
A Preliminary Sorption Study of Uranium on MnO2 (Pyrolusite) in the Presence of Siderophore Desferrioxamine B—The Mechanism of a Ternary System

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. U-DFO Solution Preparation
2.3. Sorption Experiments
2.4. Elemental and DFOB Analysis
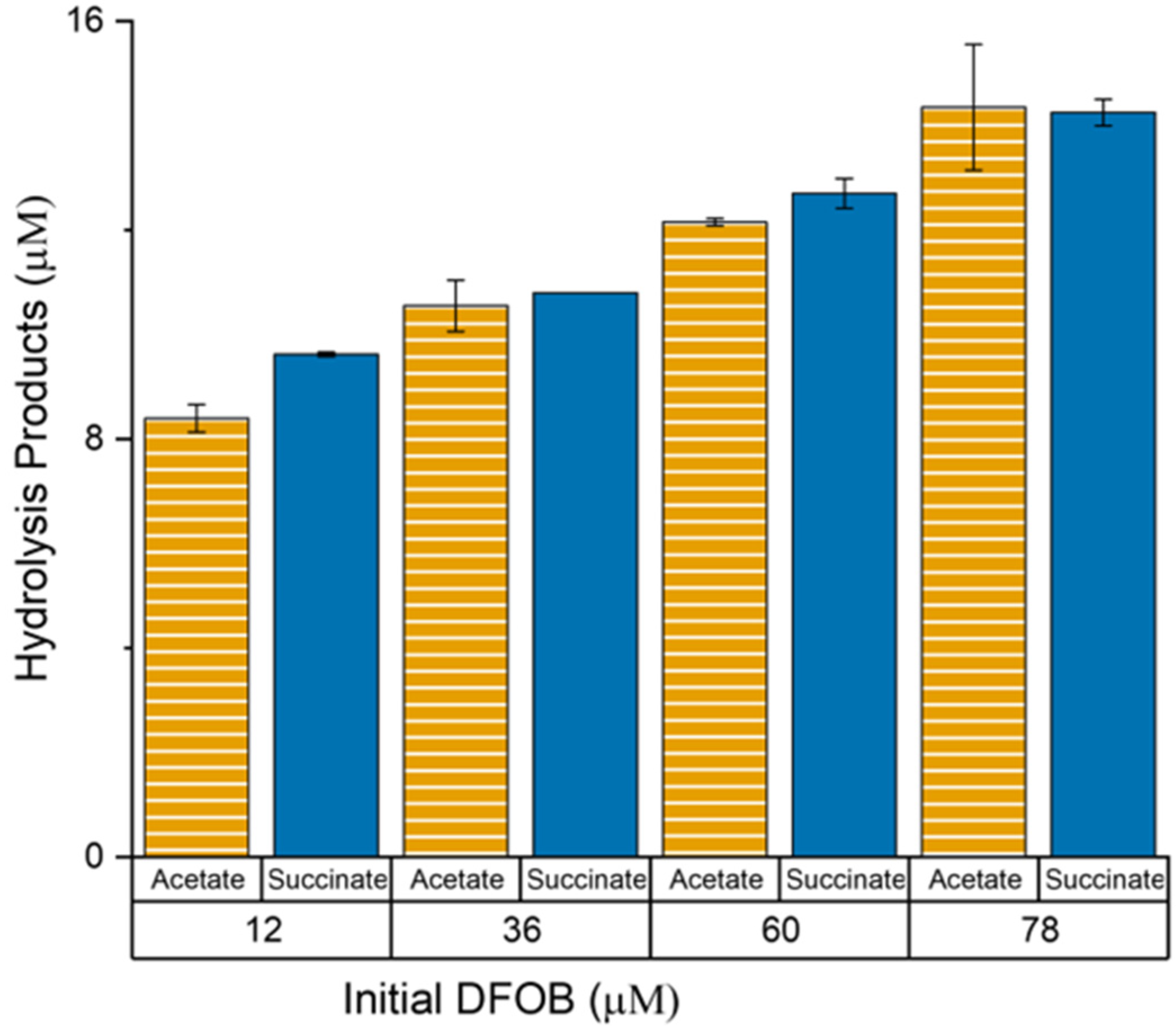
2.5. Determination of Acetate and Succinate
3. Results and Discussion
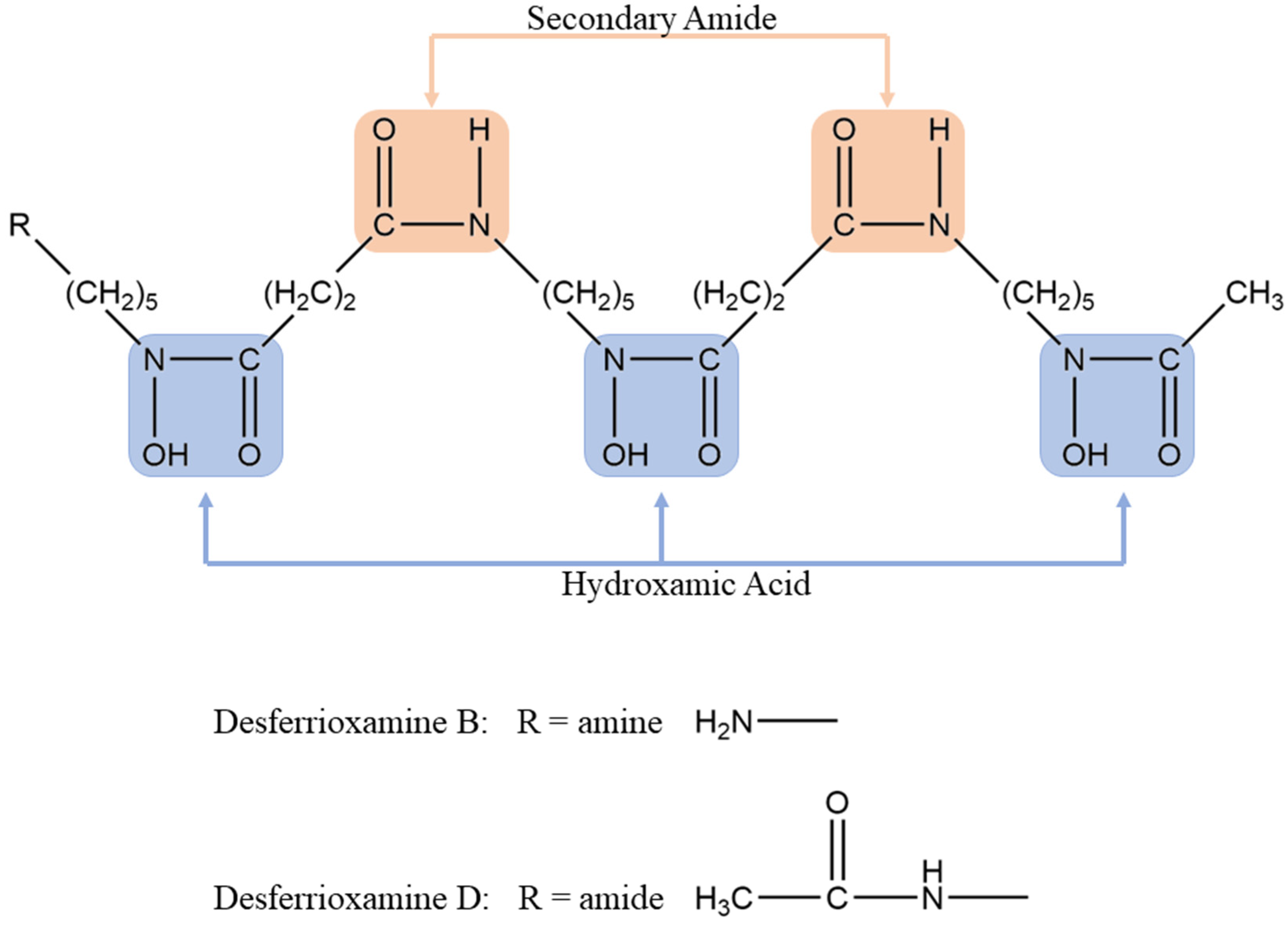
3.1. DFOB vs. DFOD
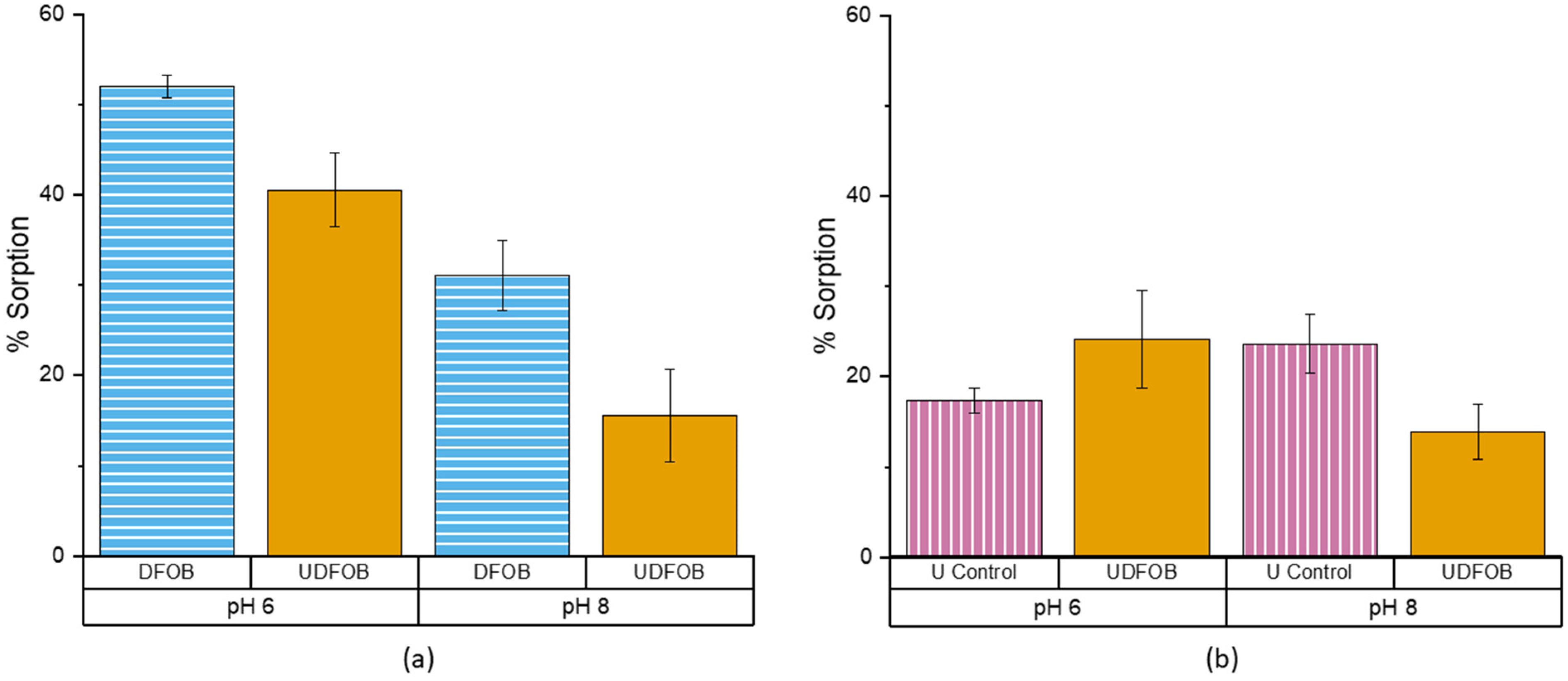
3.2. DFOB pH 6 vs. pH 8
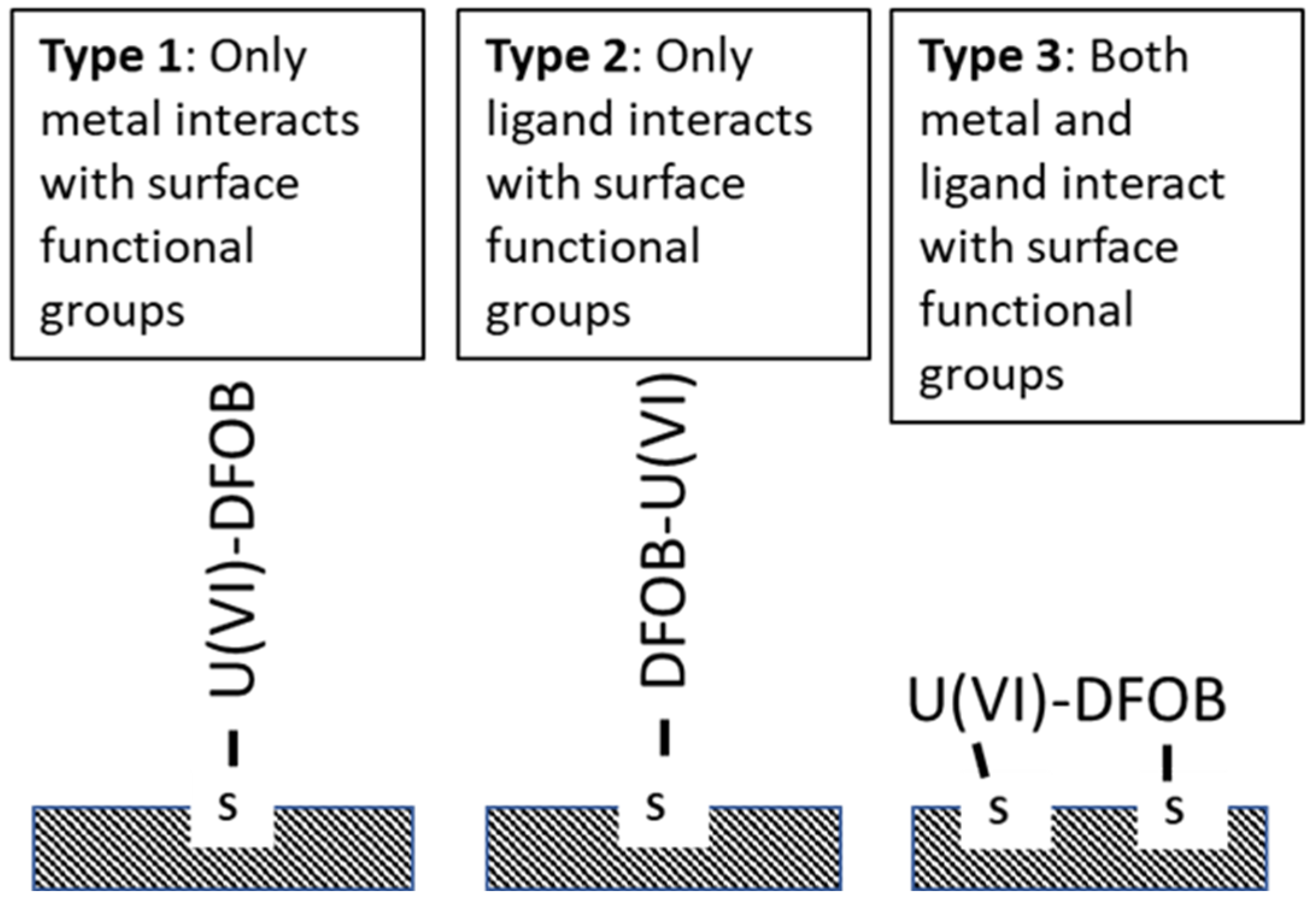
3.3. Ligand-Metal Adsorption Mechanisms
3.4. Type 2 Ligand-like Mechanism
3.5. Type 3 Ligand-like and Metal-like Mechanism
