Abstract
The Pan-Okhotsk region, which is part of the western North Pacific Ocean, is famous for its active volcanoes, which are part of the Pacific Ring of Fire and that enrich the surrounding waters with essential chemicals. Therefore, this region, including the Sea of Okhotsk and the Sea of Japan, is characterized by rich biota. Bacterioplankton plays a significant part in biological communities and is an indicator of ecosystem function. Analyzing the adaptability of three representatives of the microbiota of the Pan-Okhotsk region was the goal of our investigation. Marinomonas primoryensis KMM3633T (MP), Yersinia ruckeri KMM821 (YR), and Yersinia pseudotuberculosis 598 (YP) from the G.B. Elyakov Pacific Institute of Bioorganic Chemistry were studied by means of genomic and bioinformatic methods. The list of membrane translocator proteins, metabolism pathways, and cold shock and antifreeze proteins that were revealed in the genome of MP characterized this bacterium as being adaptable to free living in marine conditions, even at winter temperatures. The genomic potential of YR and YP makes not only survival in the environment of the Pan-Okhotsk region but also pathogenesis in eukaryotic organisms possible. The data obtained will serve as a basis for further ecosystem monitoring with the help of microbiota research.
1. Introduction
The western North Pacific Ocean is famous for the volcanoes of the Kuril–Kamchatka island arc, which include 126 known submarine volcanoes and 68 terrestrial active volcanoes, representing about 15% of the total number of active volcanoes in the Pacific Ring of Fire and producing about 20% of the world’s total volume of erupted material [1].
The water of the western subarctic Pacific Ocean is rich in nitrate, phosphate, and silicate, which are essential chemicals for producing phytoplankton, and therefore, despite the fact that the western subarctic Pacific Ocean only accounts for six percent of the world’s oceans, it produces an estimated 26 percent of the world’s marine resources [2]. As such, this area has the largest biological CO2 drawdown among the world’s oceans [3].
The Sea of Okhotsk and the Sea of Japan are marginal seas of the western Pacific Ocean and are actively used for fishing, shipments, and deep-sea mining [4]. These processes, along with the warming effects, produce marine biological resources [4]. Some academic institutions of Russia and Japan research this region. The Pan-Okhotsk Research Center was established in 2004 and explores not only the Sea of Okhotsk but also Siberia to the west, the North Pacific to the east, the Arctic region to the north, and the subtropic regions to the south [5]. One of the important research directions in this region is devoted to the study of the biogeochemistry and ecosystems of marginal seas [6]. Bacterioplankton is a significant part of biological communities and an indicator of ecosystem function. The study of Kraternaya Bight focused on bacteria that are extremely thermophilic [7,8] due to the hydrothermal activity of the Ushishir Volcano. On the other hand, a cold and dry northwesterly wind (The Siberian High) blows from Siberia to the ocean and freezes the Sea of Okhotsk and the Sea of Japan in the winter. As such, it is important to study psychrophilic and psychrotolerant bacteria too.
The Sea of Okhotsk covers an area of 1,583,000 km2 and has a mean depth of 859 m and a maximum depth of 3372 m. It is connected to the Sea of Japan on either side of Sakhalin. The Sea of Japan has a smaller surface area of about 1,050,000 km2, but a deeper mean depth of 1752 m and a deeper maximum depth of 4568 m. The average water salinity in the Sea of Japan is 34.1‰, and 33.8‰ in the Sea of Okhotsk. In the Sea of Japan, winter temperatures are 0 °C or below in the north at the latitude of the Peter the Great Gulf, so ice may start forming in the bays as early as in October, and its remains may even be seen in June in some instances [9]. Amursky Bay is a part of Peter the Great Gulf, the ice from which was researched by the G.B. Elyakov Pacific Institute of Bioorganic Chemistry (PIBOC FEB RAS). More than half of the surface of the Sea of Okhotsk is covered with ice for 6–7 months of the cold season. However, the southeastern part of the sea usually does not freeze. It was this part of the sea near Iturup Island that became the object of this research.
Marinomonas primoryemsis was isolated form the ice of Amursky Bay by Romanenko et al. [10] and became the type strain of this species according to the List of Prokaryotic names with Standing in Nomenclature (LPSN) [11]. The bacterium was postulated to be psychrophilic because it grew on Marine Agar 2216 at 4–30 °C but not at 35 °C [10]. A second strain of M. primoryensis was isolated from Ace Lake (East Antarctica, Princess Elizabeth Land) and showed better growth at 15 °C [12]. Moving from the Antarctic to the Arctic across the Pacific Ocean, there are several places with extreme conditions where Marinomonas bacteria have also been found. M. profundi was isolated from seawater collected at a depth of 1045 m in the Mariana Trench and grew at −4 to 37 °C, but the optimum temperature was determined to be 25 to 30 °C [13]. M. arctica was isolated in the most northern region: the Canadian Basin of the Arctic Ocean (78.39 N 149.12 W); it grew from 0 to 37 °C (optimum growth at 25–27 °C) [14]. Two definitions were considered for the mentioned bacteria: psychrophilic and psychrotolerant, and we took into account all of the accepted criteria: capability of growing at or close to zero [15,16], growth rate at 0 °C [15], and the optimum and upper temperature limits for growth [15,16]. The following ecological characteristics were taken into account as well: permanently cold habitats or environments that undergo thermal fluctuations [16]. Based on these criteria, we came to the conclusion that the term ‘psychrotolerant’ would be the most appropriate for these bacteria.
Another representative of the bioplankton found in the Pan-Okhotsk region, Yersinia ruckeri KMM821, was isolated from the waters of the Sea of Okhotsk by PIBOC FEB RAS staff. This is a rare example in which the source of this bacterium is not associated with sick animals belonging to freshwater and marine fish species (carp, catfish, sturgeon, salmon) or non-fish species—muskrat (Ondatra zibethica), turtles (Cheloniidae), sea gulls (Laridae), etc. [17]. The first isolation of Y. ruckeri (strain ATCC 29473™), which received its current name in 1978 [18], was associated with a study that first documented incidences of enteric redmouth disease (ERM) in rainbow trout in Idaho, Colorado, Nevada, California, and Arizona in the western United States in the 1960s [19]. Psychrotolerant properties of the seawater isolated Y. ruckeri were suggested not only by ecology, but also by its capability to grow on 2 × YT medium at 4 °C [20].
Volcanic activity not only influences the biological resources of the marginal seas in the Pan-Okhotsk region, but also the land relief resources in the Far East region (FER). The climatic and physical-geographical features of the region contribute to the formation of regional isolated animal genotypes and even the emergence of endemic animals (Panthera pardus orientalis) and plants (Microbióta and Larix olgensis). The specificity of the genotypes of the animals from the FER was demonstrated by Abramson et al. in a wide-range phylogeographic study of the gray red-backed vole (Craseomyc rufocanus) [21]. The presence of particular local bacteria genotypes in the FER was revealed in the investigation of the Burkholderia cepacia complex, Listeria monocytogenes, and Leptospira spp. [22]. Far East scarlet-like fever (FESLF), a rare and poorly studied disease caused by Yersinia pseudotuberculosis, was first described in 1959 in Vladivostok [23]. A total of 17 outbreaks of FESLF were registered in the FER between 1973 and 2014 [24]. An environmental study in the Primorsky Territory conducted by the Somov G.P. Institute of Epidemiology and Microbiology detected Y. pseudotuberculosis in various water sources [25] and modeled the marine ecosystem to study the survival of Y. pseudotuberculosis in seawater [26]. Later, Y. pseudotuberculosis was found in water sources in neighboring countries: in South Korea in mountain spring water [27], as well as in Japan in a river in the mountains of the eastern part of the Shimane Prefecture [28]. The psychrotolerance of the Y. pseudotuberculosis isolated in the FER was confirmed by Timchenko et al., who showed that low temperatures (8–10 °C) were necessary for the growth of the bacterium and that they facilitated the activation of invasive and toxic pathogenicity factors [29]. In the environment, as shown by Inoue et al., the Y. pseudotuberculosis contamination rate in water from the northern areas of the Okayama Prefecture was higher than that in southern urban regions [30].
As such, bacteria that were isolated from the marginal seas and from a patient in Primorye that were stored in the Collection of Marine Microorganisms at PIBOC FEB RAS were the objects of our research, and we aimed to identify the potential for these bacteria to adapt to different ecological niches in the Pan-Okhotsk region.
2. Materials and Methods
Strains: M. primoryensis KMM3633T was isolated by Romanenko L.A. in March 2001 from a coastal sea ice sample that was taken at a depth of 0.8 m in Amursky Bay in the Sea of Japan. Y. ruckeri KMM821 was isolated in 2000 from seawater from the Sea of Okhotsk near Iturup Island. Y. pseudotuberculosis 598 was isolated by Timchenko N.F. in the 1980s from a person with FESLF in Primorye. The cultures of the bacterial strains were prepared and provided by the staff of the Collection of Marine Microorganisms at PIBOC FEB RAS.
DNA isolation: Preparation of the genomic DNA for whole genome sequencing (WGS) was performed according to previously published protocols [31].
Genome sequencing and assembly: Two kits were used for library preparation: the KAPA HyperPlus (Roche, Basel, Switzerland) and Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA, USA). Libraries were verified on a bioanalyzer (Agilent, Santa Clara, CA, USA) and sequenced on an Illumina MiSeq instrument using a paired-end protocol. Long reads obtained by MinION (Oxford Nanopore Technologies, Oxford, UK) were used in combination with short reads to complete the genome sequences.
Genome assembly was performed with the CLC Genomic Workbench v.20.0.4 (QIAGEN, Germantown, MD, USA) and SPAdes v.3.13.0 (St. Petersburg genome assembler, Russia, URL: http://cab.spbu.ru/software/spades/ (accessed on 10 January 2022)).
The circularized chromosomes were finally rearranged to start at the dnaA/repA start position. Replication origin of the chromosomes and plasmids was verified based on the GC skew (http://stothard.afns.ualberta.ca/cgview_server/ (accessed on 10 January 2022)).
Genome annotation: Genomes were annotated by means of the Rapid Annotations using Subsystems Technology (RAST) server [32,33].
