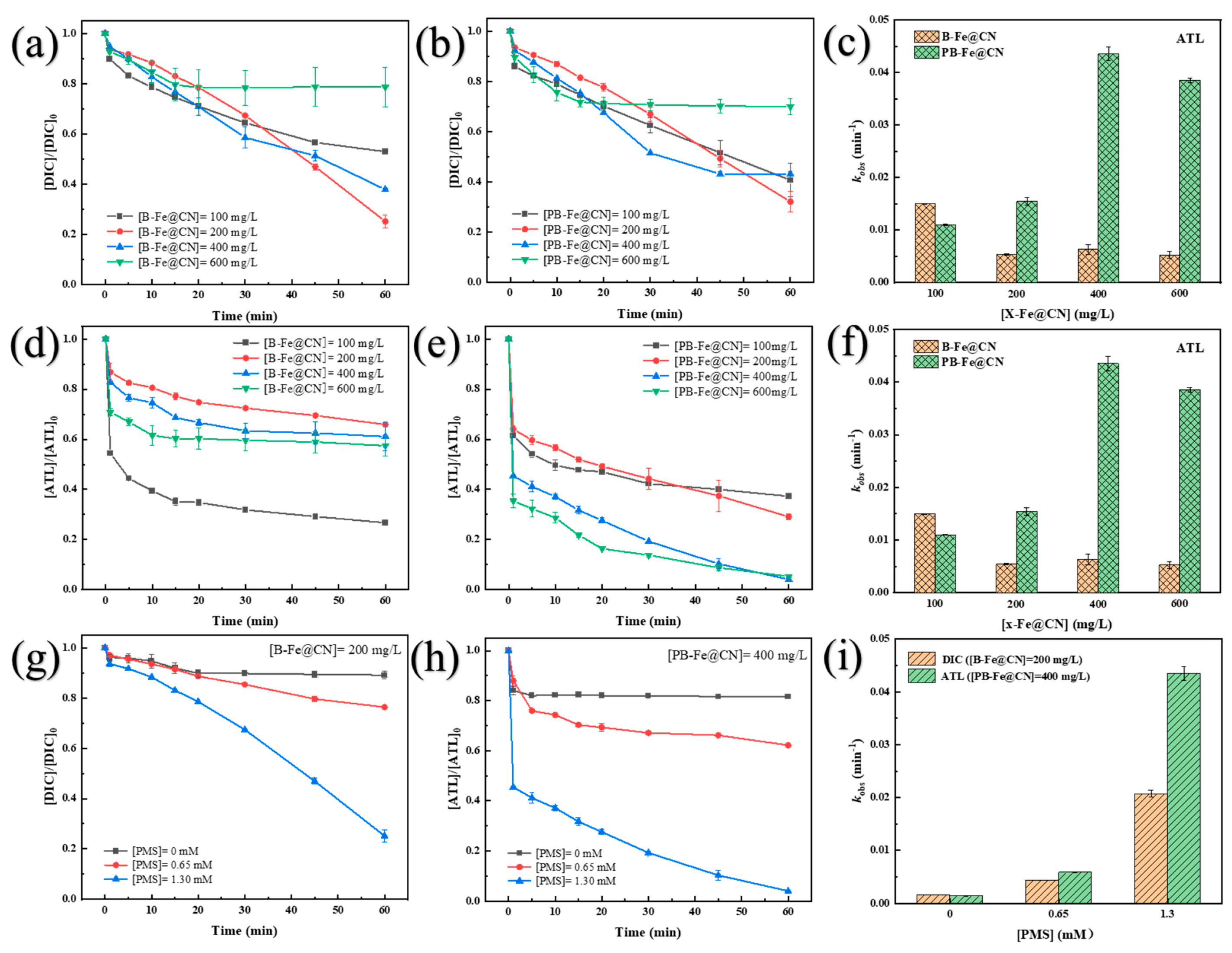
3.2. Removal Performance of TOrCs by X-Fe@CN Activating PMS
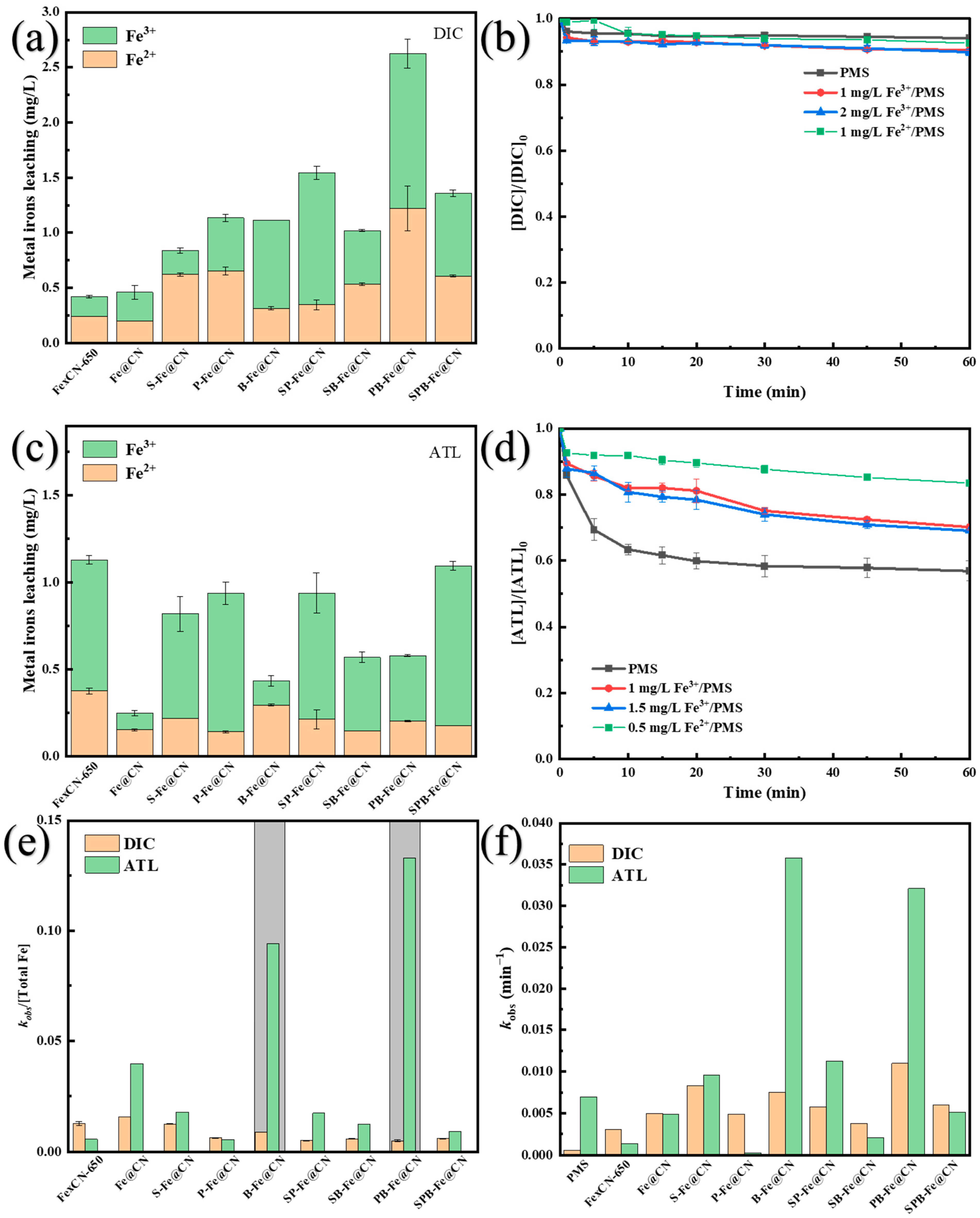
3.3. Safety Assessment of X-Fe@CN
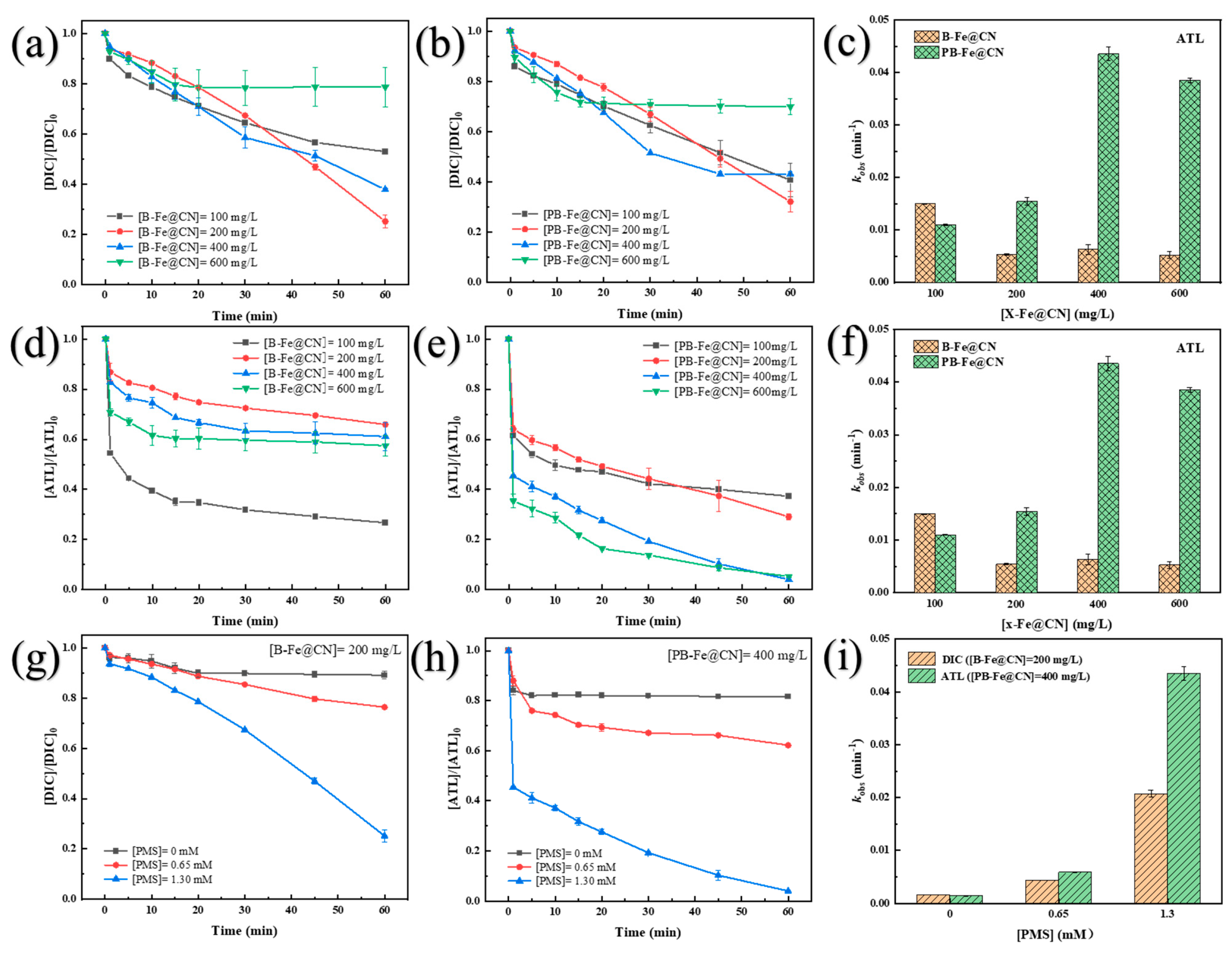
3.4. Effects of Initial Reaction Factors
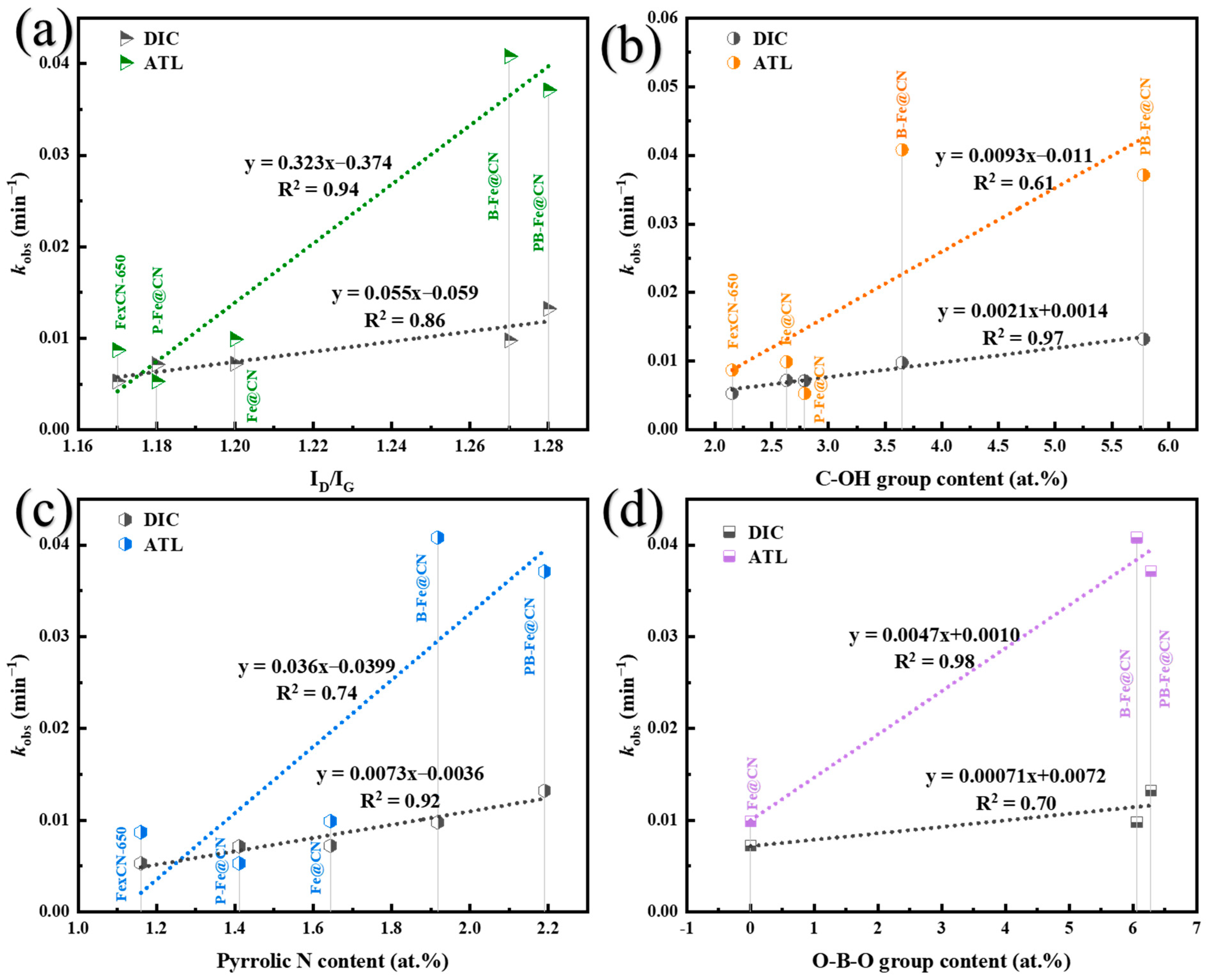
3.5. Proposed Reaction Mechanism Due to Heteroatom Doping
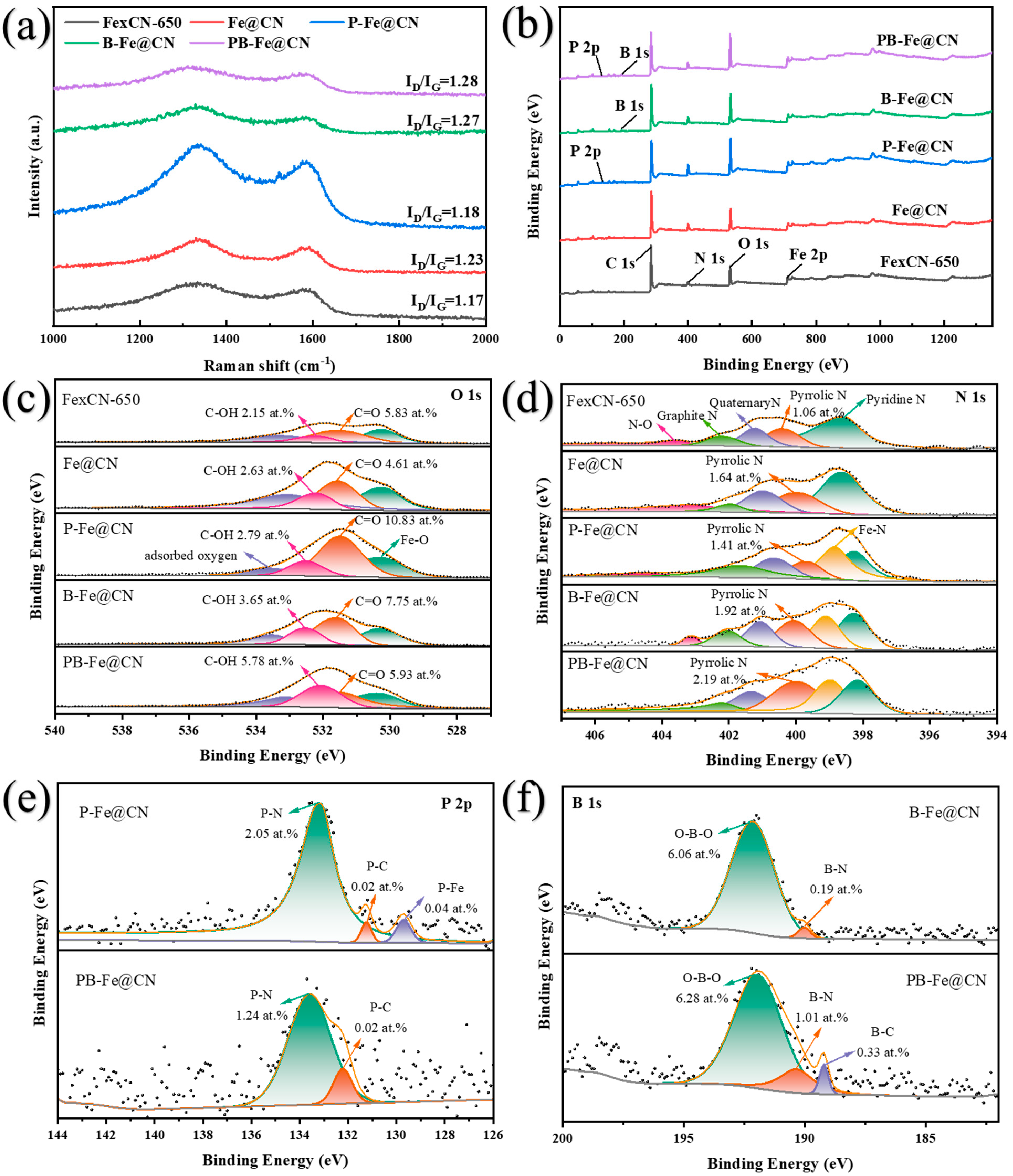
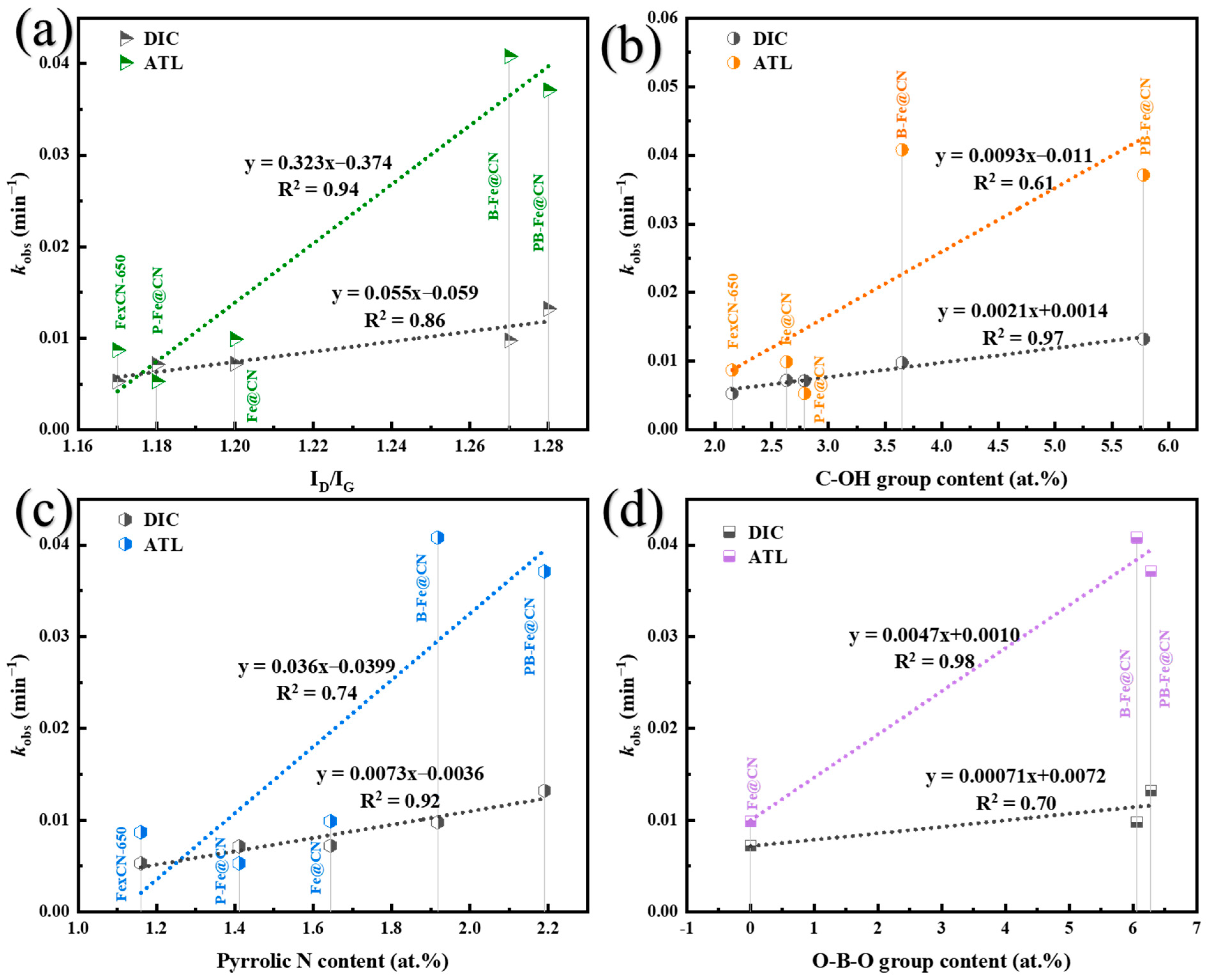
3.5.1. Identification of Surface-Active Sites
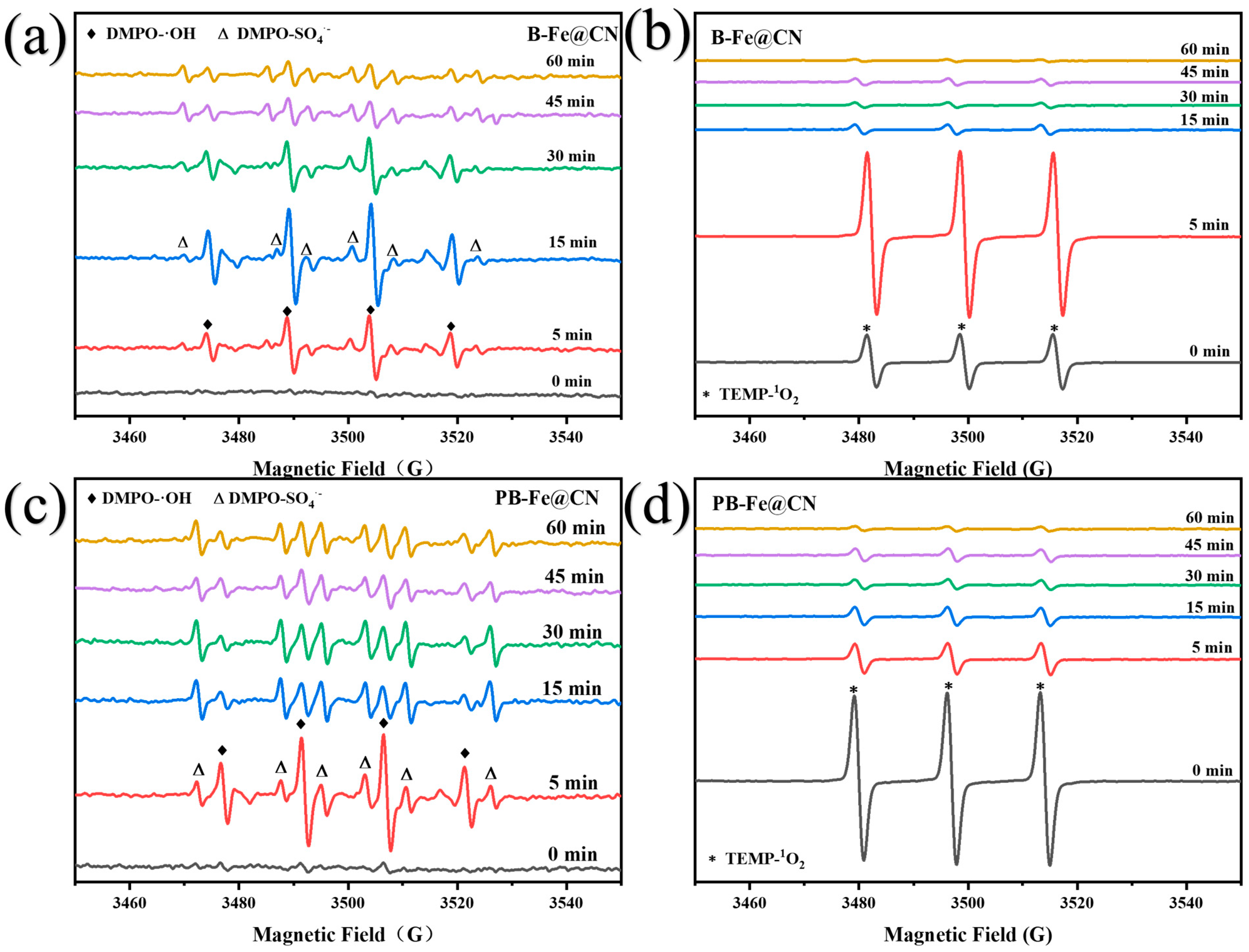
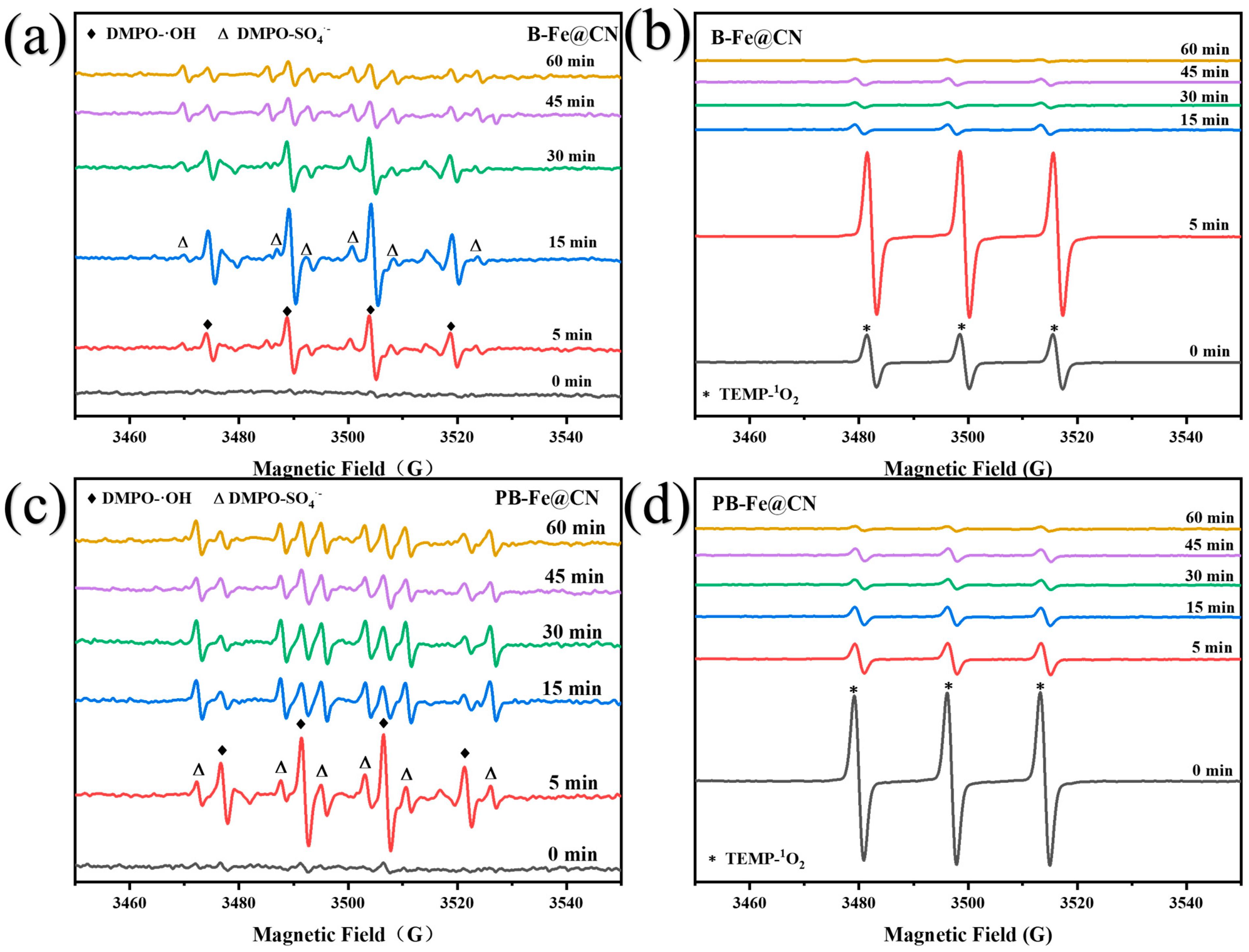
3.5.2. Identification of Reactive Oxygen Species
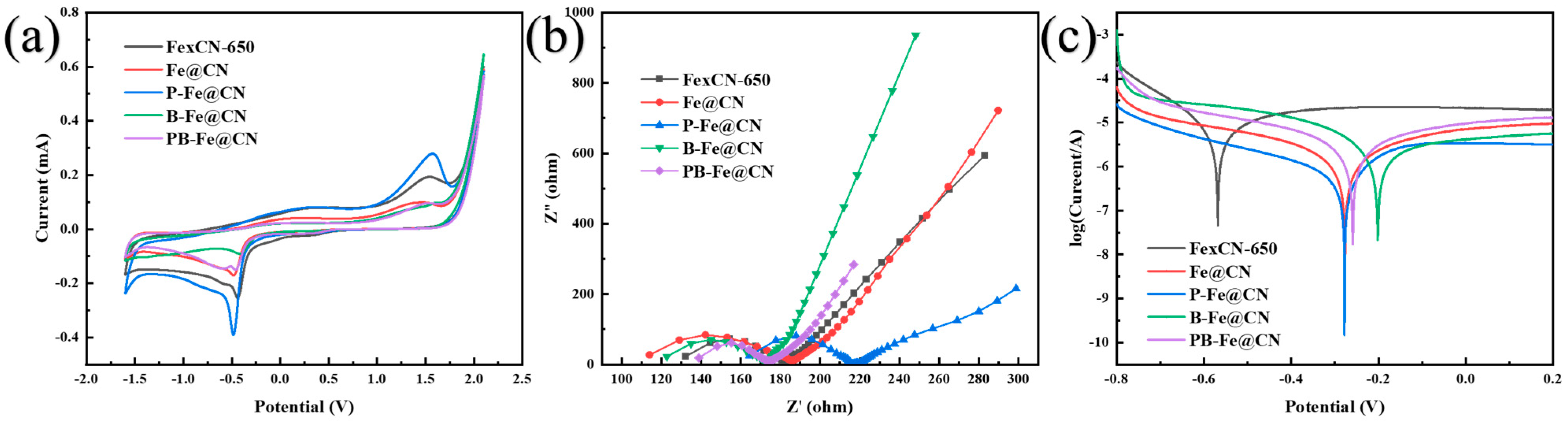
3.5.3. Identification of Electronic Transfers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Spahr, S.; Teixidó, M.; Gall, S.S.; Pritchard, J.C.; Hagemann, N.; Helmreich, B.; Luthy, R.G. Performance of biochars for the elimination of trace organic contaminants and metals from urban stormwater. Environ. Sci. Water Res. Technol. 2022, 8, 1287–1299. [Google Scholar] [CrossRef]
- Muller, A.; Osterlund, H.; Marsalek, J.; Viklander, M. The pollution conveyed by urban runoff: A review of sources. Sci. Total Environ. 2020, 709, 136125. [Google Scholar] [CrossRef] [PubMed]
- Fuchte, H.E.; Beck, N.; Bieg, E.; Bayer, V.J.; Achten, C.; Krauss, M.; Schaffer, A.; Smith, K.E.C. A look down the drain: Identification of dissolved and particle bound organic pollutants in urban runoff waters and sediments. Environ. Pollut. 2022, 302, 119047. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, L. Bioassays as a tool for evaluating advanced oxidation processes in water and wastewater treatment. Water Res. 2011, 45, 4311–4340. [Google Scholar] [CrossRef]
- Barbosa, M.O.; Moreira, N.F.F.; Ribeiro, A.R.; Pereira, M.F.R.; Silva, A.M.T. Occurrence and removal of organic micropollutants: An overview of the watch list of EU Decision 2015/495. Water Res. 2016, 94, 257–279. [Google Scholar] [CrossRef]
- Miklos, D.B.; Remy, C.; Jekel, M.; Linden, K.G.; Drewes, J.E.; Hubner, U. Evaluation of advanced oxidation processes for water and wastewater treatment—A critical review. Water Res. 2018, 139, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qu, Y.; Liu, X.; Zhai, C.; Liu, Y. Preparation of three-dimensional Fe–N co-doped open-porous carbon networks as an efficient ORR electrocatalyst in both alkaline and acidic media. Int. J. Hydrogen Energy 2021, 46, 18364–18375. [Google Scholar] [CrossRef]
