
New Sets of Primers for DNA Identification of Non-Indigenous Fish Species in the Volga-Kama Basin (European Russia)

 ,
, 

Abstract
:1. Introduction
- (1)
- Ponto-Caspian marine faunistic complexes expanding their distribution ranges north: Benthophilus stellatus (Sauvage, 1874), Clupeonella cultriventris (Nordmann, 1840), Knipowitschia longecaudata (Kessler, 1877), Neogobius fluviatilis (Pallas, 1814), Neogobius melanostomus (Pallas, 1814), Ponticola gorlap (Iljin, 1949), Ponticola syrman (Nordmann, 1840), Proterorhinus semipellucidus (Kessler, 1877), Syngnathus abaster Risso, 1827;
- (2)
- Arctic freshwater faunistic complexes expanding their distribution ranges south: Osmerus eperlanus (Linnaeus, 1758), Coregonus albula (Linnaeus, 1758), and
- (3)
- escaped aquaculture specimens or deliberate introductions: Acipenser spp., Anguilla anguilla (Linnaeus, 1758), Ctenopharyngodon idella (Valenciennes, 1844), Hypophthalmichthys molitrix (Valenciennes, 1844), Ictalurus punctatus (Rafinesque, 1818), Perccottus glenii Dybowski, 1877.
2. Materials and Methods
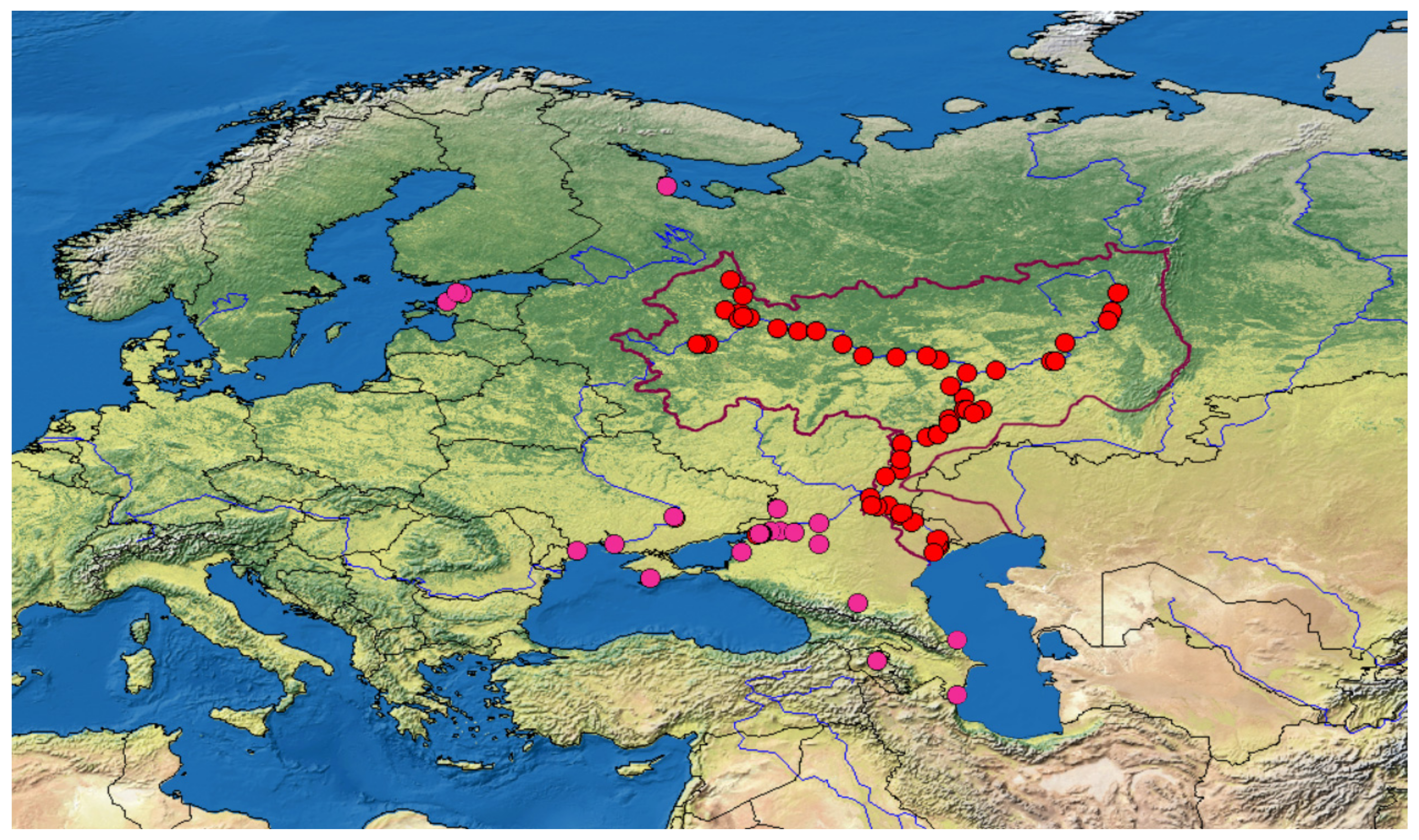
2.1. Sampling
2.2. Primer Design
2.3. DNA Extraction, PCR Amplification and Sequencing
2.4. Alignment, Nucleotide Diversity, and Phylogenetic Analysis