GenBank NCBI submission: The obtained genome sequences were submitted to the NCBI SRA database under BioProject number PRJNA622482. Complete genomes were submitted to GenBank under the accession numbers presented in Table 1.

Table 1.
Genome data of studied bacteria.
The DNA sequence of Porin_4 was translated, complementarily annotated in NCBI by means of conserved domains detection, and submitted to GenBank with the accession number MK820371 (Protein Accession—QES04118).
Bioinformatic analysis: Prophage regions were identified using PHASTER (an extended version of the PHAge search tool, https://phaster.ca/ (accessed on 10 January 2022)) [34]. IS finder (https://isfinder.biotoul.fr/ (accessed on 10 January 2022)) [35] was used to search for insertion sequence (IS) elements.
The presence of transmembrane domains in the polypeptides was checked using the TMHMM-2.0 server (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0 (accessed on 10 January 2022)). Secreted proteins were predicted by determining the presence of a signal peptide using SignalP 5.0 (https://services.healthtech.dtu.dk/service.php?SignalP-5.0 (accessed on 10 January 2022)) [36] as well as the Phobius server (https://phobius.sbc.su.se/ (accessed on 10 January 2022)) [37]. The PSORTb program was used to clarify the subcellular localization of the polypeptides (https://www.psort.org/psortb/ (accessed on 10 January 2022)) [38]. The physical and chemical capabilities were calculated in the ProtParam program (https://web.expasy.org/protparam/ (accessed on 10 January 2022)) [39].
The TBCD (Transporter Classification Database, URL: http://www.tcdb.org/ (accessed on 10 January 2022)) [40] was used for transporter classification. The search for the promoter regions of genes of interest, adjacent reading frames, and the sigma factor binding sites was performed with the iPromoter-2L program (URL: http://bliulab.net/iPromoter-2L (accessed on 10 January 2022)). The iPro70-FMWin program (URL: http://ipro70.pythonanywhere.com/ (accessed on 10 January 2022)) [41] was used to sort the selected regions according to their likelihood of belonging to a promoter region.
The virulence factors were analyzed using the VFDB (Virulence Factors Database, http://www.mgc.ac.cn/VFs/ (accessed on 10 January 2022)) and the VF analyzer (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi?func=VFanalyzer (accessed on 10 January 2022)) [42]. Functional gene annotation was performed using KAAS (KEGG Automatic Annotation Server, https://www.genome.jp/tools/kaas/ (accessed on 10 January 2022)) with GHOSTX (a homology search tool that can detect remote homologs) and the BBH (bi-directional best hit) assignment method [43].Determining whether the genes belonged to a COG category was carried out with NCBI Blastp and COG database/Conserved Protein Domain Family database [44].
The Yersinia ruckeri typing database (https://pubmlst.org/ (accessed on 10 January 2022)) was used to determine the ST (sequnce type) on the basis of the genome data of Y. ruckeri KMM821. The Genome data of Y. pseudotuberculosis 598 were analyzed with help of the Yersinia locus/sequence definitions database from the Institut Pasteur (https://bigsdb.pasteur.fr/ (accessed on 10 January 2022)).
3. Results
3.1. Genomic Data of the Studied Bacteria
The genomes of the three microorganisms: M. primoryensis KMM3633T, Y. ruckeri KMM821, and Y. pseudotuberculosis 598 were deposited under bioproject PRJNA622482 “Psychrophilic and psychrotolerant gammaproteobacteria of the Russian Far East” with the accession numbers shown in Table 1. The genome of M. primoryensis (MP) is represented by one chromosome. The genomes of Y. ruckeri (YR) and Y. pseudotuberculosis (YP) were represented by two plasmids in addition to the chromosome. All three bacterial genomes were comparable in size. The number of CDSs (coding sequences) ranged from 3413 (YR) to 4316 (YP) and was proportional to the genome size. A relatively smaller number of pseudogenes (40), a lower GC composition (43.4%), and a larger number of rRNAs (9-5S, 8-16S, 8-23S) distinguished the MP genome. The analysis of the mobile genetic elements (prophages and IS) in the genomes of the studied bacteria also showed a significantly lower number of them in the MR: there were 3.0- and 2.7-times fewer prophages and 2.2- and 3.5-times fewer IS compared to YR and YP (Table 1).
3.2. Bacterial Transporter Systems
All bacteria must scavenge and take up nutrients from their environment to survive. The cellular repertoire of transporter proteins is responsible for both the uptake of essential nutrients such as carbohydrates, amino acids, and metals into the cell as well as for the efflux of toxins and antimicrobial agents out of the cell [45]. Transporter proteins therefore play key roles in bacterial colonization, pathogenesis, and antimicrobial resistance [46]. In contrast to channel proteins, which catalyze the high flux of molecules down a concentration gradient, transporters can couple uphill substrate translocation with the movement of ions down their electrochemical gradient (secondary active transporters) or by using processes such as ATP hydrolysis (primary active transporters). These processes enable bacteria to scavenge nutrients that may be scarce [47].
Three microorganisms, the genomes of which were investigated within the framework of the project, are Gram-negative Gammaproteobacteria that belong to the Proteobacteria phylum (in October 2021, the valid name became Pseudomonadota [48]). MP belongs to the Oceanospirillales order, and YP and YR belong to the Enterobacterales order. The two membranes of the Gram-negative bacterial cells are permeated by the various transport systems (Figure 1). Through the outer membrane, nutrient transport is mainly carried out in a passive way by means of porins and substrate-specific channels. The outer membrane also contains the TonB-dependent receptors of certain inner membrane transporters [49]. The active transport of substances is carried out through the inner membrane of the cell by the ABC (ATP-binding cassette), MFS (major facilitator superfamily), TRAP (tripartite ATP-independent periplasmic system), TTT (tripartite tricarboxylate transporters), and IT (ion transporter) transporter systems [50].
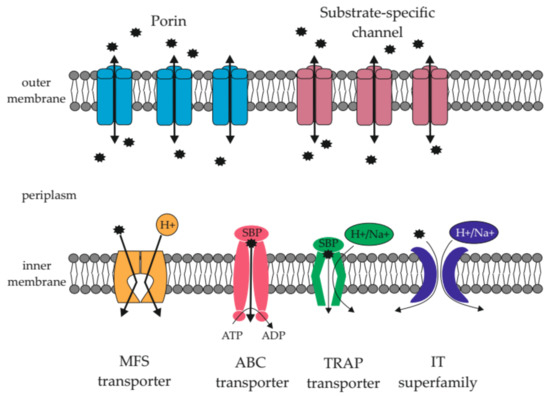
Figure 1.
Some transporter systems of Gram-negative bacteria. MFS—major facilitator superfamily; ABC—ATP-binding cassette; TRAP—tripartite; ATP—independent periplasmic (TRAP) transporters; IT—ion transporter; SBP—solute binding protein.
3.2.1. Passive Diffusion Transporters: Porins
The porins, according to the TCBD [40], belong to the class 1 Channels, subclass 1.B: β-barrel porins. The porins that were revealed in the analyzed bacteria are presented in Table 2. The number of porins was strikingly different among the studied bacteria. As such, the Yersinia genomes encoded from 10 to 13 porins belonging to different families, whereas the Marinomonas genome contained only one porin that belonged to the Porin_4 superfamily (MP3633_2986) as well as maltoporin from the sugar porin (SP) family. The first four families are represented in Yersinia in equal amounts, but YP has more maltoporins. Moreover, YP has additional porins for the transport of oligogalacturonate and N-acetyl-glucosamine. YR, in turn, is characterized by the presence of a porin for DNA transport.
Table 2.
β-barrel porins of Y. ruckeri KMM821, Y. pseudotuberculosis 598, and M. primoryensis KMM3633T.
A significant difference in the number of porins—i.e., outer membrane channels—which allow molecules to move simultaneously in both directions—both into the periplasm of the cell, and out of it—emphasizes the need to limit the membrane permeability of free living inhabitants in saltwater, which is MP in this study.
3.2.2. Primary Active Transporters: ABC Transporters and Transporters of Carboxylates (TRAP, TTT)
These groups of transporters are united by the presence of a protein that binds the dissolved substrate in the periplasmic space and are characterized by a high affinity for the ligand. ABC transporters, the primary active transporters, create an electrochemical potential for substrate translocation across the membrane, hydrolyzing ATP. In the case of TRAP, the secondary active transporters, a preexisting electrochemical cation gradient works via a symport mechanism [51].
ABC Transporters
The classical structure of the ABC transporter is represented by three subunit types: periplasmic solute binding protein (SBP); two transmembrane pore-forming permeases; and two ATPases localized in the cytoplasm of the cell. In the bacterial genome, the genes encoding the ABC transporter proteins are usually combined into operons [52]. However, in the genomes considered here, the classic operon consisting of five genes was rare. An example is the ABC operon of the oligopeptide transporter MP: MP3633_3749-MP3633_3753. More often, operons consist of three genes: ATPase, permease, and SBP. In the operons encoding eukaryotic-type ABC transporters, one gene can encode a fusion protein of permease and ATPase [53].
The classification of the identified ABC transporters is presented in Table 3. All of the identified transporters belong to the 3.A.1 ATP-binding cassette (ABC) superfamily of the subclass of P-P-bond-hydrolysis-driven transporters of the Class 3 primary active transporters [40]. To classify the ABC transporters based on their predicted function, we used the GenomeNet server (Kyoto University Bioinformatics Center, https://www.genome.jp/ (accessed on 10 January 2022)) on the KEGG pathway section (Kyoto Encyclopedia of Genes and Genomes) [54].
Table 3.
ABC transporters in the studied genomes.
As can be seen from Table 3, the total number of both prokaryotic- and eukaryotic-type ABC transporters is higher in YP and significantly lower in YR; MP occupies an intermediate position. In part, these differences can be explained by slightly different genome sizes. The YP genome is 20% larger than the MP genome, and 27% larger than the YR genome. However, the differences can be explained by adaptation to different living conditions. YR was isolated from the coastal waters of the warmest southeastern part of the Sea of Okhotsk, near Iturup Island, whereas MP was isolated from the ice in the Amur Bay, located in the coldest northwestern part of the Sea of Japan. YP is a clinical isolate from a patient with FESF (pseudotuberculosis) in Primorye, where vegetables were a confirmed source of infection [24]. Thus, YP’s habitat is in soil and groundwater [25,28].
