
Application of Magnesium Oxide Media for Remineralization and Removal of Divalent Metals in Drinking Water Treatment: A Review




Abstract
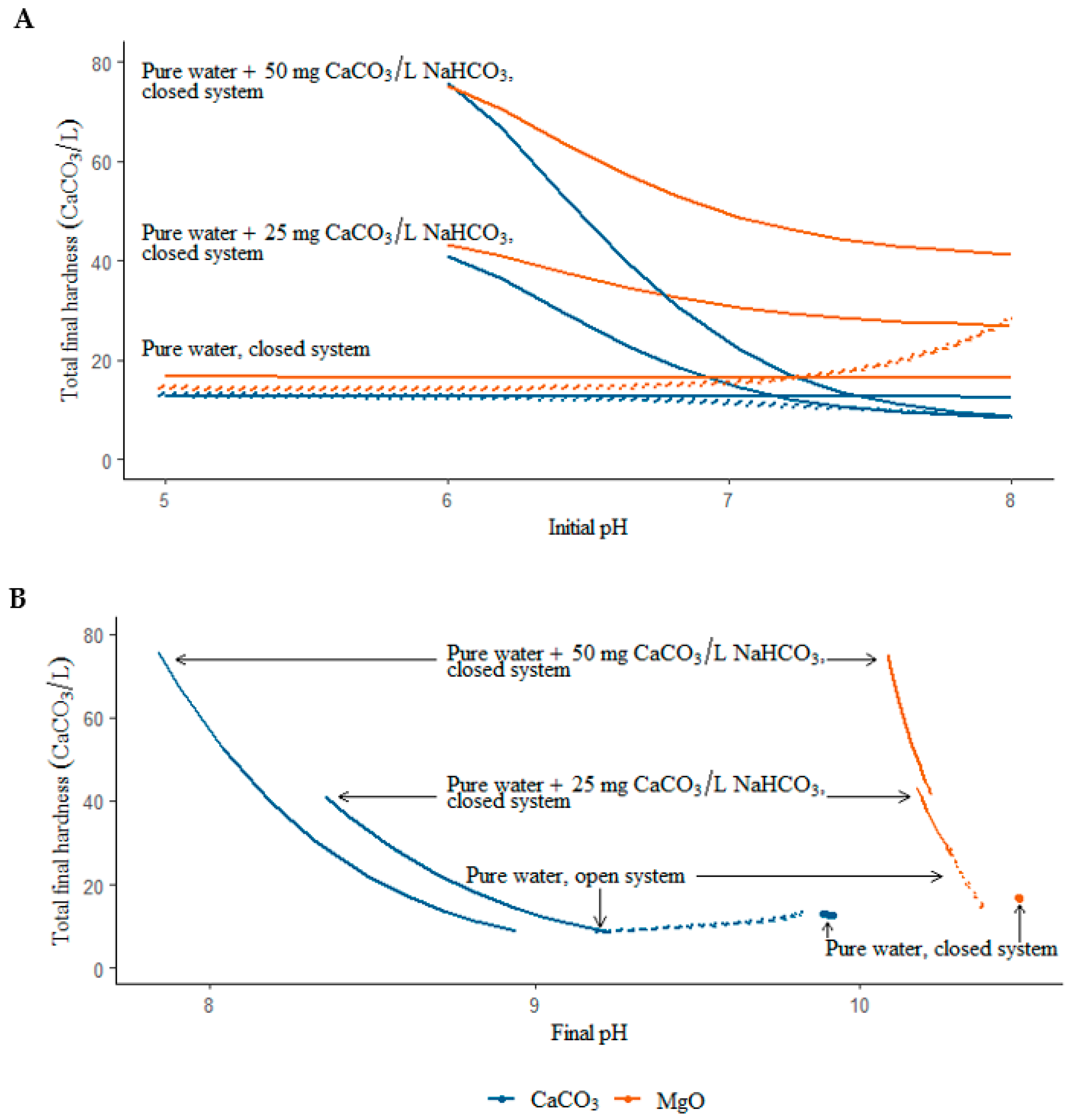
:1. Background and Objectives
- The direct dosage of chemicals such as CO2, NaOH, Ca(OH)2, NaHCO3, Na2CO3, CaCl2, MgCl2, and MgSO4;
- Blending of hard water with a soft effluent;
- Mineral dissolution (e.g., calcite); or
- A combination of the abovementioned methods.