2.5. Species Delimitation
3. Results
3.1. Comparison of the Effectiveness of Different Methods of DNA Extraction and Purification of the PCR Products, and the Amplification Efficiency of Different Primer Sets
3.2. Polymorphism and Nucleotide Diversity of the Studied Loci
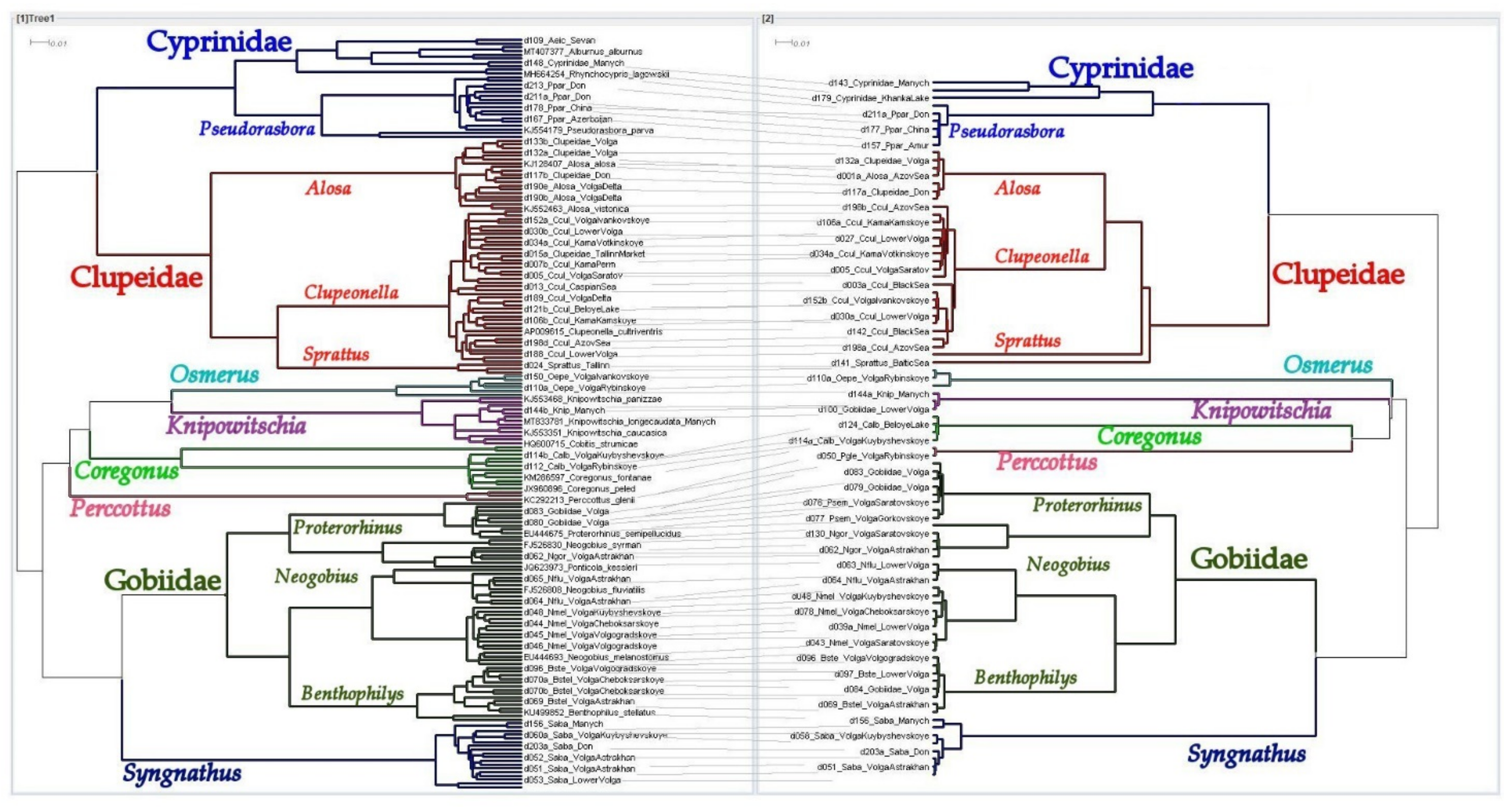
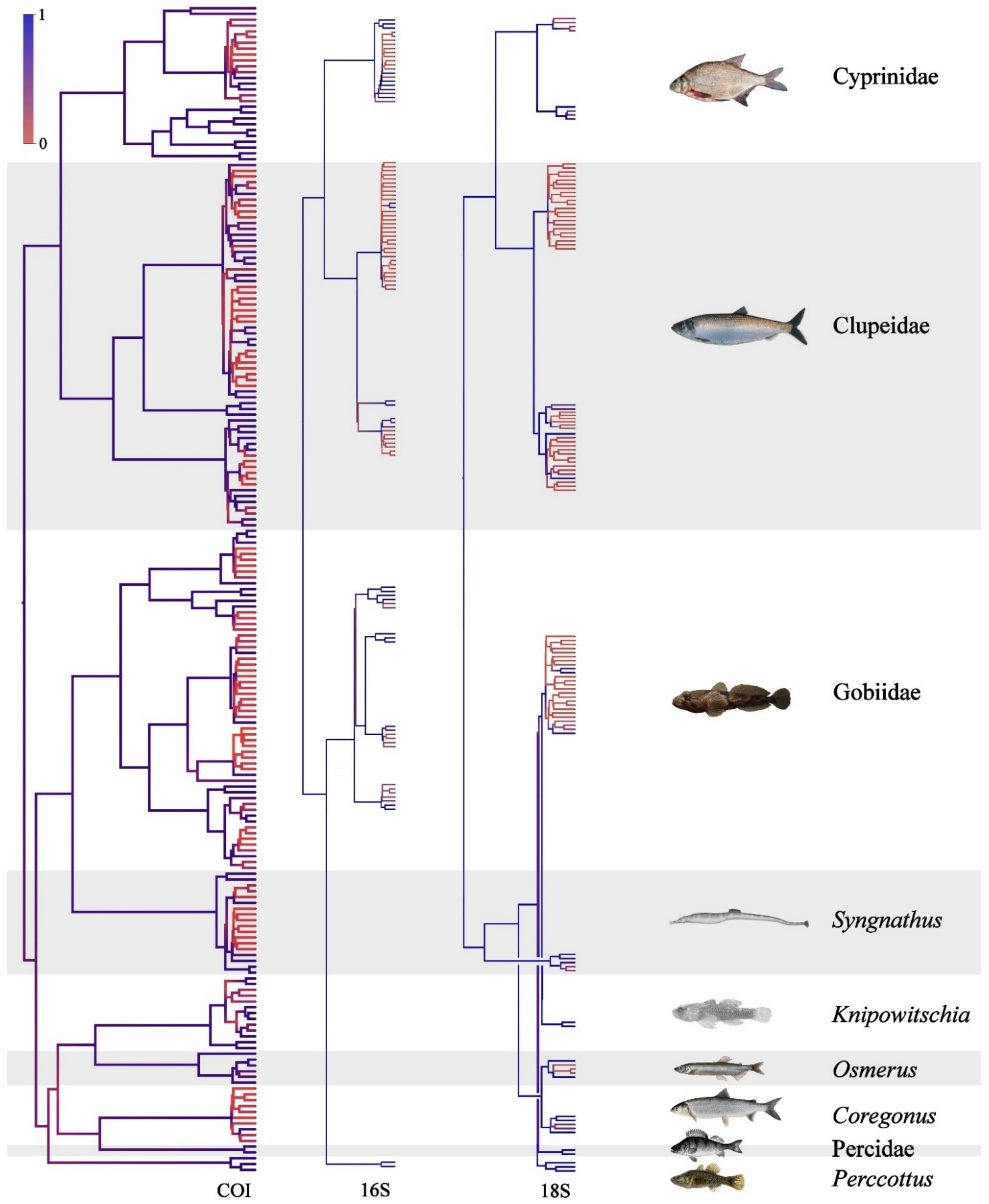
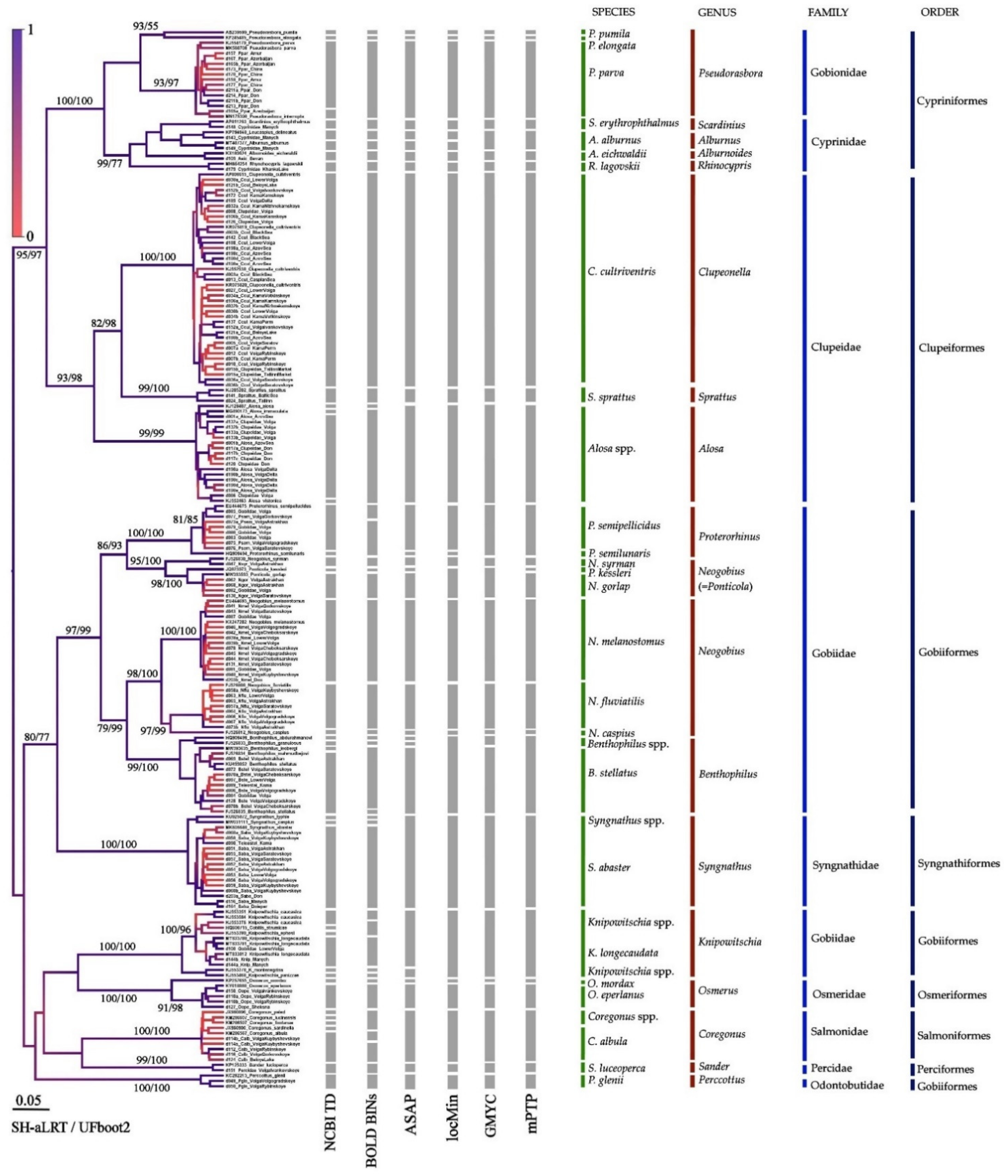
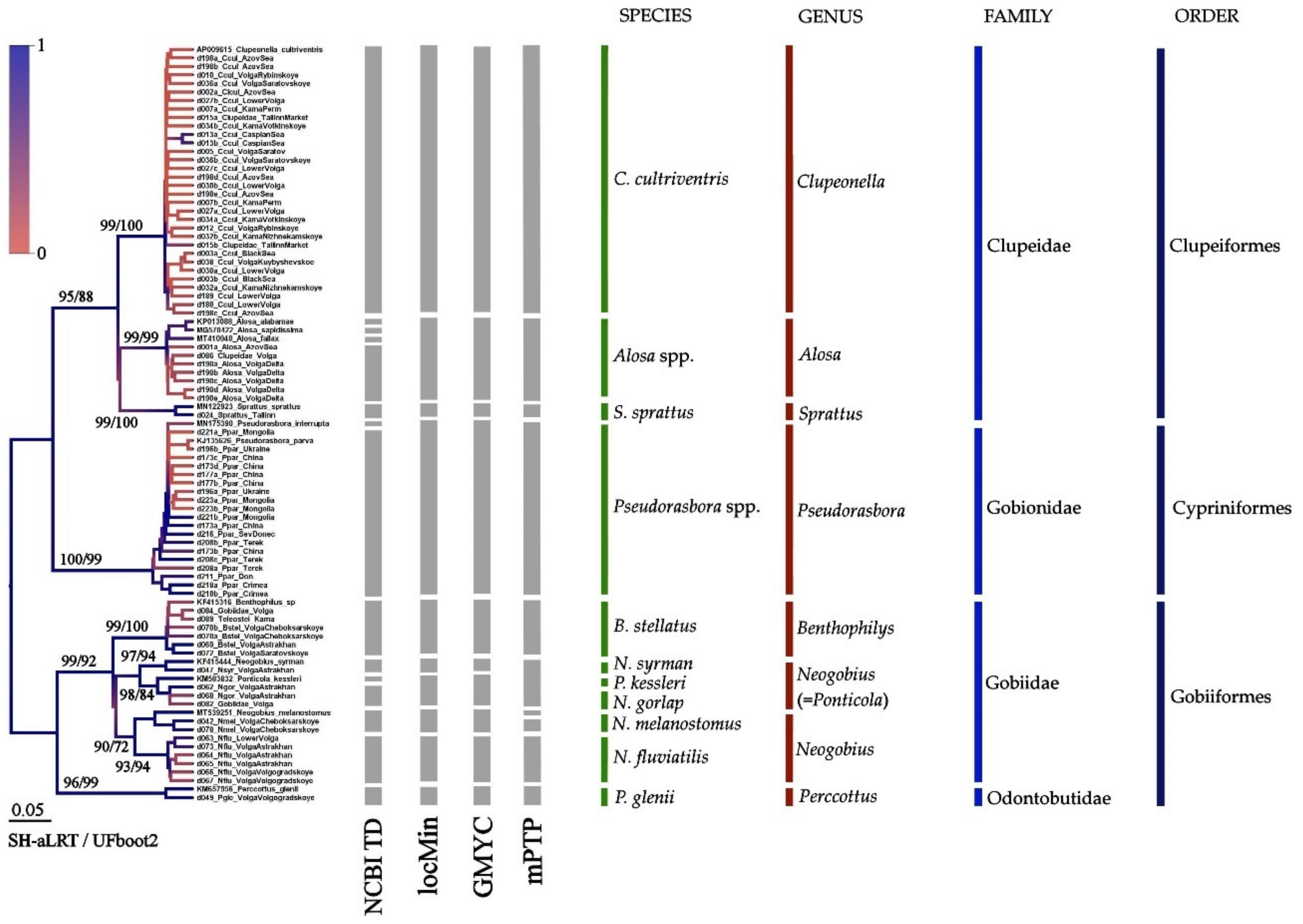
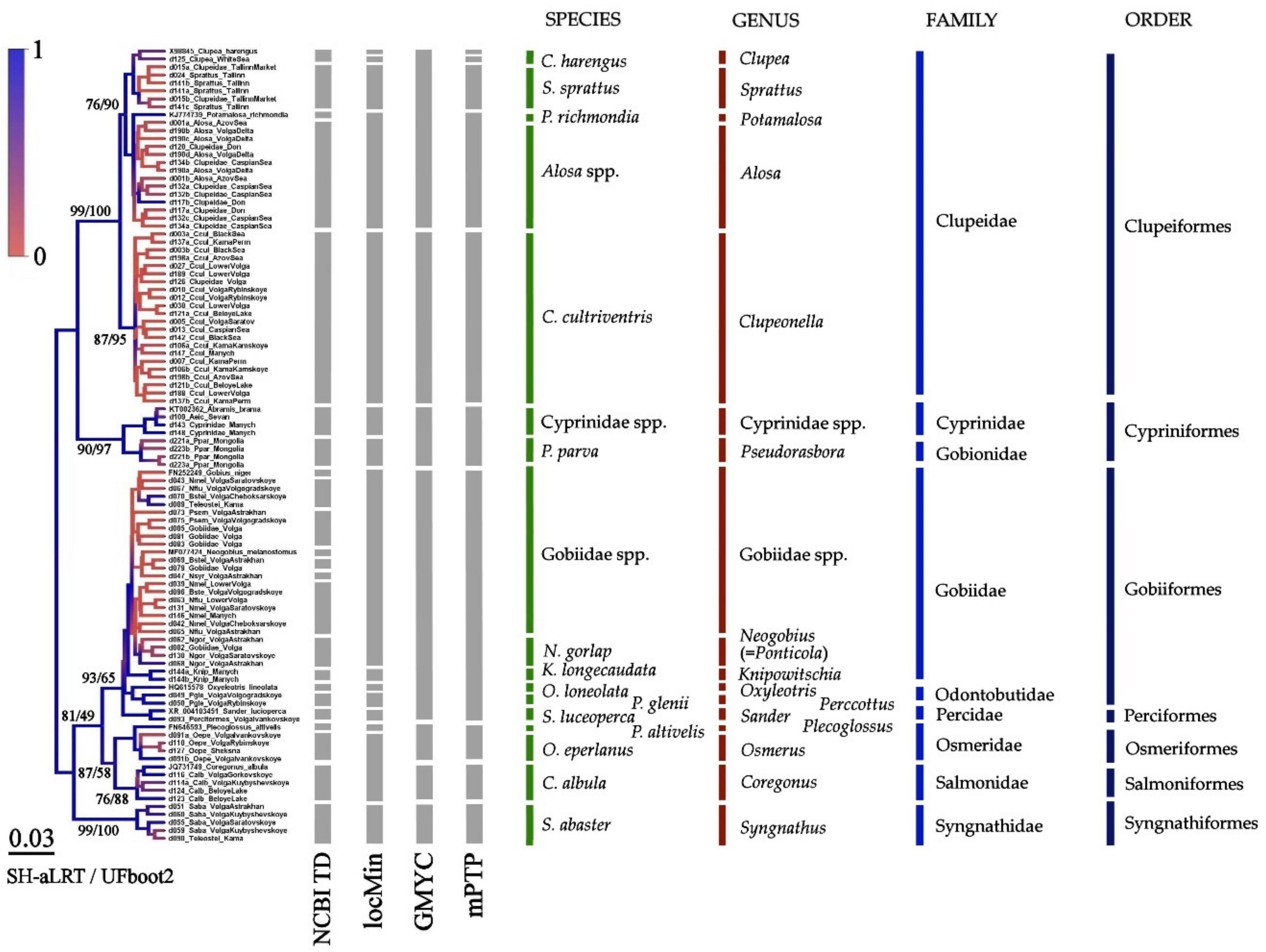
3.3. Results of Species Differentiation Based on DNA Analysis
4. Discussion
4.1. Primers’ Efficiency
4.2. Application of New Primer Sets for the Detection of Non-Indigenous Fish Species in the Volga-Kama Basin
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. DNA Extraction Protocol
Appendix A.2. PCR Product Purification Protocol
Appendix B
References
- Elton, C.S. The Ecology of Invasions by Animals and Plants: New Edition; University of Chicago Press: Chicago, IL, USA; London, UK, 2000; ISBN 0226206386. [Google Scholar]
- Robertson, P.A.; Mill, A.; Novoa, A.; Jeschke, J.M.; Essl, F.; Gallardo, B.; Geist, J.; Jarić, I.; Lambin, X.; Musseau, C.; et al. A proposed unified framework to describe the management of biological invasions. Biol. Invasions 2020, 22, 2633–2645. [Google Scholar] [CrossRef]