Transporters of Carboxylates (TRAP, TTT)
Low-molecular-weight carbon sources such as carboxylates, amino acids, and carbohydrates (mono-, di-, and oligosaccharides) are important for the efficient growth of aquatic microorganisms, which include the bacteria analyzed here. Moreover, carboxylates are preferred for bacteria that inhabit the aquatic environment [55].
TRAP, TAXI-TRAP, and (Dcu)AB transport dicarboxylic acids [51,56]. TTT transports tricarboxylic acids [51]. The difference between TRAP and TAXI-TRAP is in the periplasmic protein that binds the dissolved substrate [51]. In the TAXI-TRAP transporters, SBP was named after the immunogenic protein of Brucella, which determined the name of the subclass (TRAP associated extracytoplasmic immunogenic (TAXI)) [51].
TRAP, TAXI-TRAP, and TTT are used to transport carboxylates by means of the symport of hydrogen and sodium cations [56]. The (Dcu)AB family of transporters carries out an electrically neutral antiport of fumarate and succinate [56], which is important for anaerobic respiration, which is mainly facultative, where fumarate serves as an electron acceptor with high redox potential.
As seen in Table 4, the number of carboxylate transporters in MP is 10-times greater than that in Yersinia. This result was not unexpected for the bacteria that were isolated from the sea since the prevalence of TRAP transporters is known for marine bacteria [51]. The TRAP transporters are found in abundance among aquatic bacteria such as Chromohalobacter salexigens (Gammaproteobacteria, Oceanospirillales), Ruegeria pomeroyi and Jannaschia spp. (Alphaproteobacteria, Rhodobacterales), and Aurantimonas coralicida (Alphaproteobacteria, Hyphomicrobiales) [57].
Table 4.
Carboxylate transporters in the studied genomes.
Apparently, on the one hand, in waters that are rich in phytoplankton, low molecular carbon sources are easily accessible, and on the other hand, the inhabitants of saltwater do not have a shortage of the sodium ions that are necessary for creating electrochemical potential. The presence of a (Dcu)AB transporter in anaerobic and facultatively anaerobic bacteria capable of fumarate respiration is noteworthy [56,58,59]. So far, (Dcu)AB transporters are well-studied in three bacteria that can grow via anaerobic respiration with fumarate as the terminal electron acceptor: a rumen bacterium Wolinella succinogenes (Epsilonproteobacteria, Campylobacterales, Helicobacteraceae) [58] and two bacteria that can cause gastroenteritis: Campylobacter jejuni (Epsilonproteobacteria, Campylobacterales, Campylobacteraceae) [60] and Escherichia coli (Gammaproteobacteria, Enterobacterales, Enterobacteriaceae) [59]. However, (Dcu)AB transporters are absent in Rhodobacter capsulatus (Alphaproteobacteria, Rhodobacterales, Rhodobacteraceae), which is a non-pathogenic, free-living inhabitant of the aquatic environment [58].
3.2.3. Secondary Transporters with a Substrate-Binding Site in the Channel-Forming Protein
In the analyzed genomes, the following transporters belonged to two classes according to TBCD [40]:
- 1.
- Channels/pores;
- 2.
- Electrochemical potential-driven transporters.
All of the class 1 transporters that were found belonged to subclass 1.A: Alpha-Type Channels (Table 5).
Table 5.
Alpha-type channels in the studied genomes.
The number of transporters of subclass 1.A is greater in MP, and they represent five families/superfamilies that are responsible for cation and anion transport. MP contains more ion channels that are sensitive to voltage (VIC) and mechanical stress on the cell (MscS) than Yersinia. YP, in turn, has more Fluc channels that are responsible for the efflux of fluoride ions, which inhibit the enzymes that are necessary for glycolysis and nucleic acid synthesis [63].
Porters: uniporters, symporters, and antiporters represent subclass 2.A of the Class 2 transporters (Table 6). MFS transporters are predominant among them in all of the bacteria analyzed here. YP shows the maximum number of MFS transporters. MP is distinguished by the presence of two transporters from the SulP family that are responsible for the transport of anions—such as bicarbonate, fumarate, sulfate, chloride, oxalate, and formate—and even the anions of dicarboxylic acids (at acidic pH values) [64].
Table 6.
Porters (uniporters, symporters, antiporters).
3.2.4. Group Translocation Transporters Coupling Solute Transport with the Chemical Modification of the Solute
PTS named 4.A: phosphotransfer-driven group translocators according to TCDB [40], were found in YR and YP. Proteins from five families (L-Ascorbate; Lactose-N,N’-Diacetylchitobiose-β-glucoside; Fructose–Mannitol; Glucose–Glucoside; and Mannose–Fructose–Sorbose) were annotated by RAST and confirmed via the KEGG Automatic Annotation Server [54] (Table S1a).
PTS consist of two sugar non-specific phosphocarrier proteins (EI and HPr) and a specific enzyme II (EII) complex [65]. EII comprises three proteins or domains: IIA, IIB (cytoplasmic membrane, transfers phosphate from phosphocarrier protein to the incoming sugar), and IIC (integral membrane, permease that binds and translocates the sugar) [65,66]. EI and HPr transfer phosphoryl groups from phosphoenolpyruvate to the EIIA.
All of the PTS of YR and YP were complete and consisted of the phosphocarrier proteins and all three components of the EII complex. In total, 26 genes representing eight complete PTS (ascorbate, cellobiose, fructose, glucose, mannose, trehalose, and 2 N-acetylgalactosamine PTS) and belonging to the five families were identified. N-acetylgalactosamine PTS were characterized by two EIIC proteins, and one of them was fused with an EIIB domain. Fused EIIC proteins were also observed in the fructose, glucose, and trehalose PTS.
Unlike Yersinia, complete PTS were not revealed in MP. All of the EII complex membrane permeases were absent. Only three PTS proteins were found for MP (Table S1b). These were sugar non-specific phosphocarrier proteins (PtsP and PtsH) and the EIIA protein PtsN. According to KEGG classification, two proteins (PtsP, PtsN) were involved in nitrogen regulation.
As such, YR and YP are characterized by active sugar transport, which may indirectly indicate the possibility of these bacteria existing in different ecological niches, including in eukaryotic organisms. Additionally, active sugar intake into YR and YP cells is predicted to cause changes in metabolic activity compared to MP.
3.3. Comparison of Metabolic Pathways of Three Bacteria
The numbers of open reading frames encoding the known metabolic pathways of YR, YP, and MP were compared by use of the KEGG pathway server [54]. More active sugar intake is associated with a more active glycolysis/gluconeogenesis pathway in YR and YP compared to in MP.
Additionally, for YR and YP, the metabolic pathways that are responsible for cationic antimicrobial peptide (CAMP) resistance are supplemented by two operons. The first one, sapABCDF (YR821_1805–YR821_1809 and YP598_1824–YP598_1828), encodes an inner membrane ABC transporter. Shelton et al. proposed that there is influx of antimicrobial peptide into bacterial cells, followed by degradation by the cytoplasmic proteases [67].
A second operon called arnBCADTEF (YR821_1873–YR821_1880 and YP598_1763–YP598_1769) conditions the formation of resistance to polymyxins via covalent modifications in lipid A [68]. Two systems, PhoP/PhoQ and PmrA/PmrB, that regulate polymyxin-resistant operon activation were also annotated in YR and YP (YR821_0393–YR821_0394; YR821_1424–YR821_1425 and YP598_3691–YP598_3692; YP598_1645–YP598_1646).
MP is predicted to use carbon sources that are different from those used by YR and YP, including di- and tricarboxylates, and had a more active metabolism in 27 annotated pathways (Table 7).
Table 7.
Metabolic pathways represented by more CDS in M. primoryensis KMM3633T compared to the studied Yersinia.
MP was more able to actively metabolize a number of amino acids, porphyrin, and propionic acid derivatives; and it showed the active degradation of a large list of organic compounds compared to YR and YP. MP is distinguished by the presence of pathways for the biosynthesis of flavonoids and phenylflavonoids, something that requires additional detailed study since stilbenoids have demonstrated antimicrobial action [70]. Currently, Gram-negative microorganisms of the genus Photorhabdus are known to synthesize stilbenoids (3,5-dihydroxy-4-isopropyl-trans-stilbene) [71]. These bacteria are symbionts of entomopathogenic nematodes, and the metabolite that is synthesized by them has a pathogenic effect on insects [71]. An analysis of biosynthetic stilbenoid pathways in Photorhabdus demonstrated that phenylalanine metabolism, the branched-chain amino acid metabolism and fatty acid metabolism can contribute to stilbenoid production [71]. One CDS found by the KEGG Pathway in the MP genome encodes the KatG that is involved in phenylalanine and tryptophan metabolism and phenylpropanoid biosynthesis (https://www.kegg.jp/entry/K03782 (accessed on 10 January 2022)). Bioinformatics analyses have suggested that MP has the metabolic pathways found in Photorhabdus: phenylalanine metabolism, branched-chain amino acid metabolism, and fatty acid metabolism. However, instead of being the key gene in the synthesis of stilbenoid-encoding phenylalanine ammonium lyase [72], the MP genome contains the gene for histidine ammonium lyase, which belongs to the same aromatic amino acid lyase family, EC 4.3.1., and has the same cofactor 3,5-dihydro-5-methylidene-4H-imidazol-4-one (BRENDA Enzyme Database: https://www.brenda-enzymes.org (accessed on 10 January 2022)). To test the possibility of stilbenoid synthesis by MP, it is necessary to perform metabolite and the protein identification during the MP growth caused by liquid chromatography with tandem mass spectrometry (LC-MS-MS).
3.4. Important Pathways for Iron Acquisition
The YR and MP isolated from the marginal seas have similar iron acquisition mechanisms. A ruckerbactin gene cluster (catechol siderophore) was found in both of these bacteria. For the clinical isolate of YP, the genes that were similar to the YR ruckerbactin gene cluster were not revealed. It should be noted that IP31758 epidemic FESLF Y. pseudotuberculosis O1 strain and Y. pseudotuberculosis YPIII O3 type strain lacked the gene cluster for ruckerbactin (enterobactin), but that the sporadic Y. pseudotuberculosis IP32953 O1 strain contained this gene cluster [73].