Magnesium Oxide Sources and Properties
2. Mechanisms in MgO Dissolution
2.1. Dissolution and Hydration Theory
- (1)
- Water adsorbs at the surface and diffuses inside porous MgO particles simultaneously;
- (2)
- Oxide dissolution occurs within particles, changing porosity with time:
- (3)
- Supersaturation, nucleation, and growth of magnesium hydroxide occur at the surface of the MgO particle:
2.2. Parameters Affecting MgO Dissolution in Water Treatment
2.2.1. Media Impurities
2.2.2. Particle Size and Shape
2.2.3. Internal Particle Porosity
2.2.4. Feed Water Composition
Acidity (pH)
MgO Saturation Index
Temperature
Aqueous CO2 Content
Presence of Anions
Presence of Dissolved Metals
Precipitation
Sorption
Cation Exchange
2.3. Comparison to Calcite Dissolution
3. Water Treatment Applications
3.1. Main Applications
3.1.1. pH and Alkalinity Correction
3.1.2. Magnesium Hardness
3.1.3. Metal Removal
3.2. Process Operation
3.2.1. Scale Formation
3.2.2. Issues with Filtration Media Coating
3.2.3. Past Attempts to Model Process Performance
- (1)
- Dissolution;
- (2)
- Particle evolution and passivation;
- (3)
- Contaminant removal; and
- (4)
- Permeability loss.
3.2.4. Costs
4. Discussion and Suggestions for Further Research
- Quick hydration and the low solubility of Mg(OH)2, which provides a long-term source of alkalinity and Mg2+;
- Higher solubility than calcite;
- pH buffering range and ability to retain divalent metal contaminants as compact hydroxide and oxide precipitates;
- Health benefits associated with magnesium;
- Simpler operation than an alternative online chemical injection system (such as caustic).
Author Contributions
Funding
Conflicts of Interest
References
- McGowan, W. Water Processing: Residential, Commercial, Light-Industrial, 3rd ed.; Water Quality Association: Lisle, IL, USA, 2000. [Google Scholar]
- Health Canada. Guidelines for Canadian Drinking Water Quality: Guideline Technical Document—pH. (Catalogue No H144-28/2016E-PDF); Health Canada: Ottawa, ON, Canada, 2015; Available online: https://www.canada.ca/en/health-canada/services/publications/healthy-living/guidelines-canadian-drinking-water-quality-guideline-technical-document-ph.html (accessed on 29 December 2021).
- World Health Organization. Guidelines for Drinking-Water Quality: Fourth Edition Incorporating First; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Shemer, H.; Hasson, D.; Semiat, R. State-of-the-art review on post-treatment technologies. Desalination 2015, 356, 285–293. [Google Scholar] [CrossRef]
- Letterman, R.D.; Driscoll, C.T.; Haddad, M.; Hsu, H.A. Limestone Bed Contactors for Control of Corrosion at Small Water Utilities; Water Engineering Research Laboratory, US EPA: Washington, DC, USA, 1987. [Google Scholar]
- Birnhack, L.; Voutchkov, N.; Lahav, O. Fundamental chemistry and engineering aspects of post-treatment processes for desalinated water—A review. Desalination 2011, 273, 6–22. [Google Scholar] [CrossRef]
- Monarca, S.; Donato, F.; Zerbini, I.; Calderon, R.L.; Craun, G.F. Review of epidemiological studies on drinking water hardness and cardiovascular diseases. Eur. J. Cardiovasc. Prev. Rehabil. 2006, 13, 495–506. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Calcium and Magnesium in Drinking-Water: Public Health Significance; World Health Organization: Geneva, Switzerland, 2009; Available online: https://www.who.int/publications/i/item/9789241563550 (accessed on 29 December 2021).