- Lodge, D.M.; Williams, S.; MacIsaac, H.J.; Hayes, K.R.; Leung, B.; Reichard, S.; Mack, R.N.; Moyle, P.B.; Smith, M.; Andow, D.A.; et al. Biological invasions: Recommendations for U.S. policy and management. Ecol. Appl. 2006, 16, 2035–2054. [Google Scholar] [CrossRef] [Green Version]
- Makhrov, A.A.; Karabanov, D.P.; Koduhova, Y.V. Genetic methods for the control of alien species. Russ. J. Biol. Invasions 2014, 5, 194–202. [Google Scholar] [CrossRef]
- McGeoch, M.A.; Genovesi, P.; Bellingham, P.J.; Costello, M.J.; McGrannachan, C.; Sheppard, A. Prioritizing species, pathways, and sites to achieve conservation targets for biological invasion. Biol. Invasions 2016, 18, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Miralles, L.; Ibabe, A.; González, M.; García-Vázquez, E.; Borrell, Y.J. “If you know the enemy and know yourself”: Addressing the problem of biological invasions in ports through a new NIS invasion threat score, routine monitoring, and preventive action plans. Front. Mar. Sci. 2021, 8, e633118. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Marine Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Sharma, P.; Kobayashi, T. Are “universal” DNA primers really universal? J. Appl. Genet. 2014, 55, 485–496. [Google Scholar] [CrossRef]