- Fan, X.; Li, S.; Sun, M.; Song, C.; Xiao, J.; Du, J.; Tao, P.; Sun, T.; Shao, M.; Wang, T. Degradation of phenol by coal-based carbon membrane integrating sulfate radicals-based advanced oxidation processes. Ecotoxicol. Environ. Saf. 2019, 185, 109662. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shen, Q.; Nan, T.; Zhou, M.; Xia, Y.; Hu, G.; Zheng, Q.; Wu, Y.; Bian, T.; Wei, T.; et al. Cobalt-based catalysts for heterogeneous peroxymonosulfate (PMS) activation in degradation of organic contaminants: Recent advances and perspectives. J. Alloys Compd. 2023, 958, 170370. [Google Scholar] [CrossRef]
- Fang, Y.; Yang, Y.; Yang, Z.; Li, H.; Roesky, H.W. Advances in design of metal-organic frameworks activating persulfate for water decontamination. J. Organomet. Chem. 2021, 954–955, 122070. [Google Scholar] [CrossRef]
- Li, H.; Tian, J.; Zhu, Z.; Cui, F.; Zhu, Y.-A.; Duan, X.; Wang, S. Magnetic nitrogen-doped nanocarbons for enhanced metal-free catalytic oxidation: Integrated experimental and theoretical investigations for mechanism and application. Chem. Eng. J. 2018, 354, 507–516. [Google Scholar] [CrossRef]
- Li, M.; Li, Z.; Yu, X.; Wu, Y.; Mo, C.; Luo, M.; Li, L.; Zhou, S.; Liu, Q.; Wang, N.; et al. FeN4-doped carbon nanotubes derived from metal organic frameworks for effective degradation of organic dyes by peroxymonosulfate: Impacts of FeN4 spin states. Chem. Eng. J. 2022, 431, 133339. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Y.; Cheng, X.; Wang, J.; Yang, B.; Guo, H. Amino-modified metal–organic frameworks as peroxymonosulfate catalyst for bisphenol AF decontamination: ROS generation, degradation pathways, and toxicity evaluation. Sep. Purif. Technol. 2022, 282, 119967. [Google Scholar] [CrossRef]
- Luo, J.; Dai, Y.; Xu, X.; Liu, Y.; Yang, S.; He, H.; Sun, C.; Xian, Q. Green and efficient synthesis of Co-MOF-based/g-C3N4 composite catalysts to activate peroxymonosulfate for degradation of the antidepressant venlafaxine. J. Colloid Interface Sci. 2022, 610, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Hu, F.; Liu, Y.; Nie, G.; Xiao, L. Core-shell FeMn@NG derived from cellulose supported Prussian blue analogs for peroxymonosulfate activation: Non-radical mechanism and ultra-low metal leaching. J. Environ. Chem. Eng. 2022, 10, 108523. [Google Scholar] [CrossRef]