- World Health Organization (WHO). Nutrients in Drinking Water; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Hasson, D.; Semiat, R.; Shemer, H.; Priel, M.; Nadav, N. Simple process for hardening desalinated water with Mg2+ ions. Desalination Water Treat. 2013, 51, 924–929. [Google Scholar] [CrossRef]
- Caraballo, M.A.; Rötting, T.S.; Silva, V. Implementation of an MgO-based metal removal step in the passive treatment system of Shilbottle, UK: Column experiments. J. Hazard. Mater. 2010, 181, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Cortina, J.-L.; Lagreca, I.; De Pablo, J.; Cama, J.; Ayora, C. Passive in situ remediation of metal-polluted water with caustic magnesia: Evidence from column experiments. Environ. Sci. Technol. 2003, 37, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Courcelles, B.; Modaressi-Farahmand-Razavi, A.; Gouvenot, D.; Esnault-Filet, A. Influence of precipitates on hydraulic performance of permeable reactive barrier filters. Int. J. Géoméch. 2011, 11, 142–151. [Google Scholar] [CrossRef]
- Macías, F.; Caraballo, M.A.; Rötting, T.S.; Pérez-López, R.; Nieto, J.M.; Ayora, C. From highly polluted Zn-rich acid mine drainage to non-metallic waters: Implementation of a multi-step alkaline passive treatment system to remediate metal pollution. Sci. Total. Environ. 2012, 433, 323–330. [Google Scholar] [CrossRef]
- Navarro, A.; Chimenos, J.M.; Muntaner, D.; Fernández, A.I. Permeable reactive barriers for the removal of heavy metals: Lab-Scale experiments with low-grade magnesium oxide. Ground Water Monit. Remediat. 2006, 26, 142–152. [Google Scholar] [CrossRef]
- Oustadakis, P.; Agatzini-Leonardou, S.; Tsakiridis, P. Nickel and cobalt precipitation from sulphate leach liquor using MgO pulp as neutralizing agent. Miner. Eng. 2006, 19, 1204–1211. [Google Scholar] [CrossRef]
- Rötting, T.S.; Cama, J.; Ayora, C.; Cortina, J.-L.; De Pablo, J. Use of caustic magnesia to remove cadmium, nickel, and cobalt from water in passive treatment systems: Column experiments. Environ. Sci. Technol. 2006, 40, 6438–6443. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.E.; Tallman, D.N.; Khalafalla, S.E. Mineral processing water treatment using magnesium oxide. Environ. Prog. 1984, 3, 136–141. [Google Scholar] [CrossRef]
- Teringo Iii, J. Magnesium hydroxide reduces sludge/improves filtering. Pollut. Eng. 1987, 19, 78–83. [Google Scholar]
- Borgohain, X.; Boruah, A.; Sarma, G.K.; Rashid, H. Rapid and extremely high adsorption performance of porous MgO nanostructures for fluoride removal from water. J. Mol. Liq. 2020, 305, 112799. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z.; Bai, Y.; Dong, K.; Li, Y.; Zhuang, L. Efficient arsanilic acid removal from water via reversible phase transition in a cyclic adsorption process based on reactivated MgO. J. Hazard. Mater. Lett. 2020, 1, 100006. [Google Scholar] [CrossRef]
- Abuhatab, S.; El-Qanni, A.; Al-Qalaq, H.; Hmoudah, M.; Al-Zerei, W. Effective adsorptive removal of Zn2+, Cu2+, and Cr3+ heavy metals from aqueous solutions using silica-based embedded with NiO and MgO nanoparticles. J. Environ. Manag. 2020, 268, 110713. [Google Scholar] [CrossRef]
- Chen, T.; Wei, Y.; Yang, W.; Liu, C. Highly efficient As(III) removal in water using millimeter-sized porous granular MgO-biochar with high adsorption capacity. J. Hazard. Mater. 2021, 416, 125822. [Google Scholar] [CrossRef]
- Zhou, J.; Xia, Y.; Gong, Y.; Li, W.; Li, Z. Efficient natural organic matter removal from water using Nano-MgO coupled with microfiltration membrane separation. Sci. Total Environ. 2019, 711, 135120. [Google Scholar] [CrossRef]