- Jacquot, S.; Chartoire, N.; Piguet, F.; Hérault, Y.; Pavlovic, G. Optimizing PCR for mouse genotyping: Recommendations for reliable, rapid, cost effective, robust and adaptable to high-throughput genotyping protocol for any type of mutation. Curr. Protoc. Mouse Biol. 2019, 9, e65. [Google Scholar] [CrossRef] [Green Version]
- Kadri, K. Polymerase Chain Reaction (PCR): Principle and applications. In Synthetic Biology—New Interdisciplinary Science; Nagpal, M.L., Boldura, O.-M., Balta, C., Enany, S., Eds.; IntechOpen: London, UK, 2020; pp. 1–17. ISBN 978-1-78984-089-6. [Google Scholar]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D.N. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B 2005, 360, 1847–1857. [Google Scholar] [CrossRef]
- Ivanova, N.V.; Zemlak, T.S.; Hanner, R.H.; Hebert, P.D.N. Universal primer cocktails for fish DNA barcoding. Mol. Ecol. Notes 2007, 7, 544–548. [Google Scholar] [CrossRef]
- Lemmon, G.H.; Gardner, S.N. Predicting the sensitivity and specificity of published real-time PCR assays. Ann. Clin. Microbiol. Antimicrob. 2008, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpartida-Cardenas, K.; Rodriguez-Manzano, J.; Yu, L.