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sprynskyy, M.; Kowalkowski, T.; Tutu, H.; Cukrowska, E.M.; Buszewski, B. Adsorption Performance of Talc for Uranium Removal from Aqueous Solution. Chem. Eng. J. 2011, 171, 1185–1193. [Google Scholar] [CrossRef]
- Maher, K.; Bargar, J.R.; Brown, G.E. Environmental Speciation of Actinides. Inorg. Chem. 2013, 52, 3510–3532. [Google Scholar] [CrossRef]
- Walther, C.; Denecke, M.A. Actinide Colloids and Particles of Environmental Concern. Chem. Rev. 2013, 113, 995–1015. [Google Scholar] [CrossRef]
- Anagnostopoulos, V.; Katsenovich, Y.; Lee, B.; Lee, H.M. Biotic Dissolution of Autunite under Anaerobic Conditions: Effect of Bicarbonates and Shewanella Oneidensis MR1 Microbial Activity. Environ. Geochem. Health 2020, 42, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Salbu, B.; Kashparov, V.; Lind, O.C.; Garcia-Tenorio, R.; Johansen, M.P.; Child, D.P.; Roos, P.; Sancho, C. Challenges Associated with the Behaviour of Radioactive Particles in the Environment. J. Environ. Radioact. 2018, 186, 101–115. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry ATSDR. Toxicological Profile for Plutonium; Department of Health and Human Services: Atlanta, GA, USA, 2010.
- Agency for Toxic Substances and Disease Registry ATSDR. Toxicological Profile for Uranium; Department of Health and Human Services: Atlanta, GA, USA, 2013.
- Szlamkowicz, I.; Stanberry, J.; Lugo, K.; Murphy, Z.; Ruiz Garcia, M.; Hunley, L.; Qafoku, N.P.; Anagnostopoulos, V. Role of Manganese Oxides in Controlling Subsurface Metals and Radionuclides Mobility: A Review. ACS Earth Space Chem. 2023, 7, 1–10. [Google Scholar] [CrossRef]
- Gavrilescu, M.; Pavel, L.V.; Cretescu, I. Characterization and Remediation of Soils Contaminated with Uranium. J. Hazard. Mater. 2009, 163, 475–510. [Google Scholar] [CrossRef] [PubMed]
- Environmental Management Support, Inc. Innovative Remediation Remediation Technologies: Field-Scale Demonstration Projects in North America, 2nd ed.; Environmental Protection Agency: Washington, DC, USA, 2000.