The ruckerbactin gene cluster of YR comprises 13 genes that are responsible for ruckerbactin biosynthesis (entC), secretion by the MFS enterobactin exporter (entS), ferrichrome–iron reception (fepA), periplasmic binding (fepB), transport to the cytoplasm (by ABC transporter fepDGC), and other functions (Figure 2).
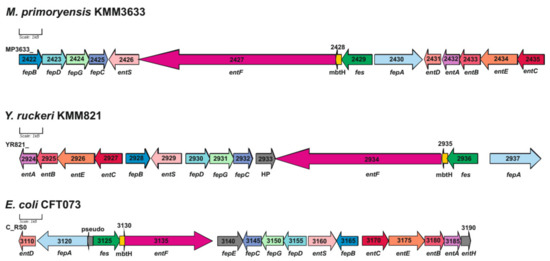
Figure 2.
Genetic organization of the Y. ruckeri KMM821 ruckerbactin biosynthetic uptake cluster compared to the M. primoryensis KMM3633T and E. coli CFT073 enterobactin gene clusters. entA—2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase; entB—isochorismatase; entC—isochorismate synthase; entD—4’-phosphopantetheinyl transferase EntD; entE—2,3-dihydroxybenzoate-AMP ligase; entF—enterobactin synthetase component F; entH—proofreading thioesterase EntH; entS—major facilitator superfamily (MFS) enterobactin exporter EntS; fepA—ferrichrome–iron receptor; fepB—iron–enterobactin ABC transporter substrate-binding protein; fepC—iron–enterobactin ABC transporter ATP-binding protein; fepD—Fe(3+)-siderophore ABC transporter permease; fepE—LPS O-antigen length regulator; fepG—iron–enterobactin ABC transporter permease; fes—enterobactin esterase; mbtH—MbtH family protein; HP—uncharacterized protein.
The YR ruckerbactin gene cluster is similar to that of enterobactin in E. coli. However, the E. coli cluster includes more genes and is complemented by fepE (LPS O-antigen length regulator), entH (proofreading thioesterase EntH), and entD (4’-phosphopantetheinyl transferase). The order of the genes in the ruckerbactin cluster in YR and of the enterobactin in E. coli was the same.
A similar gene cluster was found in the free-living MP strain examined herein. The MP cluster contained one additional entD gene and was characterized by a different gene order. At the same time, the location of genes playing one functional role remains unchanged, for example, in the fepDGC encoding an ABC transporter.
Fernandez et al., analyzing Y. ruckeri strain 150 (Y. ruckeri 150), which was isolated during naturally occurring outbreaks of ERM disease at a Danish fish farm [74], showed that the expression of the Y. ruckeri 150 ruckerbactin gene cluster was higher at 18 °C than at the optimal bacterial growth temperature of 28 °C [75]. In addition, the expression of other Y. ruckeri 150 genes also increased at lower temperatures. These genes encode pathogenic factors of Y. ruckeri 150, including type IV secretion system pili, metalloprotease, and hemolysin [76].
3.5. Major Virulence Factors in the Analyzed Yersinia
This study has shown that the isolate Y. pseudotuberculosis 598 is characterized by serotype O1b (Figure S1), genotype ST2 and VST1 (Virulence Sequence Type: inv, yadA; yopE, cnf [24]). In turn, Y. ruckeri KMM821 belongs to serotype O1 and is a DLV (Double Locus Variant: thrA, recA) of ST1.
It is well known that the vast majority of YR epizootics in salmonid fish farms are caused by motile serotype O1 [77]. As previously shown, there are 98 genes with different functions that are exclusively shared by serotype O1 strains. Thus, some of them are associated with restriction–modification and toxin–antitoxin systems, and some also affect global gene expression, resulting in altered adaptive phenotypes. Some of the genes of serotype O1 strains are involved in the biosynthesis of legionaminic acid, a nine-carbon diamino monosaccharide that is found coating the surface of various bacterial human pathogens and is a component of the O antigen of Y. ruckeri O1 [78]. Other genes that are exclusive to O1 serotype strains code for a bacteriocin that is similar to colicin Ib in E. coli (WP_062877260) and virulence factors such as a type IV secretion system, an invasin that is present in other Enterobacteriaceae such as Y. pestis and Y. pseudotuberculosis [77]. From 98 genes, which were revealed by Cascales et al. as being exclusively shared by serotype O1 strains, in the YR genome, all but one encoded protein had 100% similarity with the reference Y. ruckeri 150. One gene encoded a chromosome segregation ATPase, which showed 61% similarity to the reference (Y. ruckeri 150) protein. Most of these genes were located on the chromosome, and 33 genes were encoded on one of the two plasmids.
The genome analysis made it possible to identify the entire spectrum of known virulence factors in the studied microorganisms. A total of 178 virulence factors were identified in the YP genome, and 73 virulence factors were revealed in the YR genome (Tables S2 and S3). A total of 67 virulence factors coexisted between the two bacteria (Tables S2 and S3, blue highlight), for example, the genes involved in adherence psaC, type IV pili, exoenzyme-yplA, invasion factor-invC, and flagella genes. The rest of the factors were specific to each of the Yersinia species (82 for YP and 6 for YR).
The YP genome contains the specific adhesin genes yadA (YP598_4279), yapC (YP598_3050), yapE (YP598_0106), yapJ (YP598_0716, YP598_0718), and yapK (YP598_0717, YP598_3860); 21 chromosomal genes for the T3SS (type III secretion system); 40 T3SS plasmid ysc/yop genes; and 28 plasmid genes belonging to the icm/dot type IVB locus as well as ypmA genes for the YPM superantigenic toxin. An icm/dot type IVB secretion system that was only shared with the intracellular persisting pathogens of the order Legionellales was found on the larger plasmid and could contribute to scarlatinoid fever symptoms in patients due to the introduction of immunomodulatory and immunosuppressive capabilities [79]. In turn, the YR genome contains specific yersiniabactin iron-uptake genes (irp2, YR821_1976, YR821_2934), the aprA gene for alkaline protease (YR821_1446), 12 plasmid genes for Yst1 T2SS, 12 chromosomal genes for Ysa T3SS, as well as the gene for thermostable hemolysin (YR821_0991), and the adaptation gene sodCI (YR821_1766).
Type III Secretion System
One of the main virulence factors of pathogenic Gram-negative bacteria is a T3SS that injects effector proteins into host cells [80]. For the clinical isolate YP, two different T3SS were found via RAST annotation and the VFDB [42] (Table S3).
First, a Ysc family T3SS was plasmid encoded. The Ysc family is one of the main families of T3SS and is named after the archetypal Ysc injectisome from Yersinia spp. [81]. The Ysc family includes the Psc system of P. aeruginosa, the Lsc system of P. luminescens, the Asc system of Aeromonas spp., and the Vsc system of Vibrio parahaemolyticus. The Bsc system of Bordetella spp. and the Dsc T3S system of Desulfovibrio spp. could form a subgroup within this family [81]. That is why the T3SS of the Ysc family is indicative of an extracellular pathogen [80,81]. Pathogenic Yersinia use T3SS along with other virulence factors to penetrate the intestinal epithelial layer [82].
All of the components encoding the pore complex, needle, basal body, ATPase complex, and regulator proteins of this T3SS were annotated (Figure 3). Additionally, seven effector proteins (YopE, YopH, YopT, YopM, YopK, YopJ, and YpkA) were YP plasmid encoded. They were shown to be translocated into eukaryotic cells by T3SS and to modulate the eukaryotic signaling pathways to counteract innate and adaptive immune responses to the pathogen [83].
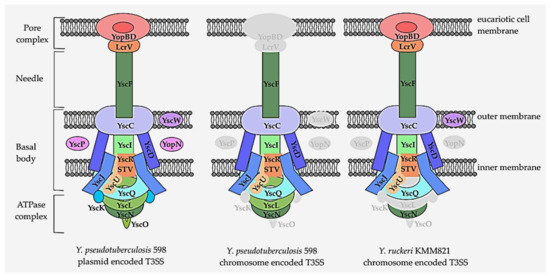
Figure 3.
Comparison of key structural elements of the type III secretion system encoded by the chromosome of Y. ruckeri KMM821 and the chromosome and plasmid of Y. pseudotuberculosis 598. Gray indicates the missing parts of T3SS. Basal body: YscC—secretin, outer membrane ring; YscW—secretin pilotin, required for the proper localization of the secretin YscC in the outer membrane; YscD—inner membrane ring; YscJ—inner membrane ring; YscF—needle; YscI—inner rod. Export apparatus: YscU—autoprotease; YscV—export gate; YscR—inner membrane component; YscS—inner membrane component; YscT—inner membrane component. Cytoplasmic ring: YscQ. ATPase complex: YscN–ATPase; YscL–stator; YscO–stalk; YscK–ATPase cofactor. Regulators: YscP—needle-length regulator; YopN—switch regulator. Translocators: YopD—translocation pore; YopB—translocation pore; LcrV—needle tip or filament.
The second T3SS was chromosome-encoded and belonged to the Ssa-Esc family [81]. The T3SS of this family, which is found in Salmonella enterica, allows the bacterium to survive in the macrophages by preventing endocytic trafficking and phagosome maturation [81].
However, no genes encoding the stalk of the ATPase complex, pore complex, secretin pilotin, and two regulator proteins were found for the chromosome-encoded T3SS of YP. The stalk protein YscO is not required for the assembly of the ATPase YscN but is essential for export [84]. No effector proteins for the YP chromosome-encoded T3SS were found.
Only one incomplete T3SS was revealed for YR (Table S3). It was chromosome-encoded and belongs to the Inv-Mxi-Spa family. It is likely that the bacteria that belong to this family and that have complete T3SS have an invasive phenotype [81]. However, no effector proteins were found in the YR genome. Moreover, the T3SS of YR lacked some parts of the ATPase complex and the regulator proteins YscP and YopN (Figure 3). We researched the genome of the fish-pathogenic strain Y. ruckeri SC09 (Accession Number CP025800.1), isolated from Ictalurus punctatus [85], using the VF analyzer (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi?func=VFanalyzer (accessed on 10 January 2022)) [42] and showed that this strain also lacks these T3SS genes.