-S.; Delves, M.J.; Nguon, C.; Chotivanich, K.; Baum, J.; Georgiou, P. Allele-specific isothermal amplification method using unmodified self-stabilizing competitive primers. Anal. Chem. 2018, 90, 11972–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.C.; Turner, R.J. Lessons and considerations for the creation of universal primers targeting non-conserved, horizontally mobile genes. Appl. Environ. Microbiol. 2020, 87, e02181-20. [Google Scholar] [CrossRef]
- Frolova, N.L.; Agafonova, S.A.; Kireeva, M.B.; Povalishnikova, E.S.; Pakhomova, O.M. Recent changes of annual flow distribution of the Volga basin rivers. Geogr. Environ. Sustain. 2017, 10, 28–39. [Google Scholar] [CrossRef]
- Schletterer, M.; Shaporenko, S.I.; Kuzovlev, V.V.; Minin, A.E.; van Geest, G.J.; Middelkoop, H.; Gorski, K. The Volga: Management issues in the largest river basin in Europe. River Res. Appl. 2019, 35, 510–519. [Google Scholar] [CrossRef]
- Mineeva, N.; Lazareva, V.; Litvinov, A.; Stepanova, I.; Chuiko, G.; Papchenkov, V.; Korneva, L.; Scherbina, G.; Pryanichnikova, E.; Perova, S.; et al. The Volga River. In Rivers of Europe, 2nd ed.; Tockner, K., Zarfl, C., Robinson, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 27–79. ISBN 9780081026120. [Google Scholar]