- Choong, Z.-Y.; Lin, K.-Y.A.; Lisak, G.; Lim, T.-T.; Oh, W.-D. Multi-heteroatom-doped carbocatalyst as peroxymonosulfate and peroxydisulfate activator for water purification: A critical review. J. Hazard. Mater. 2022, 426, 128077. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Indrawirawan, S.; Sun, H.; Wang, S. Effects of nitrogen-, boron-, and phosphorus-doping or codoping on metal-free graphene catalysis. Catal. Today 2015, 249, 184–191. [Google Scholar] [CrossRef]
- Chen, X.; Oh, W.-D.; Lim, T.-T. Graphene- and CNTs-based carbocatalysts in persulfates activation: Material design and catalytic mechanisms. Chem. Eng. J. 2018, 354, 941–976. [Google Scholar] [CrossRef]
- Ye, J.; Shao, Q.; Wang, X.; Wang, T. Effects of B, N, P and B/N, B/P pair into zigzag single-walled carbon nanotubes: A first-principle study. Chem. Phys. Lett. 2016, 646, 95–101. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; Zou, Y.; Lin, L.; Liang, H.; Lei, H.; Li, B.; Li, X.-y. Catalytic activity enhancement by P and S co-doping of a single-atom Fe catalyst for peroxymonosulfate-based oxidation. Chem. Eng. J. 2023, 453, 139890. [Google Scholar] [CrossRef]
- Xie, J.; Pan, X.; Jiang, C.; Zhao, L.; Gong, X.; Liu, Y. Enhanced conversion of superoxide radical to singlet oxygen in peroxymonosulfate activation by metal-organic frameworks derived heteroatoms dual-doped porous carbon catalyst. Environ. Res. 2023, 236, 116745. [Google Scholar] [CrossRef] [PubMed]
- Choong, Z.-Y.; Gasim, M.F.; Zhou, T.; Hamidon, T.S.; Hussin, M.H.; Khoerunnisa, F.; Oh, W.-D. The influence of B heteroatom concentrations on the physiochemical properties of N, B-co-doped biochar for peroxymonosulfate activation in ciprofloxacin removal. J. Water Process Eng. 2023, 51, 103468. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, H.; Wang, X.; Ni, W.; Gao, F.; Wang, J.; Liu, M.; Li, Y. HPLC and GC methods development for the analysis of key intermediate for synthesis of dicamba. Chin. J. Chem. Eng. 2021, 33, 112–117. [Google Scholar] [CrossRef]