As such, the contribution of the T3SS encoded by the YP and YR chromosomes during pathogenesis remains unclear. Pujol et al. were the first to point out that the T3SS that is encoded by the chromosome did not appear to be required for replication in macrophages, and they concluded that the function of the chromosomal T3SS in Yersinia pathogenesis remains a mystery [86]. Reuter et al. studied the genomic data of 241 Yersinia isolates from various species, including 31 Y. pseudotuberculosis isolates, and indicated that the chromosomal T3SS appears to be in the process of being lost in the more acutely pathogenic lineages of Y. pestis/Y. pseudotuberculosis and Y. enterocolitica [87]. This process was always coincident with the acquisition of an alternative T3SS (the virulence plasmid, bearing the Yop T3SS) [88]. However, the role of the remaining genes of the chromosomal T3SS during pathogenesis remains unexplored.
3.6. Possibilities of Resistance to Cold Shock in Psychrophilic and Psychrotolerant Microorganisms
Cold environments are predominant all over the Earth [16]. In the response to cold conditions, microorganisms use different adaptation strategies, such as the structural adjustment of enzymes, the maintenance of membrane fluidity, the expression of cold shock proteins, and the presence of compatible solutes [89]. Microorganisms that exist in cold conditions also express antifreeze proteins that inhibit the growth of ice crystals in solution [90]. For the analyzed psychrotolerant strains, two important strategies for adaptation to the cold were considered: the induction of cold shock proteins that affect intracellular processes and the ice-binding antifreeze protein located on the cell surface.
3.6.1. Cold Shock Proteins
The induction of the cold shock proteins (Csps) is one of the main strategies of psychrophilic and psychrotolerant bacteria that helps them to survive and multiply at low temperatures [91]. The same proteins are synthesized in response to stresses such as starvation. Csps are involved in RNA metabolism, protein folding, and the synthesis of membrane lipid A [92].
The Csps that are 65–75 amino acid (aa) in size [89] have two conserved RNA-binding motifs: RNP1 and RNP2. On the basis of protein sequences, the Csps of E. coli were divided into nine homologous (CspA-CspI) groups [92]. CspA, CspB, CspE, CspG, and CspI are generally recognized as being cold induced. CspC and CspE are involved in the regulation of alternative sigma factor σS and universal stress protein UspA. CspD is induced during the early stationary phase during nutritional starvation [89,92].
For YR and YP, almost equal numbers of Csps (10 and 9) were found. In the free-living MP, only six Csps were detected.
The MEGAX software [93] was used for the phylogenetic analysis of Csps of three bacteria. The phylogenetic tree (Figure 4) was constructed by the maximum likelihood method. As shown in Figure 4, the cold shock protein sequence homologs of E. coli CspD formed a single phylogenetic group for all of the analyzed bacteria, including the Y. pseudotuberculosis IP32953 reference. This suggests that similar adaptation mechanisms exist for the stress condition where there is a lack of nutrients for both opportunistic Yesinia and free-living Marinomonas bacteria. The five non-CspD Csps of MP formed a separate phylogenetic group, which confirms the differences in the adaptation potential at low temperatures for MP and Yesinia.
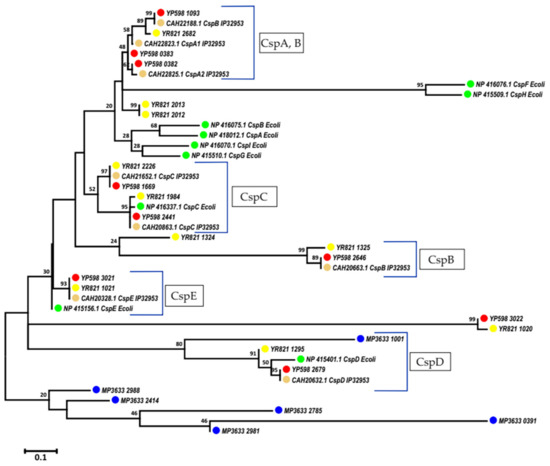
Figure 4.
Phylogenetic tree of the cold shock proteins (Csps). Green mark—Escherichia coli K-12 W3110; orange—Y. pseudotuberculosis IP32953; blue—M. primoryensis KMM3633T; red—Y. pseudotuberculosis 598; yellow—Y. ruckeri KMM821.
Besides CspD, only the CspE and CspC proteins of Yesinia were homologs to E. coli. It should be noted that the regulator protein CspC had a second copy among all of the analyzed Yesinia genomes (YR, YP, and Y. pseudotuberculosis IP32953). The low-temperature-induced Yesinia proteins CspA and CspB formed a separate phylogenetic group, which included two copies of CspA for Y. pseudotuberculosis. Additionally, the second copies of the CspB protein of three Yesinia strains were in a separate branch, and branch Csp YR821_1324 was found to be adjacent to it. The proteins YP598_3022 иYR821_1020 constituted another branch. They were longer—159 aa, with 62 aa matching the Csps sequence. Any conserved domains were not identified in the remaining parts of these proteins.
3.6.2. Antifreeze Proteins of M. primoryensis
Antifreeze proteins have been studied in Antarctic bacteria, especially in the inhabitants of Ace Lake (East Antarctica, Princess Elizabeth Land, 68.28 S 78.11 E) [90]. The lowest water temperature in this lake is -1 °C, and the lake has a salinity of 11.2–20.9‰ [94]. In the winter, the water temperature of the Sea of Japan is the same, but the salinity is higher—33.7–34.3‰.
Antifreeze proteins attach to the surface of the ice and lower the freezing point of the seawater and also help bacteria remain in the upper layers of water, which are enriched with oxygen and nutrients [90,94].
M. primoryensis AceL, which was isolated from Ace Lake in Antarctica in November 2000, has an antifreeze protein of 416,269.89 Da (4145 aa) [95] that has the ability to bind ice and is responsible for binding M. primoryensis AceL to diatoms [95]. This antifreeze protein is similar to adhesin protein LpaA in Pseudomonas fluorescens, which is 518,401.28 Da (5211 aa) and that attaches bacterial cells to various biotic and abiotic substrates and contributes to the tenacity of bacterial biofilms [96]. Despite the high (98.42%) similarity of the genome sequences of M. primoryensis AceL (accession number CP016181) and MP, the MP genome is 516,944 bp shorter. One of the regions deleted in the MP genome—the region at 20,690 bp, includes the gene for an antifreeze protein and six other open reading frames. However, a smaller antifreeze protein that is 250,165.24 Da (2411 aa) was encoded by another genome region and contained all of the functional domains typical of M. primoryensis AceL antifreeze proteins [97] and that are necessary to bind ice [95] (Figure 5). The MP protein has two domains that are predicted to bind calcium ions, which determine the correct folding of the molecule and the presentation of adhesion domains, including the ice-binding RIV domain. The M. primoryensis AceL had an analogous antifreeze protein with a 92.19% identity. Thus, among the bacteria analyzed in this research, MP is proposed to be the most adapted to the conditions of cold salt water.
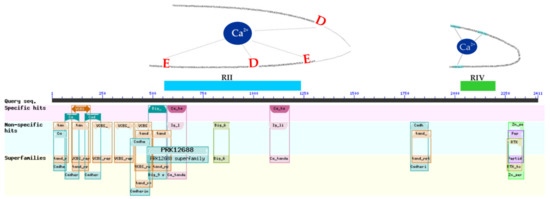
Figure 5.
Domain organization of antifreeze protein M. primoryensis KMM3633T (MP3633_2272). VCBS (Vibrio, Colwellia, Bradyrhizobium, and Shewanella)—VCBS repeat; Big_9—bacterial immunoglobulin-like domain; Ca_ta—Ca2+-stabilized repeat; Cadher—cadherin-like domain; tan—95 aa tandem repeat; RTX—RTX family hemolysin; Big_6—bacterial immunoglobulin-like domain; PRK12688—flagellin. Blue rectangle—kernel structure forming Ca2+-binding RII region; green rectangle—ice-binding RIV region. The Ca2+ binding residues in the RII region are marked in red, and those in the RIV region are marked in blue.
4. Discussion
Three bacteria representing the marine and terrestrial freshwater microbiota of the Pan-Okhotsk region were included in our analysis. This study of marine microorganisms was a continuation of a series of works by the PIBOC FEB RAS researchers, who are devoted to studying the adaptability of marine bacteria [98,99]. In these works, porins were the objects of study. A separate study was also dedicated to Porin_4 in M. primoryensis KMM3633T, and this study determined its physicochemical properties and pore-forming activity [100]. In the present work, we studied different classes of the bacterial translocators and some of the principal factors that contribute to the adaptability of psychrophilic and psychrotolerant bacteria to different ecological niches.
Significant differences were revealed in the list of porins (translocators in the outer membrane) between opportunistic Yersinia and free-living MP. Only one gene encoding a β-barrel outer membrane protein, which is predicted to allow the permeation of hydrophilic molecules with masses of less than 600 Da [101], was found in the MP genome. A second gene was found for the substrate-specific Maltoporin. Since porins cover ~70% of the membrane surface and form local regular lattices [102], the genes that code porins in MP presumably have very active transcription and translation and effective regulation. Interestingly, MP has all of the essential genes for the β-barrel assembly machinery complex (lptD-MP3633_0735, bamE MP3633_0422, bamD-MP3633_2979, bamB-MP3633_2689, bamA-MP3633_2764), and one of them, bamD, is located in close proximity to the ORF of Porin_4 in the genome along with the ORF of two Csps (MP2981, MP2988) and an anti-sigma factor (MP2985).
A second distinctive feature of the MP genome is the presence of genes for the caboxylates transporters (TRAP, TTT) in a 10-fold excess compared to in Yersinia. This fact is consistent with MP living in seawater that is rich in phytoplankton. In turn, for opportunistic Yersinia, carbohydrates are the main source of carbons, a finding that is supported by the presence of a large number of various PTS in these bacteria. The number of PTS could vary in the genomes of isolates belonging to the same Yersinia species. We compared the amount of PTS revealed in Y. ruckeri KMM821 and in the highly virulent Y. ruckeri strain SC09 that had been isolated from Ictalurus punctatus with severe septicemia [85] by analyzing genomic data using the KEGG. In the genome of Y. ruckeri SC09, we found an additional PTS operon that is responsible for sorbitol transport.