- Szlamkowicz, I.B.; Fentress, A.J.; Longen, L.F.; Stanberry, J.S.; Anagnostopoulos, V.A. Transformations and Speciation of Iodine in the Environment as a Result of Oxidation by Manganese Minerals. ACS Earth Space Chem. 2022, 6, 1948–1956. [Google Scholar] [CrossRef]
- Stanberry, J.; Szlamkowicz, I.; Purdy, L.R.; Anagnostopoulos, V. TcO2 Oxidative Dissolution by Birnessite under Anaerobic Conditions: A Solid–Solid Redox Reaction Impacting the Environmental Mobility of Tc-99. Environ. Sci. Process. Impacts 2021, 23, 844–854. [Google Scholar] [CrossRef]
- Stanberry, J.; Szlamkowicz, I.; Magno, D.; Shultz, L.; Anagnostopoulos, V. Oxidative Dissolution of TcO2 by Mn(III) Minerals under Anaerobic Conditions: Implications on Technetium-99 Remediation. Appl. Geochem. 2021, 127, 104858. [Google Scholar] [CrossRef]
- Wu, W.-M.; Carley, J.; Fienen, M.; Mehlhorn, T.; Lowe, K.; Nyman, J.; Luo, J.; Gentile, M.E.; Rajan, R.; Wagner, D.; et al. Pilot-Scale in Situ Bioremediation of Uranium in a Highly Contaminated Aquifer. 1. Conditioning of a Treatment Zone. Environ. Sci. Technol. 2006, 40, 3978–3985. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.M. Iron Oxide Dissolution and Solubility in the Presence of Siderophores. Aquat. Sci. Res. Across Boundaries 2004, 66, 3–18. [Google Scholar] [CrossRef]
- Liermann, L.J.; Kalinowski, B.E.; Brantley, S.L.; Ferry, J.G. Role of Bacterial Siderophores in Dissolution of Hornblende. Geochim. Cosmochim. Acta 2000, 64, 587–602. [Google Scholar] [CrossRef]
- Saha, M.; Sarkar, S.; Sarkar, B.; Sharma, B.K.; Bhattacharjee, S.; Tribedi, P. Microbial Siderophores and Their Potential Applications: A Review. Environ. Sci. Pollut. Res. 2016, 23, 3984–3999. [Google Scholar] [CrossRef]
- Saha, R.; Saha, N.; Donofrio, R.S.; Bestervelt, L.L. Microbial Siderophores: A Mini Review: Microbial Siderophores. J. Basic Microbiol. 2013, 53, 303–317. [Google Scholar] [CrossRef]
- Haas, H. Molecular Genetics of Fungal Siderophore Biosynthesis and Uptake: The Role of Siderophores in Iron Uptake and Storage. Appl. Microbiol. Biotechnol. 2003, 62, 316–330. [Google Scholar] [CrossRef]
- Braud, A.; Geoffroy, V.; Hoegy, F.; Mislin, G.L.A.; Schalk, I.J. Presence of the Siderophores Pyoverdine and Pyochelin in the Extracellular Medium Reduces Toxic Metal Accumulation in Pseudomonas Aeruginosa and Increases Bacterial Metal Tolerance: Extracellular Sequestration of Toxic Metals by Siderophores. Environ. Microbiol. Rep. 2010, 2, 419–425. [Google Scholar] [CrossRef]
- Farkas, E.; Csóka, H.; Micera, G.; Dessi, A. Copper(II), Nickel(II), Zinc(II), and Molybdenum(VI) Complexes of Desferrioxamine B in Aqueous Solution. J. Inorg. Biochem. 1997, 65, 281–286. [Google Scholar] [CrossRef]
- Aoki, J.; Oonuma, C.; Sudowe, R.; Takagai, Y. Adsorption Behavior of Pu(IV), Am(III), Cm(III), and U(VI) on Desferrioxamine B-Immobilized Micropolymer and Its Applications in the Separation of Pu(IV). Anal. Sci. 2021, 37, 1641–1644. [Google Scholar] [CrossRef]
- Kenney, J.P.L.; Ellis, T.; Nicol, F.S.; Porter, A.E.; Weiss, D.J. The Effect of Bacterial Growth Phase and Culture Concentration on U(VI) Removal from Aqueous Solution. Chem. Geol. 2018, 482, 61–71. [Google Scholar] [CrossRef]