- Haddad, M.; Barbeau, B. Hybrid hollow fiber nanofiltration–calcite contactor: A novel point-of-entry treatment for removal of dissolved Mn, Fe, NOM and hardness from domestic groundwater supplies. Membranes 2019, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Withers, A. Options for recarbonation, remineralisation and disinfection for desalination plants. Desalination 2005, 179, 11–24. [Google Scholar] [CrossRef]
- Shand, M.A. Physical and chemical properties of magnesium oxide. In The Chemistry and Technology of Magnesia; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 121–131. [Google Scholar] [CrossRef]
- Ropp, R.C. Chapter 3-Group 16 (O, S, Se, Te) Alkaline Earth Compounds. In Encyclopedia of the Alka-Line Earth Compounds; Ropp, R.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 105–197. [Google Scholar]
- Birchal, V.; Rocha, S.D.F.; Ciminelli, V. The effect of magnesite calcination conditions on magnesia hydration. Miner. Eng. 2000, 13, 1629–1633. [Google Scholar] [CrossRef]
- Aphane, M.E.; Van Der Merwe, E.M.; Strydom, C.A.; Van Der Merwe, E. Influence of hydration time on the hydration of MgO in water and in a magnesium acetate solution. J. Therm. Anal. Calorim. 2009, 96, 987–992. [Google Scholar] [CrossRef]
- Canterford, J.H. Magnesia—An important industrial mineral: A review of processing options and uses. Miner. Process. Extr. Met. Rev. 1985, 2, 57–104. [Google Scholar] [CrossRef]
- Eubank, W.R. Calcination studies of magnesium oxides. J. Am. Ceram. Soc. 1951, 34, 225–229. [Google Scholar] [CrossRef]
- Kato, Y.; Yamashita, N.; Kobayashi, K.; Yoshizawa, Y. Kinetic study of the hydration of magnesium oxide for a chemical heat pump. Appl. Therm. Eng. 1996, 16, 853–862. [Google Scholar] [CrossRef]
- Liu, B.; Thomas, P.S.; Ray, A.S.; Guerbois, J.P. A TG analysis of the effect of calcination conditions on the properties of reactive magnesia. J. Therm. Anal. Calorim. 2007, 88, 145–149. [Google Scholar] [CrossRef]
- Mo, L.; Deng, M.; Tang, M. Effects of calcination condition on expansion property of MgO-type expansive agent used in cement-based materials. Cem. Concr. Res. 2010, 40, 437–446. [Google Scholar] [CrossRef]
- Vermilyea, D.A. The Dissolution of MgO and Mg(OH)2 in Aqueous Solutions. J. Electrochem. Soc. 1969, 116, 1179–1183. [Google Scholar] [CrossRef]
- Birchal, V.S.; Rocha, S.D.F.; Mansur, M.B.; Ciminelli, V.S.T. A simplified mechanistic analysis of the hydration of magnesia. Can. J. Chem. Eng. 2001, 79, 507–511. [Google Scholar] [CrossRef]
- Kitamura, A.; Onizuka, K.; Tanaka, K. Hydration Characteristics of Magnesia. Taikabutsu Overseas 1996, 16, 3–11. [Google Scholar]
- Smithson, G.L.; Bakhshi, N.N. The kinetics and mechanism of the hydration of magnesium oxide in a batch reactor. Can. J. Chem. Eng. 1969, 47, 508–513. [Google Scholar] [CrossRef]
- Rocha, S.D.; Mansur, M.B.; Ciminelli, V.S. Kinetics and mechanistic analysis of caustic magnesia hydration. J. Chem. Technol. Biotechnol. 2004, 79, 816–821. [Google Scholar] [CrossRef]
- Feitknecht, W.; Braun, H. Der Mechanismus der Hydratation von Magnesiumoxid mit Wasserdampf. Helv. Chim. Acta 1967, 50, 2040–2053. [Google Scholar] [CrossRef]
- Glasson, D.R. Reactivity of lime and related oxides. IX. Hydration of magnesium oxide. J. Appl. Chem. 1963, 13, 119–123. [Google Scholar] [CrossRef]
- Tang, X.; Guo, L.; Chen, C.; Liu, Q.; Li, T.; Zhu, Y. The analysis of magnesium oxide hydration in three-phase reaction system. J. Solid State Chem. 2014, 213, 32–37. [Google Scholar] [CrossRef]
- Shand, M.A. Water and Wastewater Applications for Magnesia Products; John Wiley & Sons: Hoboken, NJ, USA, 2006; pp. 155–177. [Google Scholar] [CrossRef]