- Amruth, N.V.; Chinnaiyan, P.; Sridhar, N.K.; Akshaya, R.B.; Prasitha, P. Modelling of AOP removal of β-blocker atenolol from wastewater. Mater. Today Proc. 2022, 49, 2301–2305. [Google Scholar] [CrossRef]
- Babu, P.; Mohanty, S.; Naik, B.; Parida, K. Synergistic Effects of Boron and Sulfur Co-doping into Graphitic Carbon Nitride Framework for Enhanced Photocatalytic Activity in Visible Light Driven Hydrogen Generation. ACS Appl. Energy Mater. 2018, 1, 5936–5947. [Google Scholar] [CrossRef]
- Jin, J.; Wu, H.; Wang, S.; Ding, Y.; Yin, S. Heteroatoms doped C3N4 as high performance catalysts for the oxygen reduction reaction. Int. J. Hydrogen Energy 2017, 42, 20579–20588. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, Y.; Zhang, W.; Peng, Z.; Xie, H.; Wang, Y.; Guo, X.; Zhang, M.; Li, R.; Huang, Y. Template-free preparation of non-metal (B, P, S) doped g-C3N4 tubes with enhanced photocatalytic H2O2 generation. J. Mater. Sci. Technol. 2021, 95, 127–135. [Google Scholar] [CrossRef]
- Liu, C.; Liu, L.; Tian, X.; Wang, Y.; Li, R.; Zhang, Y.; Song, Z.; Xu, B.; Chu, W.; Qi, F.; et al. Coupling metal–organic frameworks and g-CN to derive Fe@N-doped graphene-like carbon for peroxymonosulfate activation: Upgrading framework stability and performance. Appl. Catal. B Environ. 2019, 255, 117763. [Google Scholar] [CrossRef]
- Long, J.; Xie, X.; Xu, J.; Gu, Q.; Chen, L.; Wang, X. Nitrogen-Doped Graphene Nanosheets as Metal-Free Catalysts for Aerobic Selective Oxidation of Benzylic Alcohols. ACS Catal. 2012, 2, 622–631. [Google Scholar] [CrossRef]
- Lan, H.; Li, L.; An, X.; Liu, F.; Chen, C.; Liu, H.; Qu, J. Microstructure of carbon nitride affecting synergetic photocatalytic activity: Hydrogen bonds vs. structural defects. Appl. Catal. B Environ. 2017, 204, 49–57. [Google Scholar] [CrossRef]
- Dou, J.; Cheng, J.; Lu, Z.; Tian, Z.; Xu, J.; He, Y. Biochar co-doped with nitrogen and boron switching the free radical based peroxydisulfate activation into the electron-transfer dominated nonradical process. Appl. Catal. B Environ. 2022, 301, 120832. [Google Scholar] [CrossRef]
- Wu, Z.; Tong, Z.; Xie, Y.; Sun, H.; Gong, X.; Qin, P.; Liang, Y.; Yuan, X.; Zou, D.; Jiang, L. Efficient degradation of tetracycline by persulfate activation with Fe, Co and O co−doped g−C3N4: Performance, mechanism and toxicity. Chem. Eng. J. 2022, 434, 134732. [Google Scholar] [CrossRef]