- Bellotti, D.; Remelli, M. Deferoxamine B: A Natural, Excellent and Versatile Metal Chelator. Molecules 2021, 26, 3255. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Retamal-Morales, G.; Tischler, D. Metal Binding Ability of Microbial Natural Metal Chelators and Potential Applications. Nat. Prod. Rep. 2020, 37, 1262–1283. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Holmström, S.J.M. Siderophores in Environmental Research: Roles and Applications: Siderophores in Environmental Research. Microb. Biotechnol. 2014, 7, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Snow, G.A. Metal Complexes of Mycobactin P and of Desferrisideramines. Biochem. J. 1969, 115, 199–205. [Google Scholar] [CrossRef]
- Dhungana, S.; White, P.S.; Crumbliss, A.L. Crystal Structure of Ferrioxamine B: A Comparative Analysis and Implications for Molecular Recognition. J. Biol. Inorg. Chem. 2001, 6, 810–818. [Google Scholar] [CrossRef]
- Siebner-Freibach, H.; Hadar, Y.; Yariv, S.; Lapides, I.; Chen, Y. Thermospectroscopic Study of the Adsorption Mechanism of the Hydroxamic Siderophore Ferrioxamine B by Calcium Montmorillonite. J. Agric. Food Chem. 2006, 54, 1399–1408. [Google Scholar] [CrossRef]
- Tebo, B.M.; Bargar, J.R.; Clement, B.G.; Dick, G.J.; Murray, K.J.; Parker, D.; Verity, R.; Webb, S.M. BIOGENIC MANGANESE OXIDES: Properties and Mechanisms of Formation. Annu. Rev. Earth Planet. Sci. 2004, 32, 287–328. [Google Scholar] [CrossRef]
- Post, J.E.; McKeown, D.A.; Heaney, P.J. Raman Spectroscopy Study of Manganese Oxides: Tunnel Structures. Am. Mineral. 2020, 105, 1175–1190. [Google Scholar] [CrossRef]
- Birkner, N.; Navrotsky, A. Thermodynamics of Manganese Oxides: Sodium, Potassium, and Calcium Birnessite and Cryptomelane. Proc. Natl. Acad. Sci. USA 2017, 114, E1046–E1053. [Google Scholar] [CrossRef]
- Ma, W.; Kwan, K.W.; Wu, R.; Ngan, A.H.W. High-Performing, Linearly Controllable Electrochemical Actuation of c-Disordered δ-MnO2/Ni Actuators. J. Mater. Chem. A 2021, 9, 6261–6273. [Google Scholar] [CrossRef]
- Bi, X.; Tang, T.; Meng, X.; Gou, M.; Liu, X.; Zhao, P. Aerobic Oxidative Dehydrogenation of N-Heterocycles over OMS-2-Based Nanocomposite Catalysts: Preparation, Characterization and Kinetic Study. Catal. Sci. Technol. 2020, 10, 360–371. [Google Scholar] [CrossRef]
- Yao, N.; Zhao, H.; Liu, X.; Serol Ertürk, A.; Elmaci, G.; Zhao, P.; Meng, X. Synergistic Adsorption and Oxidative Degradation of Polyvinyl Alcohol by Acidified OMS-2: Catalytic Mechanism, Degradation Pathway and Toxicity Evaluation. Sep. Purif. Technol. 2022, 302, 122047. [Google Scholar] [CrossRef]
- Wang, X.; Bi, X.; Yao, N.; Elmaci, G.; Serol Ertürk, A.; Lv, Q.; Zhao, P.; Meng, X. Doping Strategy-Tuned Non-Radical Pathway on Manganese Oxide for Catalytic Degradation of Parabens. Chem. Eng. J. 2022, 442, 136180. [Google Scholar] [CrossRef]
- Minakshi, M.; Mitchell, D.; Prince, K. Incorporation of TiB2 Additive into MnO2 Cathode and Its Influence on Rechargeability in an Aqueous Battery System. Solid State Ion. 2008, 179, 355–361. [Google Scholar] [CrossRef]
- Jenne, E.A. Trace Inorganics in Water; Advances in Chemistry; Baker, R.A., Ed.; American Chemical Society: Washington, DC, USA, 1968; Volume 73, ISBN 978-0-8412-0074-6. [Google Scholar]
- Post, J.E. Manganese Oxide Minerals: Crystal Structures and Economic and Environmental Significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, O.W.; Sposito, G. Siderophore−Manganese(III) Interactions II. Manganite Dissolution Promoted by Desferrioxamine B. Environ. Sci. Technol. 2005, 39, 6045–6051. [Google Scholar] [CrossRef]
- Mullen, L.; Gong, C.; Czerwinski, K. Complexation of Uranium (VI) with the Siderophore Desferrioxamine B. J. Radioanal. Nucl. Chem. 2007, 273, 683–688. [Google Scholar] [CrossRef]