- Langmuir, D. Aqueous Environmental Geochemistry; Prentice Hall: Upper Saddle River, NJ, USA, 1997. [Google Scholar]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Johnson, W.A.; Mehl, R.F. Reaction kinetics in processes of nucleation and growth. American Institute of Mining and Metallurgical Engineers–Transactions. Am. Inst. Min. Metal. Petro. Eng. 1939, 135, 416–442. [Google Scholar]
- Kolmogorov, A.N. On the statistical theory of the crystallization of metals. Bull. Acad. Sci. USSR Math. Ser. 1937, 1, 355–359. [Google Scholar]
- Jägle, E.A.; Mittemeijer, E. The kinetics of grain-boundary nucleated phase transformations: Simulations and modelling. Acta Mater. 2011, 59, 5775–5786. [Google Scholar] [CrossRef]
- Cahn, J.W. The kinetics of grain boundary nucleated reactions. Acta Met. 1956, 4, 449–459. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Tian, Q.; Zhang, S. Modeling hydration process of magnesia based on nucleation and growth theory: The isothermal calorimetry study. Thermochim. Acta 2012, 550, 27–32. [Google Scholar] [CrossRef]
- Thomas, J. A new approach to modeling the nucleation and growth kinetics of tricalcium silicate hydration. J. Am. Ceram. Soc. 2007, 90, 3282–3288. [Google Scholar] [CrossRef]
- Thomas, J.J.; Musso, S.; Prestini, I. Kinetics and Activation Energy of Magnesium Oxide Hydration. J. Am. Ceram. Soc. 2013, 97, 275–282. [Google Scholar] [CrossRef]
- Fruhwirth, O.; Herzog, G.; Hollerer, I.; Rachetti, A. Dissolution and hydration kinetics of MgO. Surf. Technol. 1985, 24, 301–317. [Google Scholar] [CrossRef]
- Jones, C.F.; Segall, R.L.; Smart, R.S.C.; Turner, P.S. Initial dissolution kinetics of ionic oxides. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1981, 374, 141–153. [Google Scholar] [CrossRef]
- Jordan, G.; Higgins, S.R.; Eggleston, C.M. Dissolution of the periclase (001) surface; a scanning force microscope study. Am. Miner. 1999, 84, 144–151. [Google Scholar] [CrossRef]
- Segall, R.L.; Smart, R.S.C.; Turner, P.S. Ionic oxides: Distinction between mechanisms and surface roughening effects in the dissolution of magnesium oxide. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1978, 74, 2907–2912. [Google Scholar] [CrossRef]
- Wogelius, R.A.; Refson, K.; Fraser, D.G.; Grime, G.W.; Goff, J.P. Periclase surface hydroxylation during dissolution. Geochim. Cosmochim. Acta 1995, 59, 1875–1881. [Google Scholar] [CrossRef]
- Eisenlohr, L.; Meteva, K.; Gabrovšek, F.; Dreybrodt, W. The inhibiting action of intrinsic impurities in natural calcium carbonate minerals to their dissolution kinetics in aqueous H2O–CO2 solutions. Geochim. Cosmochim. Acta 1999, 63, 989–1001. [Google Scholar] [CrossRef]
- Rotting, T.S.; Ayora, C.; Carrera, J. Improved passive treatment of high Zn and Mn Concentrations using caustic magnesia (MgO): Particle size effects. Environ. Sci. Technol. 2008, 42, 9370–9377. [Google Scholar] [CrossRef]
- Mejias, J.A.; Berry, A.; Refson, K.; Fraser, D. The kinetics and mechanism of MgO dissolution. Chem. Phys. Lett. 1999, 314, 558–563. [Google Scholar] [CrossRef]
- Kameda, T.; Yamamoto, Y.; Kumagai, S.; Yoshioka, T. Effect of the specific surface area of MgO on the treatment of boron and fluorine. Appl. Water Sci. 2020, 10, 104. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Nield, D.A.; Bejan, A. Convection in Porous Media, 5th ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Jin, F.; Al-Tabbaa, A. Characterisation of different commercial reactive magnesia. Adv. Cem. Res. 2014, 26, 101–113. [Google Scholar] [CrossRef]
- Blesa, M.A. Chemical Dissolution of Metal Oxides; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar] [CrossRef]