- Yang, Y.; Jin, H.; Zhang, C.; Gan, H.; Yi, F.; Wang, H. Nitrogen-deficient modified P–Cl co-doped graphitic carbon nitride with enhanced photocatalytic performance. J. Alloys Compd. 2020, 821, 153439. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, X.; Jiang, Z.; Chen, H.-S.; Yang, P. Phosphorus and sulphur co-doping of g-C3N4 nanotubes with tunable architectures for superior photocatalytic H2 evolution. Int. J. Hydrogen Energy 2019, 44, 20042–20055. [Google Scholar] [CrossRef]
- Li, G.-L.; Cheng, G.-C.; Chen, W.-W.; Liu, C.-D.; Yuan, L.-F.; Yang, B.-B.; Hao, C. N/S/B-doped graphitized carbon encased Fe species as a highly active and durable catalyst towards oxygen reduction reaction. J. Colloid Interface Sci. 2018, 514, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, H.; Han, Q.; Jiang, W.; Liu, C.; Che, G. Optimized design of BiVO4/NH2-MIL-53(Fe) heterostructure for enhanced photocatalytic degradation of methylene blue and ciprofloxacin under visible light. J. Phys. Chem. Solids 2021, 154, 110027. [Google Scholar] [CrossRef]
- He, X.; Wang, L.; Sun, S.; Yang, X.; Tian, H.; Xia, Z.; Li, X.; Yan, X.; Pu, X.; Jiao, Z. Self-assembled synthesis of recyclable g-C3N4/NH2-MIL-53(Fe) aerogel for enhanced photocatalytic degradation of organic pollutants. J. Alloys Compd. 2023, 946, 169391. [Google Scholar] [CrossRef]
- Yu, Z.; Ma, J.; Huang, X.; Lv, Y.; Liu, Y.; Lin, C.; Dou, R.; Ye, X.; Shi, Y.; Liu, M. Insights into enhanced peroxydisulfate activation with S doped Fe@C catalyst for the rapid degradation of organic pollutants. J. Colloid Interface Sci. 2022, 610, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Z.; Sun, X.; Han, J.; Guo, R. Fe, P, N- and FeP, N-doped carbon hollow nanospheres: A comparison study toward oxygen reduction reaction electrocatalysts. J. Colloid Interface Sci. 2021, 602, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, A.; Yang, A.; Zuo, G.; Dai, J.; Zheng, Y. ZnS, Fe, and P co-doped N enriched carbon derived from MOFs as efficient electrocatalyst for oxygen reduction reaction. Int. J. Hydrogen Energy 2020, 45, 31863–31870. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, G.; Lei, T.; Wu, Y.; Zhang, X. Oxidative removal of carbazole and alkylated derivatives by FeOx/OMS-2 nanorods activated peroxymonosulfate (PMS) and their utilizing for real wastewater. J. Water Process Eng. 2023, 55, 104120. [Google Scholar] [CrossRef]