- Kraemer, D.; Kopf, S.; Bau, M. Oxidative Mobilization of Cerium and Uranium and Enhanced Release of “Immobile” High Field Strength Elements from Igneous Rocks in the Presence of the Biogenic Siderophore Desferrioxamine B. Geochim. Cosmochim. Acta 2015, 165, 263–279. [Google Scholar] [CrossRef]
- Takahashi, A.; Igarashi, S. Solvent Extraction of Uranium (IV) with Desferrioxamine B and Its Application to the Homogenous Liquid-Liquid Extraction Method Using a Fluorosurfactant. Solvent Extr. Res. Dev. 1999, 6, 61–71. [Google Scholar]
- Hafez, M.B.; Hafez, N. Spectrophotometric Determination of Ce(IV), Fe(III) and U(VI) Using Desferrioxamine B (Desferal). J. Chem. Technol. Biotechnol. 2007, 54, 267–269. [Google Scholar] [CrossRef]
- Neubauer, U.; Nowack, B.; Furrer, G.; Schulin, R. Heavy Metal Sorption on Clay Minerals Affected by the Siderophore Desferrioxamine B. Environ. Sci. Technol. 2000, 34, 2749–2755. [Google Scholar] [CrossRef]
- Kirby, M.E.; Watson, J.S.; Najorka, J.; Kenney, J.P.L.; Krevor, S.; Weiss, D.J. Experimental Study of PH Effect on Uranium (UVI) Particle Formation and Transport through Quartz Sand in Alkaline 0.1 M Sodium Chloride Solutions. Colloids Surf. A Physicochem. Eng. Asp. 2020, 592, 124375. [Google Scholar] [CrossRef]
- Kirby, M.; Weiss, D.J. A Pilot Study on the Effect of Desferrioxamine B on Uranium VI Precipitation and Dissolution in pH 11.5, 0.1 M NaCl Solutions. J. Radioanal. Nucl. Chem. 2022, 331, 1779–1784. [Google Scholar] [CrossRef]
- Kraemer, S.M.; Cheah, S.-F.; Zapf, R.; Xu, J.; Raymond, K.N.; Sposito, G. Effect of Hydroxamate Siderophores on Fe Release and Pb(II) Adsorption by Goethite. Geochim. Cosmochim. Acta 1999, 63, 3003–3008. [Google Scholar] [CrossRef]
- Simanova, A.A.; Persson, P.; Loring, J.S. Evidence for Ligand Hydrolysis and Fe(III) Reduction in the Dissolution of Goethite by Desferrioxamine-B. Geochim. Cosmochim. Acta 2010, 74, 6706–6720. [Google Scholar] [CrossRef]
- Frazier, S.W.; Kretzschmar, R.; Kraemer, S.M. Bacterial Siderophores Promote Dissolution of UO2 under Reducing Conditions. Environ. Sci. Technol. 2005, 39, 5709–5715. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Rao, R.A.K.; Siddiqui, B.A. Adsorption Studies and the Removal of Dissolved Metals Using Pyrolusite as Adsorbent. Environ. Monit. Assess. 1995, 38, 25–35. [Google Scholar] [CrossRef]
- Liu, J.; Lippold, H.; Wang, J.; Lippmann-Pipke, J.; Chen, Y. Sorption of Thallium(I) onto Geological Materials: Influence of PH and Humic Matter. Chemosphere 2011, 82, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.R.; Huang, C.P. Adsorption Characteristics of Metal-EDTA Complexes onto Hydrous Oxides. J. Colloid Interface Sci. 1986, 110, 575–590. [Google Scholar] [CrossRef]
- Nowack, B.; Sigg, L. Adsorption of EDTA and Metal–EDTA Complexes onto Goethite. J. Colloid Interface Sci. 1996, 177, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.A.; Huang, C.P. The Adsorption Characteristics of Cu(II) in the Presence of Chelating Agents. J. Colloid Interface Sci. 1979, 70, 29–45. [Google Scholar] [CrossRef]
- Benjamin, M.M.; Leckie, J.O. Conceptual Model for Metal-Ligand-Surface Interactions during Adsorption. Environ. Sci. Technol. 1981, 15, 1050–1057. [Google Scholar] [CrossRef]
- Davis, J.A.; Leckie, J.O. Effect of Adsorbed Complexing Ligands on Trace Metal Uptake by Hydrous Oxides. Environ. Sci. Technol. 1978, 12, 1309–1315. [Google Scholar] [CrossRef]