- Gorichev, I.G.; Kipriyanov, N.A. Regular kinetic features of the dissolution of metal oxides in acidic media. Russ. Chem. Rev. 1984, 53, 1039–1061. [Google Scholar] [CrossRef]
- Stumm, W. Reactivity at the mineral-water interface: Dissolution and inhibition. Colloids Surf. A Physicochem. Eng. Asp. 1997, 120, 143–166. [Google Scholar] [CrossRef]
- Bratton, R.J.; Brindley, G.W. Kinetics of vapour phase hydration of magnesium oxide. Part 2.—Dependence on temperature and water vapour pressure. Trans. Faraday Soc. 1965, 61, 1017–1025. [Google Scholar] [CrossRef]
- Layden, G.K.; Brindley, G.W. Kinetics of Vapor-Phase hydration of magnesium oxide. J. Am. Ceram. Soc. 1963, 46, 518–522. [Google Scholar] [CrossRef]
- Amaral, L.; Oliveira, I.; Salomão, R.; Frollini, E.; Pandolfelli, V. Temperature and common-ion effect on magnesium oxide (MgO) hydration. Ceram. Int. 2010, 36, 1047–1054. [Google Scholar] [CrossRef]
- Fedoročková, A.; Raschman, P. Effects of pH and acid anions on the dissolution kinetics of MgO. Chem. Eng. J. 2008, 143, 265–272. [Google Scholar] [CrossRef]
- Evans, R.L.; Clair, H.W.S. Carbonation of aqueous suspensions containing magnesium oxides or hydroxides. Ind. Eng. Chem. 1949, 41, 2814–2817. [Google Scholar] [CrossRef]
- Brown, G.E.; Henrich, V.E.; Casey, W.H.; Clark, D.L.; Eggleston, C.; Felmy, A.; Goodman, D.W.; Grätzel, M.; Maciel, G.; McCarthy, M.I.; et al. Metal oxide surfaces and their interactions with aqueous solutions and microbial organisms. Chem. Rev. 1998, 99, 77–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokrovsky, O.; Schott, J.; Castillo, A. Kinetics of brucite dissolution at 25 °C in the presence of organic and inorganic ligands and divalent metals. Geochim. Cosmochim. Acta 2005, 69, 905–918. [Google Scholar] [CrossRef]
- Pourahmad, H.; Haddad, M.; Claveau-Mallet, D.; Barbeau, B. Impact of media coating on simultaneous manganese removal and remineralization of soft water via calcite contactor. Water Res. 2019, 161, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.T.; Jones, M.H.; Flann, R.C.; Hart, R.L.; Strode, P.R.; Urban, A.J.; Tassios, S. Application of Caustic Calcined Magnesia to Effluent Treatment. In Transactions of the Institutions of Mining and Metallurgy; Section C; 1990; Volume 161, pp. 117–124. Available online: https://publications.csiro.au/rpr/pub?list=BRO&pid=procite:ee52c57d-be71-4599-8c89-090481a3a270 (accessed on 29 December 2021).
- Balintova, M.; Petrilakova, A. Study of pH influence on selective precipitation of heavy metals from acid mine drainage. Chem. Eng. Trans. 2011, 25, 1–6. [Google Scholar]
- Cao, C.-Y.; Qu, J.; Wei, F.; Liu, H.; Song, W.-G. Superb adsorption capacity and mechanism of flowerlike magnesium oxide nanostructures for lead and cadmium ions. ACS Appl. Mater. Interfaces 2012, 4, 4283–4287. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Woo, N.C.; Jang, M.; Cannon, F.S.; Snyder, S.A. Magnesium oxide impregnated polyurethane to remove high levels of manganese cations from water. Sep. Purif. Technol. 2014, 136, 184–189. [Google Scholar] [CrossRef]
- Plummer, L.N.; Wigley, T.M.L.; Parkhurst, D.L. The kinetics of calcite dissolution in CO2—Water systems at 5 degrees to 60 degrees C and 0.0 to 1.0 atm CO 2. Am. J. Sci. 1978, 278, 179–216. [Google Scholar] [CrossRef]
- Yamauchi, V.; Tanaka, K.; Hattori, K.; Kondo, M.; Ukawa, N. Remineralization of desalinated water by limestone dissolution filter. Desalination 1987, 66, 365–383. [Google Scholar] [CrossRef]
- Buhmann, D.; Dreybrodt, W. The kinetics of calcite dissolution and precipitation in geologically relevant situations of karst areas: 1. Open system. Chem. Geol. 1985, 48, 189–211. [Google Scholar] [CrossRef]