Since fishing makes a significant contribution to the economy of the countries in the Pan-Okhotsk region, we paid attention to Y. ruckeri, which can cause the enteric redmouth disease (ERM) that is predominantly found in salmonid fish [19]. Y. ruckeri is a facultative intracellular bacterium that capable of surviving inside the macrophages of the fish, and ERM infections can be transmitted by direct contact between infected and non-infected specimens [103]. The Y. ruckeri KMM821 isolated from the seawater from the Sea of Okhotsk near Iturup Island demonstrated pathogenic potential. It was a serotype O1 strain with 100% similarity with the reference genome in terms of 97 genes that are exclusively shared by serotype O1 on the basis of the research conducted by Cascales et al. [77]. In the genome of this strain, there are plasmids harboring 8 of the 73 virulence factors revealed in the YR genome and that are known to be upregulated at 18 °C. These data suggest the possibility that Y. ruckeri KMM821 can cause disease in fish. Only the effectiveness of the invasion of this bacterium into eukaryotic cells is questionable since there are no genes in the genome for some parts of the ATPase complex and regulator proteins YscP and YopN as well as for the effector proteins of T3SS. At the same time, an analysis of the pathogenic and invasive strain Y. ruckeri SC09 showed that it also lacks these genes.
The pathogenic properties of terrestrial Y. pseudotuberculosis are beyond doubt. Y. pseudotuberculosis 598 was isolated from a person with FESLF in Primorye, presumably in the 1980s. Genomic research has suggested that this strain is of the O1b serotype and genotypes ST2 and VST1, which coincides with the description of the most dangerous isolates [24]. The plasmids; 173 virulence factors; two T3SS with genes for effectors; the presence of a (Dcu)AB transporter, which is important for anaerobic respiration; and the multiple Fluoride Exporter (Fluc) Family transporters that remove the fluoride ions that inhibit the enzymes that are necessary for glycolysis and nucleic acid synthesis are all characteristics inherent in pathogenic strains [79]. Additional evidence of the high pathogenicity of Y. pseudotuberculosis 598 was the identification of 28 plasmid-encoded genes belonging to the icm/dot type IVB secretion system. Previously, T4SS was detected in the FESLF-causing strain Y. pseudotuberculosis IP31758 and was absent in Y. pseudotuberculosis IP32953, which caused classical gastrointestinal symptoms [79].
In addition to the possibility of the parasitism in eukaryotic organisms, the researched Yersinia can survive in the cold environmental conditions of the Pan-Okhotsk region, and its survival is presumably facilitated by cold shock proteins. However, the arsenal of proteins that are resistant to the cold appear to be greater in the MP, which has the gene for an antifreeze protein other than Csps.
The present research on the bacterial genomes of three representatives of the microbiota from the Pan-Okhotsk region showed that the genomic potential of these bacteria is consistent with the adaptability of M. primoryensis KMM3633T to the nutrient and climatic characteristics of this environment and the ability of Y. ruckeri KMM821 and Y. pseudotuberculosis 598 to not only survive in this environment, but to also parasitize eukaryotic organisms.
5. Conclusions
The Pan-Okhotsk region ecosystem, a reservoir of genetic microbial diversity, represents a virtually unlimited source of microorganisms that are able to interact with human and animal beings. Despite the continuous exploration of Pan-Okhotsk habitats and the descriptions of their microbial communities, bacterial phenotypes that are commonly associated with pathogenicity have rarely been reported upon [25,27,28,104].
In this work, we compared the genomes of three bacteria isolated from the marginal seas and from a patient in Primorye. The analysis of genomic data revealed distinctive features showing the metabolic and pathogenic potential of M. primoryensis KMM3633T, Y. ruckeri KMM821, and Y. pseudotuberculosis 598. The clinical strain Y. pseudotuberculosis 598 showed clear pathogenic potential.
Y. ruckeri KMM821 is characterized by its pathogenic potential, and it is distinguished by its pronounced ability to adapt to various ecological niches. In turn, the metabolic features of non-pathogenic M. primoryensis KMM3633T—namely, the more intensive metabolism of a number of amino acids, porphyrin, and propionic acid derivatives; the active degradation of a large list of organic compounds; and the presence of biosynthetic pathways for flavonoids and phenylflavonoids, can find applications in biotechnology, as can adhesive properties of the antifreeze protein.
Our research contributes knowledge that is useful for the prediction and preparation of responses to the uncountable consequences of the significantly changing climate. In the future, combining various ‘-omics’ molecular approaches with measures of microbial productivity, activity, geochemical, and physical characterization, and modeling efforts in low-temperature environments will provide the next critical steps toward a better understanding of the function of low-temperature microbial ecosystems and their pathogenic potential in terms of survival in the environment and aggressiveness inside eukaryotic host cells.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/w14071107/s1, Figure S1: Y. pseudotuberculosis 598 O-antigen gene cluster; Table S1a: Characteristics of phosphotransferase system EIIC proteins of Y. pseudotuberculosis 598 and Y. ruckeri KMM821; Table S1b: Characteristics of phosphotransferase system components of M. primoryensis KMM3633T; Table S2: Comparison of the major virulence factors excluding the T3SS encoded by the chromosome of Y. ruckeri. KMM821 and the chromosome and plasmid of Y. pseudotuberculosis 598; Table S3: Comparison of the key structural elements of the type III secretion system encoded by the chromosome of Y. ruckeri KMM821 and the chromosome and plasmid of Y. pseudotuberculosis 598.
Author Contributions
Conceptualization, O.L.V.; Methodology, O.L.V., N.N.R., M.S.K. and E.I.A.; Software, N.N.R. and M.S.K.; Validation, O.L.V., M.S.K. and N.N.R.; Formal analysis, E.I.A.; Investigation, N.N.R., M.S.K., E.I.A. and O.L.V.; Resources, O.L.V.; Data curation, M.S.K., N.N.R. and O.L.V.; Writing—original draft preparation, O.L.V., N.N.R., M.S.K. and E.I.A.; Writing—review and editing, O.L.V. and N.N.R.; Visualization, N.N.R. and M.S.K.; Supervision, O.L.V.; Project administration, O.L.V. and M.S.K.; Funding acquisition, O.D.N. and A.L.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Foundation for Basic Research grant no. 19-04-00318.
Data Availability Statement
Genomic data are available at GenBank NCBI (BioProject PRJNA622482).
Acknowledgments
We thank Romanenko L.A. and the staff of the Collection of Marine Microorganisms at PIBOC FEB RAS for the cultures of the microorganisms that were provided for genomic analysis and Portnyagina O.Yu. for coordinating the interaction between the research teams that participated in the RFBR grant.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IVS FEB RAS Geoportal. Available online: http://geoportal.kscnet.ru/volcanoes/ (accessed on 9 December 2021).
- Nishioka, J.; Obata, H.; Ogawa, H.; Ono, K.; Yamashita, Y.; Lee, K.; Takeda, S.; Yasuda, I. Subpolar marginal seas fuel the North Pacific through the intermediate water at the termination of the global ocean circulation. Proc. Natl. Acad. Sci. USA 2020, 117, 12665–12673. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Sutherland, S.C.; Sweeney, C.; Poisson, A.; Metzl, N.; Tilbrook, B.; Bates, N.; Wanninkhof, R.; Feely, R.A.; Sabine, C.; et al. Global sea–air CO2 flux based on climatological surface ocean pCO2, and seasonal biological and temperature effects. Deep. Sea Res. Part II Top. Stud. Oceanogr. 2002, 49, 1601–1622. [Google Scholar] [CrossRef]
- PICES Scientific Report No. 36. Proceedings of the Fourth Workshop on the Okhotsk Sea and Adjacent Areas; Kashiwai, M., Kantakov, G.A., Eds.; PICES: Sidney, BC, Canada, 2009; Available online: http://www.pices.int (accessed on 10 January 2022).