- Haack, E.A.; Johnston, C.T.; Maurice, P.A. Mechanisms of Siderophore Sorption to Smectite and Siderophore-Enhanced Release of Structural Fe3+. Geochim. Cosmochim. Acta 2008, 72, 3381–3397. [Google Scholar] [CrossRef]
- Rosenberg, D.R.; Maurice, P.A. Siderophore Adsorption to and Dissolution of Kaolinite at PH 3 to 7 and 22 °C. Geochim. Cosmochim. Acta 2003, 67, 223–229. [Google Scholar] [CrossRef]
- Groenewold, G.S.; Van Stipdonk, M.J.; Gresham, G.L.; Chien, W.; Bulleigh, K.; Howard, A. Collision-Induced Dissociation Tandem Mass Spectrometry of Desferrioxamine Siderophore Complexes from Electrospray Ionization of UO22+, Fe3+ and Ca2+ Solutions. J. Mass Spectrom. 2004, 39, 752–761. [Google Scholar] [CrossRef]
- Winkelmann, G.; Busch, B.; Hartmann, A.; Kirchhof, G.; Süßmuth, R.; Jung, G. Degradation of Desferrioxamines by Azospirillum Irakense: Assignment of Metabolites by HPLC/Electrospray Mass Spectrometry. BioMetals 1999, 12, 255–264. [Google Scholar] [CrossRef]
- Norén, K.; Persson, P. Adsorption of Monocarboxylates at the Water/Goethite Interface: The Importance of Hydrogen Bonding. Geochim. Cosmochim. Acta 2007, 71, 5717–5730. [Google Scholar] [CrossRef]
- Duckworth, O.W.; Sposito, G. Siderophore-Promoted Dissolution of Synthetic and Biogenic Layer-Type Mn Oxides. Chem. Geol. 2007, 242, 497–508. [Google Scholar] [CrossRef]
- Peña, J.; Duckworth, O.W.; Bargar, J.R.; Sposito, G. Dissolution of Hausmannite (Mn3O4) in the Presence of the Trihydroxamate Siderophore Desferrioxamine B. Geochim. Cosmochim. Acta 2007, 71, 5661–5671. [Google Scholar] [CrossRef]
- Duckworth, O.W.; Sposito, G. Siderophore−Manganese(III) Interactions. I. Air-Oxidation of Manganese(II) Promoted by Desferrioxamine B. Environ. Sci. Technol. 2005, 39, 6037–6044. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [DFOB]supernatant (µM) | [Acetate] (µM) | [U(VI)]sorbed (µM) | [DFOB]supernatant + [Acetate] + [U(VI)]sorbed (µM) | [DFOB]total (µM) |
|---|---|---|---|---|
| 37.44 | 14.35 | 24.4 | 76.19 (±4.00) | 78 |
| 26.206 | 12.15 | 21.95 | 60.31 (±4.45) | 60 |
| 14.01 | 10.55 | 15.0 | 39.56 (±0.75) | 37 |
| 2.02 | 8.40 | 8.59 | 19.01 (±0.36) | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snyder, M.; Hunley, L.; Stanberry, J.; Szlamkowicz, I.; Jones, B.; Anagnostopoulos, V. A Preliminary Sorption Study of Uranium on MnO2 (Pyrolusite) in the Presence of Siderophore Desferrioxamine B—The Mechanism of a Ternary System. Water 2023, 15, 3241. https://doi.org/10.3390/w15183241
Snyder M, Hunley L, Stanberry J, Szlamkowicz I, Jones B, Anagnostopoulos V. A Preliminary Sorption Study of Uranium on MnO2 (Pyrolusite) in the Presence of Siderophore Desferrioxamine B—The Mechanism of a Ternary System. Water. 2023; 15(18):3241. https://doi.org/10.3390/w15183241
Chicago/Turabian StyleSnyder, Morgan, Lucy Hunley, Jordan Stanberry, Ilana Szlamkowicz, Brandon Jones, and Vasileios Anagnostopoulos. 2023. "A Preliminary Sorption Study of Uranium on MnO2 (Pyrolusite) in the Presence of Siderophore Desferrioxamine B—The Mechanism of a Ternary System" Water 15, no. 18: 3241. https://doi.org/10.3390/w15183241
APA StyleSnyder, M., Hunley, L., Stanberry, J., Szlamkowicz, I., Jones, B., & Anagnostopoulos, V. (2023). A Preliminary Sorption Study of Uranium on MnO2 (Pyrolusite) in the Presence of Siderophore Desferrioxamine B—The Mechanism of a Ternary System. Water, 15(18), 3241. https://doi.org/10.3390/w15183241