- Chou, L.; Garrels, R.M.; Wollast, R. Comparative study of the kinetics and mechanisms of dissolution of carbonate minerals. Chem. Geol. 1989, 78, 269–282. [Google Scholar] [CrossRef]
- Dreybrodt, W.; Lauckner, J.; Zaihua, L.; Svensson, U.; Buhmann, D. The kinetics of the reaction CO2 + H2O → H+ + HCO3−; as one of the rate limiting steps for the dissolution of calcite in the system H2O CO2 CaCO3. Geochim. Cosmochim. Acta 1996, 60, 3375–3381. [Google Scholar] [CrossRef]
- Svensson, U.; Dreybrodt, W. Dissolution kinetics of natural calcite minerals in CO2-water systems approaching calcite equilibrium. Chem. Geol. 1992, 100, 129–145. [Google Scholar] [CrossRef]
- Hasson, D.; Bendrihem, O. Modeling remineralization of desalinated water by limestone dissolution. Desalination 2006, 190, 189–200. [Google Scholar] [CrossRef]
- Morse, J.W.; Arvidson, A.R.S.; Lüttge, A. Calcium carbonate formation and dissolution. Chem. Rev. 2007, 107, 342–381. [Google Scholar] [CrossRef]
- Plummer, L.N.; Parkhurst, D.L.; Wigley, T.M.L. Critical review of the kinetics of calcite dissolution and precipitation. In Chemical Modeling in Aqueous Systems; American Chemical Society: Washington, DC, USA, 1979; Volume 93, pp. 537–573. [Google Scholar]
- Ginocchio, J. Protection against corrosion in drinking water distribution systems. Anti-Corros. Methods Mater. 1985, 32, 14–16. [Google Scholar] [CrossRef]
- Luptáková, A.; Derco, J. Improving drinking water quality by remineralisation. Acta Chim. Slov. 2015, 62, 859–866. [Google Scholar] [CrossRef]
- Machel, H.G. Sedimentary Rocks|Dolomites. In Encyclopedia of Geology; Selley, R.C., Cocks, L.R.M., Plimer, I.R., Eds.; Elsevier: Oxford, UK, 2005; pp. 79–94. [Google Scholar]
- Liu, Z.; Yuan, D.; Dreybrodt, W. Comparative study of dissolution rate-determining mechanisms of limestone and dolomite. Environ. Earth Sci. 2005, 49, 274–279. [Google Scholar] [CrossRef]
- Morse, J.W.; Arvidson, R. The dissolution kinetics of major sedimentary carbonate minerals. Earth Rev. 2002, 58, 51–84. [Google Scholar] [CrossRef]
- Pishtshev, A.; Karazhanov, S.Z.; Klopov, M. Materials properties of magnesium and calcium hydroxides from first-principles calculations. Comput. Mater. Sci. 2014, 95, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Hiortdahl, K.M.; Borden, R.C. Enhanced reductive dechlorination of tetrachloroethene dense nonaqueous phase liquid with EVO and Mg(OH)2. Environ. Sci. Technol. 2013, 48, 624–631. [Google Scholar] [CrossRef]
- Kameda, T.; Takeuchi, H.; Yoshioka, T. Preparation of organic acid anion-modified magnesium hydroxides by coprecipitation: A novel material for the uptake of heavy metal ions from aqueous solutions. J. Phys. Chem. Solids 2009, 70, 1104–1108. [Google Scholar] [CrossRef]
- Caraballo, M.A.; Rötting, T.S.; Macías, F.; Nieto, J.M.; Ayora, C. Field multi-step limestone and MgO passive system to treat acid mine drainage with high metal concentrations. Appl. Geochem. 2009, 24, 2301–2311. [Google Scholar] [CrossRef]
- Schwartz, R.; Shemer, H.; Hasson, D.; Semiat, R. Design model for magnesium ions re-mineralization of desalinated water by dissolution of magnesia pellets. Desalination 2015, 373, 10–15. [Google Scholar] [CrossRef]
- Huang, H.-H.; Liu, Q. Bench-Scale chemical treatability study of the berkeley pit water. In Emerging Technologies in Hazardous Waste Management V; American Chemical Society: Washington, DC, USA, 1995; Volume 607, pp. 196–209. [Google Scholar] [CrossRef]
- Younger, P.; Banwart, S.; Hedin, R. Mine water, hydrology, pollution, remediation. In Environmental Pollution; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2002; Volume 5. [Google Scholar]