- Mitsudera, H. Environmental problems in the Pan Okhotsk Region. In Energy and Environment in Slavic Eurasia: Toward the Establishment of the Network of Environmental Studies in the Pan-Okhotsk Region; Shinichiro, T., Ed.; Slavic Research Center: Sapporo, Japan, 2008; pp. 157–166. [Google Scholar]
- Alekseev, A.V.; Baklanov, P.J.; Arzamastsev, I.S.; Blinov, Y.G.; Fedorovskii, A.S.; Kachur, A.N.; Khrapchenkov, F.F.; Medvedeva, I.A.; Minakir, P.A.; Titova, G.D.; et al. Sea of Okhotsk, GIWA Regional Assessment 30; UNEP; University of Kalmar: Kalmar, Sweden, 2006. [Google Scholar]
- Miroshnichenko, M.L.; Bonch-Osmolovskaya, E.A.; Alekseev, V.A. Extremely thermophilic bacteria from Kraternaya Bight. Biol. Morya [Mar. Biol.] 1989, 3, 77–83. (In Russian) [Google Scholar]
- Tarasov, V.G. Effects of shallow-water hydrothermal venting on biological communities of coastal marine ecosystems of the western. Pac. Adv. Mar. Biol. 2006, 50, 267–421. [Google Scholar] [CrossRef]
- Zalogin, B.S.; Kosarev, A.N. Moria Seas; Idea: Moscow, Russia, 1999; 400p. (In Russian) [Google Scholar]
- Romanenko, L.A.; Uchino, M.; Mikhailov, V.V.; Zhukova, N.V.; Uchimura, T. Marinomonas primoryensis sp. nov., a novel psychrophile isolated from coastal sea-ice in the Sea of Japan. Int. J. Syst. Evol. Microbiol. 2003, 53, 829–832. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Hill, P.J.; Dodd, C.E.R.; Laybourn-Parry, J. Demonstration of antifreeze protein activity in Antarctic lake bacteria. Microbiology 2004, 150, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, X.M.; Li, J.; Song, X.Y.; Qin, Q.L.; Su, H.N.; Chen, X.L.; Zhang, Y.Z.; Fan, S.J.; Zhang, X.Y. Marinomonas profundi sp. nov., isolated from deep seawater of the Mariana Trench. Int. J. Syst. Evol. Microbiol. 2020, 70, 5747–5752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.C.; Li, H.R.; Xin, Y.H.; Liu, H.C.; Chen, B.; Chi, Z.M.; Zhou, P.J.; Yu, Y. Marinomonas arctica sp. nov., a psychrotolerant bacterium isolated from the Arctic. Int. J. Syst. Evol. Microbiol. 2008, 58, 1715–1718. [Google Scholar] [CrossRef]
- Ingraham, J.L. Growth of psychrophilic bacteria. J. Bacteriol. 1958, 76, 75–80. [Google Scholar] [CrossRef]
- Russell, N.J. Cold adaptation of microorganisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 326, 595–608; discussion 608–611. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.; Leo, J.C.; Linke, D. Overcoming Fish Defences: The Virulence Factors of Yersinia ruckeri. Genes 2019, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- Ewing, W.H.; Ross, A.J.; Brenner, D.J.; Fanning, G.R. Yersinia ruckeri sp. nov., the redmouth (RM) bacterium. Int. J. Syst. Bacteriol. 1978, 28, 37–44. [Google Scholar] [CrossRef]
- Ross, A.J.; Rucker, R.R.; Ewing, W.H. Description of a bacterium associated with redmouth disease of rainbow trout (Salmo gairdneri). Can. J. Microbiol. 1966, 12, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Chistyulin, D.K.; Novikova, O.D.; Portnyagina, O.Y.; Khomenko, V.A.; Vakorina, T.I.; Kim, N.Y.; Isaeva, M.P.; Likhatskaya, G.N.; Solov’eva, T.F. Isolation and Characterization of OmpF like Porin from Yersinia ruckeri. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2012, 6, 235–242. [Google Scholar] [CrossRef]
- Abramson, N.I.; Petrova, T.V.; Dokuchaev, N.E.; Obolenskaya, E.V.; Lissovsky, A.A. Phylogeography of the gray red-backed vole Craseomys rufocanus (Rodentia: Cricetidae) across the distribution range inferred from nonrecombining molecular markers. Russ. J. Theriol. 2012, 11, 137–156. [Google Scholar] [CrossRef]
- Voronina, O.L.; Kunda, M.S.; Ryzhova, N.N.; Aksenova, E.I.; Semenov, A.N.; Kurnaeva, M.A.; Ananyina, Y.V.; Lunin, V.G.; Gintsburg, A.L. Regularities of the ubiquitous polyhostal microorganisms selection by the example of three taxa. Mol. Biol. 2015, 49, 430–441. [Google Scholar] [CrossRef]
- Grunin, I.I.; Somov, G.P.; Zalmover, I.I. Far Eastern scarlatinoid fever. Voen Med. Zh. 1960, 8, 626. (In Russian) [Google Scholar]
- Timchenko, N.F.; Adgamov, R.R.; Popov, A.F.; Psareva, E.K.; Sobyanin, K.A.; Gintsburg, A.L.; Ermolaeva, S.A. Far East Scarlet-Like Fever Caused by a Few Related Genotypes of Yersinia pseudotuberculosis, Russia. Emerg. Infect. Dis. 2016, 22, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.G. Vydelenie bakteriĭ roda Yersinia iz razlichnykh vodoistochnikov v Primorskom Krae [Isolation of Yersinia from various water sources in the Primor’e Territory]. Gig. Sanit. 1983, 2, 72–74. (In Russian) [Google Scholar]
- Timchenko, N.F.; Dolmatov, I.I.; Naĭdenko, T.K. Yersinia pseudotuberculosis v model’noĭ morskoĭ ékosisteme (éksperimental’noe issledovanie) [Yersinia pseudotuberculosis in a model marine ecosystem (experimental research)]. Zh. Mikrobiol. Epidemiol. Immunobiol. 1995, 5, 84–88. (In Russian) [Google Scholar]
- Koo, J.W.; Park, S.N.; Choi, S.M.; Chang, C.H.; Cho, C.R.; Paik, I.K.; Chung, C.Y. Acute renal failure associated with Yersinia pseudotuberculosis infection in children. Pediatr. Nephrol. 1996, 10, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H. Direct isolation of Yersinia pseudotuberculosis from fresh water in Japan. Appl. Environ. Microbiol. 1992, 58, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, N.F.; Somov, G.P. Patogeneticheskoe znachenie psikhrofil’nosti Yersinia pseudotuberculosis [Pathogenetic significance of the psychrophilia of Yersinia pseudotuberculosis]. Zh. Mikrobiol. Epidemiol. Immunobiol. 1986, 3, 34–38. (In Russian) [Google Scholar]
- Inoue, M.; Nakashima, H.; Ishida, T.; Tsubokura, M.; Sakazaki, R. Isolation of Yersinia pseudotuberculosis from water. Zentralbl. Bakteriol. Mikrobiol. Hyg. B 1988, 186, 338–343. [Google Scholar] [PubMed]
- Wilson, K. Unit 2.4 Preparation of genomic DNA from bacteria. In Current Protocols in Molecular Biology; Wiley Online Library: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H. Predicting Secretory Proteins with SignalP. Methods Mol. Biol. 2017, 1611, 59–73. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Aktar, U.; Jani, M.R.; Shatabda, S. iPro70-FMWin: Identifying Sigma70 promoters using multiple windowing and minimal features. Mol. Genet. Genom. 2019, 294, 69–84. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef]
- Saier, M.H., Jr. Families of transmembrane sugar transport proteins. Mol. Microbiol. 2000, 35, 699–710. [Google Scholar] [CrossRef]
- Putman, M.; Van Veen, H.W.; Degener, J.E.; Konings, W.N. Antibiotic resistance: Era of the multidrug pump. Mol. Microbiol. 2000, 36, 772–773. [Google Scholar] [CrossRef]
- Davies, J.S.; Currie, M.J.; Wright, J.D.; Newton-Vesty, M.C.; North, R.A.; Mace, P.D.; Allison, J.R.; Dobson, R.C.J. Selective Nutrient Transport in Bacteria: Multicomponent Transporter Systems Reign Supreme. Front. Mol. Biosci. 2021, 8, 699222. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, N.; Takahashi, K.; Mori, K.; Araki, T.; Fujita, M.; Higuchi, Y.; Masai, E. Bacterial catabolism of lignin-derived aromatics: New findings in a recent decade: Update on bacterial lignin catabolism. Environ. Microbiol. Rep. 2017, 9, 679–705. [Google Scholar] [CrossRef] [PubMed]
- Rosa, L.T.; Bianconi, M.E.; Thomas, G.H.; Kelly, D.J. Tripartite ATP-Independent Periplasmic (TRAP) Transporters and Tripartite Tricarboxylate Transporters (TTT): From Uptake to Pathogenicity. Front. Cell Infect. Microbiol. 2018, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Tomii, K.; Kanehisa, M. A comparative analysis of ABC transporters in complete microbial genomes. Genome Res. 1998, 8, 1048–1059. [Google Scholar] [CrossRef]
- Igarashi, Y.; Aoki, K.F.; Mamitsuka, H.; Kuma, K.; Kanehisa, M. The evolutionary repertoires of the eukaryotic-type ABC transporters in terms of the phylogeny of ATP-binding domains in eukaryotes and prokaryotes. Mol. Biol. Evol. 2004, 21, 2149–2160. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Berggren, M.; Laudon, H.; Haei, M.; Ström, L.; Jansson, M. Efficient aquatic bacterial metabolism of dissolved low-molecular-weight compounds from terrestrial sources. ISME J. 2010, 4, 408–416. [Google Scholar] [CrossRef]
- Janausch, I.G.; Zientz, E.; Tran, Q.H.; Kröger, A.; Unden, G. C4-dicarboxylate carriers and sensors in bacteria. Biochim. Biophys. Acta 2002, 1553, 39–56. [Google Scholar] [CrossRef]
- Mulligan, C.; Kelly, D.J.; Thomas, G.H. Tripartite ATP-independent periplasmic transporters: Application of a relational database for genome-wide analysis of transporter gene frequency and organization. J. Mol. Microbiol. Biotechnol. 2007, 12, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, R.; Gross, R.; Simon, J.; Unden, G.; Kröger, A. Transport of C(4)-dicarboxylates in Wolinella succinogenes. J. Bacteriol. 2000, 182, 5757–5764. [Google Scholar] [CrossRef][Green Version]