- Yu, X.; Qin, W.; Yuan, X.; Sun, L.; Pan, F.; Xia, D. Synergistic mechanism and degradation kinetics for atenolol elimination via integrated UV/ozone/peroxymonosulfate process. J. Hazard. Mater. 2021, 407, 124393. [Google Scholar] [CrossRef]
- Sun, W.; Pang, K.; Ye, F.; Pu, M.; Zhou, C.; Yang, C.; Zhang, Q. Efficient persulfate activation catalyzed by pyridinic N, C OH, and thiophene S on N,S-co-doped carbon for nonradical sulfamethoxazole degradation: Identification of active sites and mechanisms. Sep. Purif. Technol. 2022, 284, 120197. [Google Scholar] [CrossRef]
- Pang, K.; Sun, W.; Ye, F.; Yang, L.; Pu, M.; Yang, C.; Zhang, Q.; Niu, J. Sulfur-modified chitosan derived N,S-co-doped carbon as a bifunctional material for adsorption and catalytic degradation sulfamethoxazole by persulfate. J. Hazard. Mater. 2022, 424, 127270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H.; Ang, H.M.; Tadé, M.O.; Wang, S. Magnetic Fe3O4/carbon sphere/cobalt composites for catalytic oxidation of phenol solutions with sulfate radicals. Chem. Eng. J. 2014, 245, 1–9. [Google Scholar] [CrossRef]
- Zeng, T.; Yu, M.; Zhang, H.; He, Z.; Chen, J.; Song, S. Fe/Fe3C@N-doped porous carbon hybrids derived from nano-scale MOFs: Robust and enhanced heterogeneous catalyst for peroxymonosulfate activation. Catal. Sci. Technol. 2017, 7, 396–404. [Google Scholar] [CrossRef]
- Li, X.; Ye, Z.; Xie, S.; Li, H.; Lv, Y.; Wang, Y.; Wang, Y.; Lin, C. Insight into the performance and mechanism of peroxymonosulfate activation by B, N co-doped hierarchical porous carbon for phenol degradation. J. Environ. Chem. Eng. 2022, 10, 108264. [Google Scholar] [CrossRef]
- Tang, Y.; Lei, Y.; Tong, K.; Yang, T.; Fu, T.; Xiang, Y.; Zhang, S.; Si, Y.; Guo, C. Fe, N-doped graphene-wrapped carbon black nanoparticles as highly efficient catalyst towards oxygen reduction reaction. Appl. Surf. Sci. 2021, 545, 148981. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, Y.; Chen, J.; Wu, T.; Wang, J.; Zhao, X.; Sun, W.; Lin, H.; Lv, S. Graphene/Fe@N-doped carbon hybrid derived from spent MOF adsorbent as efficient persulfate activator for degradation of tetracycline hydrochloride. Chem. Eng. J. 2022, 431, 133443. [Google Scholar] [CrossRef]