- Skousen, J.; Zipper, C.; Rose, A.; Ziemkiewicz, P.F.; Nairn, R.; McDonald, L.M.; Kleinmann, R.L. Review of passive systems for acid mine drainage treatment. Mine Water Environ. 2016, 36, 133–153. [Google Scholar] [CrossRef] [Green Version]
- de Repentigny, C.; Courcelles, B.; Zagury, G.J. Spent MgO-carbon refractory bricks as a material for permeable reactive barriers to treat a nickel- and cobalt-contaminated groundwater. Environ. Sci. Pollut. Res. 2018, 25, 23205–23214. [Google Scholar] [CrossRef]
- de Repentigny, C.; Zagury, G.J.; Courcelles, B. Modeling of the clogging in a MgO column used to treat a Ni- and Co-contaminated water and performance prediction for a centripetal radial column. Chemosphere 2019, 236, 124307. [Google Scholar] [CrossRef]
- Chilingar, G.V.; Mourhatch, R.; Al-Qahtani, G.D. The Fundamentals of Corrosion and Scaling for Petroleum & Environmental Engineers; Chapter 6-Scaling; Gulf Publishing Company: Houston, TX, USA, 2008; pp. 117–139. [Google Scholar]
- Larson, T.E.; Lane, R.W.; Neff, C.H. Stabilization of magnesium hydroxide in the solids-contact process. J.-Am. Water Works Assoc. 1959, 51, 1551–1558. [Google Scholar] [CrossRef]
- Bahadori, A. Prediction of scale formation in calcium carbonate aqueous phase for water treatment and distribution systems. Water Qual. Res. J. 2010, 45, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Health Canada. Guidelines for Canadian Drinking Water Quality: Guideline Technical Document—Manganese; Health Canada: Ottawa, ON, Canada, 2019; Available online: https://www.canada.ca/en/health-canada/services/publications/healthy-living/guidelines-canadian-drinking-water-quality-guideline-technical-document-lead/guidance-document.html (accessed on 29 December 2021).
- Voutchkov, N. Desalination Engineering-Planning and Design; McGraw Hill: New York, NY, USA, 2012. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Media | Purity (%) | Specific Gravity (g/cm3) | Bulk Density (kg/cm3) | Surface Area (m2/g) |
|---|---|---|---|---|
| CorosexTM | 98.0 | 3.60 | 1200 | 0.39 |
| Mineral | Formula | Ksp Expression | −log Ksp |
|---|---|---|---|
| Brucite | Mg(OH)2 | [Mg2+] [OH−]2 | 11.16 |
| Calcite | CaCO3 | [Ca2+] [CO32−] | 8.48 |
| Authors | Modeled Phenomenon | Validation |
|---|---|---|
| Dissolution in water | ||
| 59, 14 | A, B | |
| 10, 99 | B | |
| Particle evolution and passivation | ||
| 59 | None | |
| Contaminant removal | ||
| 59, 14 | A, B | |
| 13 | B | |
| Permeability and hydraulic conductivity loss | ||
| 13 | C | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymoniak, L.; Claveau-Mallet, D.; Haddad, M.; Barbeau, B. Application of Magnesium Oxide Media for Remineralization and Removal of Divalent Metals in Drinking Water Treatment: A Review. Water 2022, 14, 633. https://doi.org/10.3390/w14040633
Szymoniak L, Claveau-Mallet D, Haddad M, Barbeau B. Application of Magnesium Oxide Media for Remineralization and Removal of Divalent Metals in Drinking Water Treatment: A Review. Water. 2022; 14(4):633. https://doi.org/10.3390/w14040633
Chicago/Turabian StyleSzymoniak, Lena, Dominique Claveau-Mallet, Maryam Haddad, and Benoit Barbeau. 2022. "Application of Magnesium Oxide Media for Remineralization and Removal of Divalent Metals in Drinking Water Treatment: A Review" Water 14, no. 4: 633. https://doi.org/10.3390/w14040633
APA StyleSzymoniak, L., Claveau-Mallet, D., Haddad, M., & Barbeau, B. (2022). Application of Magnesium Oxide Media for Remineralization and Removal of Divalent Metals in Drinking Water Treatment: A Review. Water, 14(4), 633. https://doi.org/10.3390/w14040633