- Janausch, I.G.; Kim, O.B.; Unden, G. DctA- and Dcu-independent transport of succinate in Escherichia coli: Contribution of diffusion and of alternative carriers. Arch. Microbiol. 2001, 176, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Wösten, M.M.; van de Lest, C.H.; van Dijk, L.; van Putten, J.P. Function and Regulation of the C4-Dicarboxylate Transporters in Campylobacter jejuni. Front. Microbiol. 2017, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.H.; Liu, E.; Dean, K.; Gingras, M.; DeGraff, W.; Trun, N.J. Overproduction of three genes leads to camphor resistance and chromosome condensation in Escherichia coli. Genetics 1996, 143, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Stockbridge, R.B.; Miller, C. Bacterial fluoride resistance, Fluc channels, and the weak acid accumulation effect. J. Gen. Physiol. 2014, 144, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, R.B.; Robertson, J.L.; Kolmakova-Partensky, L.; Miller, C. A family of fluoride-specific ion channels with dual-topology architecture. Elife 2013, 2, e01084. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, L.; Baars, T.L.; Fendler, K.; Michel, H. Functional characterization of solute carrier (SLC) 26/sulfate permease (SulP) proteins in membrane mimetic systems. Biochim. Biophys. Acta 2016, 1858, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Erni, B. The bacterial phosphoenolpyruvate: Sugar phosphotransferase system (PTS): An interface between energy and signal transduction. J. Iran. Chem. Soc. 2013, 10, 593–630. [Google Scholar] [CrossRef]
- Milton, H.S., Jr. The Bacterial Phosphotransferase System: New frontiers 50 years after its discovery. J. Mol. Microbiol. Biotechnol. 2015, 25, 73–78. [Google Scholar] [CrossRef]
- Shelton, C.L.; Rael, F.K.; Beatty, W.L.; Johnson, S.M.; Mason, K.M. Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog. 2011, 7, e1002360. [Google Scholar] [CrossRef] [PubMed]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [PubMed]
- Coggan, K.A.; Wolfgang, M.C. Global regulatory pathways and cross-talk control pseudomonas aeruginosa environmental lifestyle and virulence phenotype. Curr. Issues Mol. Biol. 2012, 14, 47–70. [Google Scholar] [PubMed]
- Akinwumi, B.C.; Bordun, K.M.; Anderson, H.D. Biological Activities of Stilbenoids. Int. J. Mol. Sci. 2018, 19, 792. [Google Scholar] [CrossRef]
- Hapeshi, A.; Benarroch, J.M.; Clarke, D.J.; Waterfield, N.R. Iso-Propyl stilbene: A life cycle signal? Microbiology 2019, 165, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Joyce, S.A.; Brachmann, A.O.; Glazer, I.; Lango, L.; Schwär, G.; Clarke, D.J.; Bode, H.B. Bacterial biosynthesis of a multipotent stilbene. Angew. Chem. Int. Ed. Engl. 2008, 47, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Rakin, A.; Schneider, L.; Podladchikova, O. Hunger for iron: The alternative siderophore iron scavenging systems in highly virulent Yersinia. Front. Cell Infect. Microbiol. 2012, 2, 151. [Google Scholar] [CrossRef] [PubMed]
- Secades, P.; Guijarro, J.A. Purification and characterization of an extracellular protease from the fish pathogen Yersinia ruckeri and effect of culture conditions on production. Appl. Environ. Microbiol. 1999, 65, 3969–3975. [Google Scholar] [CrossRef]
- Fernandez, L.; Marquez, I.; Guijarro, J.A. Identification of specific in Vivo-Induced (ivi) genes in Yersinia ruckeri and analysis of ruckerbactin, a catecholate siderophore iron acquisition system. Appl. Environ. Microbiol. 2004, 70, 5199–5207. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, J.A.; Cascales, D.; García-Torrico, A.I.; García-Domínguez, M.; Méndez, J. Temperature-dependent expression of virulence genes in fish-pathogenic bacteria. Front. Microbiol. 2015, 6, 700. [Google Scholar] [CrossRef] [PubMed]
- Cascales, D.; Guijarro, J.A.; García-Torrico, A.I.; Méndez, J. Comparative genome analysis reveals important genetic differences among serotype O1 and serotype O2 strains of Y. ruckeri and provides insights into host adaptation and virulence. Microbiologyopen 2017, 6, e00460. [Google Scholar] [CrossRef] [PubMed]
- Beynon, L.M.; Richards, J.C.; Perry, M.B. The structure of the lipopolysaccharide O antigen from Yersinia ruckeri serotype 01. Carbohydr. Res. 1994, 256, 303–317. [Google Scholar] [CrossRef]
- Eppinger, M.; Rosovitz, M.J.; Fricke, W.F.; Rasko, D.A.; Kokorina, G.; Fayolle, C.; Lindler, L.E.; Carniel, E.; Ravel, J. The complete genome sequence of Yersinia pseudotuberculosis IP31758, the causative agent of Far East scarlet-like fever. PLoS Genet. 2007, 3, e142. [Google Scholar] [CrossRef]
- Deng, W.; Marshall, N.C.; Rowland, J.L.; McCoy, J.M.; Worrall, L.J.; Santos, A.S.; Strynadka, N.C.J.; Finlay, B.B. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017, 15, 323–337. [Google Scholar] [CrossRef]
- Troisfontaines, P.; Cornelis, G.R. Type III secretion: More systems than you think. Physiology 2005, 20, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Böhme, K.; Heroven, A.K.; Lobedann, S.; Guo, Y.; Stolle, A.S.; Dersch, P. The Small Protein YmoA Controls the Csr System and Adjusts Expression of Virulence-Relevant Traits of Yersinia pseudotuberculosis. Front. Microbiol. 2021, 12, 706934. [Google Scholar] [CrossRef]
- Zhang, Y.; Romanov, G.; Bliska, J.B. Type III secretion system-dependent translocation of ectopically expressed Yop effectors into macrophages by intracellular Yersinia pseudotuberculosis. Infect. Immun. 2011, 79, 4322–4331. [Google Scholar] [CrossRef]
- Diepold, A.; Wiesand, U.; Amstutz, M.; Cornelis, G.R. Assembly of the Yersinia injectisome: The missing pieces. Mol. Microbiol. 2012, 85, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, K.Y.; Wang, J.; Chen, D.F.; Huang, X.L.; Ouyang, P.; Geng, Y.; He, Y.; Zhou, Y.; Min, J. Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism. Int J. Mol. Sci. 2016, 17, 557. [Google Scholar] [CrossRef] [PubMed]
- Pujol, C.; Bliska, J.B. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 2003, 71, 5892–5899. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Corander, J.; de Been, M.; Harris, S.; Cheng, L.; Hall, M.; Thomson, N.R.; McNally, A. Directional gene flow and ecological separation in Yersinia enterocolitica. Microb. Genom. 2015, 1, e000030. [Google Scholar] [CrossRef]
- Reuter, S.; Connor, T.R.; Barquist, L.; Walker, D.; Feltwell, T.; Harris, S.R.; Fookes, M.; Hall, M.E.; Petty, N.K.; Fuchs, T.M.; et al. Parallel independent evolution of pathogenicity within the genus Yersinia. Proc. Natl. Acad. Sci. USA 2014, 111, 6768–6773. [Google Scholar] [CrossRef] [PubMed]
- Phadtare, S. Recent Developments in bacterial cold-shock response. Curr. Issues Mol. Biol. 2004, 6, 125–136. [Google Scholar] [PubMed]
- Muñoz, P.A.; Márquez, S.L.; González-Nilo, F.D.; Márquez-Miranda, V.; Blamey, J.M. Structure and application of antifreeze proteins from Antarctic bacteria. Microb. Cell Fact. 2017, 16, 138. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Hao, L.; Chen, F.; Li, S.; Abdelrahman, A.M.; Zhang, Y.; Yu, H.; Liu, S.; Li, M. CspE is Overproduced by temperature downshift in the Acinetobacter johnsonii DBP-3. Curr. Microbiol. 2016, 72, 563–569. [Google Scholar] [CrossRef]
- Yu, T.; Keto-Timonen, R.; Jiang, X.; Virtanen, J.P.; Hannu Korkeala, H. Insights into the phylogeny and evolution of cold shock proteins: From enteropathogenic Yersinia and Escherichia coli to Eubacteria. Int. J. Mol. Sci. 2019, 20, 4059. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Laybourn-Parry, J.; Bell, E.M. Ace Lake: Three decades of research on a meromictic, Antarctic lake. Polar Biol. 2014, 37, 1685–1699. [Google Scholar] [CrossRef]
- Guo, S.; Stevens, C.A.; Vance, T.D.R.; Olijve, L.L.C.; Graham, L.A.; Campbell, R.L.; Yazdi, S.R.; Escobedo, C.; Bar-Dolev, M.; Yashunsky, V.; et al. Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci. Adv. 2017, 3, e1701440. [Google Scholar] [CrossRef]
- Collins, A.J.; Pastora, A.B.; Smith, T.J.; O’Toole, G.A. MapA, a Second Large RTX Adhesin Conserved across the Pseudomonads, Contributes to Biofilm Formation by Pseudomonas fluorescens. J. Bacteriol. 2020, 202, 18. [Google Scholar] [CrossRef]
- Garnham, C.P.; Campbell, R.L.; Davies, P.L. Anchored clathrate waters bind antifreeze proteins to ice. Proc. Natl. Acad. Sci. USA 2011, 108, 7363–7367. [Google Scholar] [CrossRef]
- Novikova, O.D.; Khomenko, V.A.; Frolova, G.M.; Likhatskaya, G.N.; Romanenko, L.A.; Portnyagina, O.Y.; Kuznetsova, S.M.; Solovyeva, T. F Porins from marine bacteria of the Genus Pseudoalteromonas (Gammaproteobacteria: Pseudoalteromonadaceae). Russ. J. Mar. Biol. 2013, 39, 58–64. [Google Scholar] [CrossRef]
- Khomenko, V.A.; Likhatskaya, G.N.; Romanenko, L.A.; Portnyagina, O.Y.; Solovyeva, T.F.; Novikova, O.D. Protein composition of the cell envelope of the bacterium Shewanella frigidimarina Pi 2–35 (Gammaproteobacteria: Shewanellaceae). Russ. J. Mar. Biol. 2016, 42, 73–80. [Google Scholar] [CrossRef]
- Novikova, O.D.; Khomenko, V.A.; Kim, N.Y.; Likhatskaya, G.N.; Romanenko, L.A.; Aksenova, E.I.; Kunda, M.S.; Ryzhova, N.N.; Portnyagina, O.Y.; Solov’eva, T.F.; et al. Porin from Marine Bacterium Marinomonas primoryensis KMM 3633T: Isolation, Physico-Chemical Properties, and Functional Activity. Molecules 2020, 25, 3131. [Google Scholar] [CrossRef] [PubMed]
- Cowan, S.W.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.A.; Jansonius, J.N.; Rosenbusch, J.P. Crystal structures explain functional properties of two E. coli porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Jarosławski, S.; Duquesne, K.; Sturgis, J.N.; Scheuring, S. High-resolution architecture of the outer membrane of the Gram-negative bacteria Roseobacter denitrificans. Mol. Microbiol. 2009, 74, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.; Bossier, P.; D’Herde, K.; Diez-Fraile, A.; Sorgeloos, P.; Haesebrouck, F.; Pasmans, F. Persistence of Yersinia ruckeri in trout macrophages. Fish Shellfish Immunol. 2010, 29, 648–655. [Google Scholar] [CrossRef]
- Zaytseva, E.; Ermolaeva, S.; Somov, G.P. Low genetic diversity and epidemiological significance of Listeria monocytogenes isolated from wild animals in the far east of Russia. Infect. Genet. Evol. 2007, 7, 736–742. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).