- Wang, Y.; Ao, Z.; Sun, H.; Duan, X.; Wang, S. Activation of peroxymonosulfate by carbonaceous oxygen groups: Experimental and density functional theory calculations. Appl. Catal. B Environ. 2016, 198, 295–302. [Google Scholar] [CrossRef]
- Mao, S.; Liu, C.; Wu, Y.; Xia, M.; Wang, F. Porous P, Fe-doped g-C3N4 nanostructure with enhanced photo-Fenton activity for removal of tetracycline hydrochloride: Mechanism insight, DFT calculation and degradation pathways. Chemosphere 2022, 291, 133039. [Google Scholar] [CrossRef]
- Lan, J.; Zhang, Q.; Yang, G.; Liu, Z.; Peng, F. Removal of tetracycline hydrochloride by N, P co-doped carbon encapsulated Fe2P activating PMS: A non-radical pathway dominated by singlet oxygen. J. Environ. Chem. Eng. 2023, 11, 110481. [Google Scholar] [CrossRef]
- Pan, G.; Wei, J.; Xu, M.; Li, J.; Wang, L.; Li, Y.; Cui, N.; Li, J.; Wang, Z. Insight into boron-doped biochar as efficient metal-free catalyst for peroxymonosulfate activation: Important role of -O-B-O- moieties. J. Hazard. Mater. 2023, 445, 130479. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Banerjee, G.; Brudvig, G.W.; Kim, J.-H.; Pignatello, J.J. Oxidation of Organic Compounds in Water by Unactivated Peroxymonosulfate. Environ. Sci. Technol. 2018, 52, 5911–5919. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J. Synergistic effect of PMS activation by Fe0@Fe3O4 anchored on N, S, O co-doped carbon composite for degradation of sulfamethoxazole. Chem. Eng. J. 2022, 427, 131960. [Google Scholar] [CrossRef]
- Yun, E.-T.; Yoo, H.-Y.; Bae, H.; Kim, H.-I.; Lee, J. Exploring the Role of Persulfate in the Activation Process: Radical Precursor Versus Electron Acceptor. Environ. Sci. Technol. 2017, 51, 10090–10099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, C.; Sun, X.; Gao, H.; Liu, X.; Sun, J.; Shi, W.; Ai, S. Iron nanoparticles encapsulated within nitrogen and sulfur co-doped magnetic porous carbon as an efficient peroxymonosulfate activator to degrade 1-naphthol. Sci. Total Environ. 2020, 739, 139896. [Google Scholar] [CrossRef]
- Zhu, K.; Xia, W.; He, D.; Huang, J.; He, H.; Lei, L.; Chen, W.; Liu, X. Facile fabrication of Fe/Fe3C embedded in N-doped carbon nanofiber for efficient degradation of tetracycline via peroxymonosulfate activation: Role of superoxide radical and singlet oxygen. J. Colloid Interface Sci. 2022, 609, 86–101. [Google Scholar] [CrossRef]