- Reshetnyak, O.S.; Nikanorov, A.M.; Bryzgalo, V.A.; Kosmenko, L.S. Anthropogenic transformation of the aquatic ecosystem of the Lower Volga. Water Resour. 2013, 40, 667–676. [Google Scholar] [CrossRef]
- Ellis, E.C. Anthropogenic transformation of the terrestrial biosphere. Philos. Trans. R. Soc. A 2011, 369, 1010–1035. [Google Scholar] [CrossRef]
- Mukharamova, S.; Ivanov, M.; Yermolaev, O. Assessment of anthropogenic pressure on the Volga Federal District territory using river basin approach. Geosciences 2020, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Avakyan, A.B. Volga-Kama cascade reservoirs and their optimal use. Lakes Reservoirs 1998, 3, 113–121. [Google Scholar] [CrossRef]
- Bij de Vaate, A.; Jazdzewski, K.; Ketelaars, H.A.; Gollasch, S.; van der Velde, G. Geographical patterns in range extension of Ponto-Caspian macroinvertebrate species in Europe. Can. J. Fish. Aquat. Sci. 2002, 59, 1159–1174. [Google Scholar] [CrossRef]
- Slynko, Y.V.; Korneva, L.G.; Rivier, I.K.; Papchenkov, V.G.; Scherbina, G.H.; Orlova, M.I.; Therriault, T.W. The Caspian-Volga-Baltic invasion corridor. In Invasive Aquatic Species of Europe. Distribution, Impacts and Management; Leppakoski, E., Gollasch, S., Olenin, S., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 399–411. ISBN 978-90-481-6111-9. [Google Scholar]