- Wang, D.; Kijkla, P.; Saleh, M.A.; Kumseranee, S.; Punpruk, S.; Gu, T. Tafel scan schemes for microbiologically influenced corrosion of carbon steel and stainless steel. J. Mater. Sci. Technol. 2022, 130, 193–197. [Google Scholar] [CrossRef]
- Xu, L.; Xin, Y.; Wang, J. A comparative study on IrO2–Ta2O5 coated titanium electrodes prepared with different methods. Electrochim. Acta 2009, 54, 1820–1825. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simple | Name |
|---|---|
| NH2-MIL-53(Fe) | FexCN-650 |
| g-C3N4@NH2-MIL-53(Fe) | Fe@CN |
| S-C3N4@NH2-MIL-53(Fe) | S-Fe@CN |
| P-C3N4@NH2-MIL-53(Fe) | P-Fe@CN |
| B-C3N4@NH2-MIL-53(Fe) | B-Fe@CN |
| SP-C3N4@NH2-MIL-53(Fe) | SP-Fe@CN |
| SB-C3N4@NH2-MIL-53(Fe) | SB-Fe@CN |
| PB-C3N4@NH2-MIL-53(Fe) | PB-Fe@CN |
| SPB-C3N4@NH2-MIL-53(Fe) | SPB-Fe@CN |
| Catalyst | Rs (Ω cm−2) | Rct (Ω cm−2) |
|---|---|---|
| FexCN-650 | 51.13 | 127.6 |
| Fe@CN | 39.29 | 134.3 |
| P-Fe@CN | 43.32 | 128.7 |
| B-Fe@CN | 38.55 | 130.3 |
| PB-Fe@CN | 82.01 | 79.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Ren, R.; Liu, Y.; Li, C.; Wang, Z.; Qi, F. Multi-Heteroatom Doped Fe@CN Activation Peroxomonosulfate for the Removal of Trace Organic Contaminants from Water: Optimizing Fabrication and Performance. Water 2023, 15, 4241. https://doi.org/10.3390/w15244241
Chen J, Ren R, Liu Y, Li C, Wang Z, Qi F. Multi-Heteroatom Doped Fe@CN Activation Peroxomonosulfate for the Removal of Trace Organic Contaminants from Water: Optimizing Fabrication and Performance. Water. 2023; 15(24):4241. https://doi.org/10.3390/w15244241
Chicago/Turabian StyleChen, Jiamin, Ruijun Ren, Yatao Liu, Chen Li, Zhenbei Wang, and Fei Qi. 2023. "Multi-Heteroatom Doped Fe@CN Activation Peroxomonosulfate for the Removal of Trace Organic Contaminants from Water: Optimizing Fabrication and Performance" Water 15, no. 24: 4241. https://doi.org/10.3390/w15244241
APA StyleChen, J., Ren, R., Liu, Y., Li, C., Wang, Z., & Qi, F. (2023). Multi-Heteroatom Doped Fe@CN Activation Peroxomonosulfate for the Removal of Trace Organic Contaminants from Water: Optimizing Fabrication and Performance. Water, 15(24), 4241. https://doi.org/10.3390/w15244241