- Panov, V.E.; Alexandrov, B.; Arbaciauskas, K.; Binimelis, R.; Copp, G.H.; Grabowski, M.; Lucy, F.; Leuven, R.S.E.W.; Nehring, S.; Paunović, M.; et al. Assessing the risks of aquatic species invasions via European inland waterways: From concepts to environmental indicators. Integr. Environ. Assess. Manag. 2009, 5, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Mordukhai-Boltovskoi, P.D. Caspian Polyphemids in the reservoirs of the Don and Dnieper Rivers. Tr. Inst. Biol. Vnutr. Vod AN SSSR 1965, 8, 37–43. [Google Scholar]
- Karabanov, D.P.; Garibian, P.G.; Bekker, E.I.; Sabitova, R.Z.; Kotov, A.A. Genetic signature of a past anthropogenic transportation of a Far-Eastern endemic Cladoceran (Crustacea: Daphniidae) to the Volga Basin. Water 2021, 13, 2589. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Pavlov, D.D.; Bazarov, M.I.; Borovikova, E.A.; Gerasimov, Y.V.; Kodukhova, Y.V.; Smirnov, A.K.; Stolbunov, I.A. Alien species of fish in the littoral of Volga and Kama reservoirs (Results of complex expeditions of IBIW RAS in 2005–2017). Trans. IBIW RAS 2018, 82, 67–80. [Google Scholar] [CrossRef]
- Lecaudey, L.A.; Schletterer, M.; Kuzovlev, V.V.; Hahn, C.; Weiss, S.J. Fish diversity assessment in the headwaters of the Volga River using environmental DNA metabarcoding. Aquat. Conserv. 2019, 29, 1785–1800. [Google Scholar] [CrossRef] [Green Version]
- Schenekar, T.; Schletterer, M.; Lecaudey, L.A.; Weiss, S.J. Reference databases, primer choice, and assay sensitivity for environmental metabarcoding: Lessons learnt from a re-evaluation of an eDNA fish assessment in the Volga headwaters. River Res. Appl. 2020, 36, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Schenekar, T.; Schletterer, M.; Weiss, S.J. Development of a TaqMan qPCR protocol for detecting Acipenser ruthenus in the Volga headwaters from eDNA samples. Conserv. Genet. Resour. 2020, 12, 395–397. [Google Scholar] [CrossRef] [Green Version]
- Leray, M.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Agudelo, N.; Ranwez, V.; Boehm, J.T.; Machida, R.J. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front. Zool. 2013, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Sultana, S.; Ali, M.E.; Hossain, M.A.M.; Naquiah, N.; Zaidul, I.S.M. Universal mini COI barcode for the identification of fish species in processed products. Food Res. Int. 2018, 105, 19–28. [Google Scholar] [CrossRef]
- Ando, H.; Mukai, H.; Komura, T.; Dewi, T.; Ando, M.; Isagi, Y. Methodological trends and perspectives of animal dietary studies by noninvasive fecal DNA metabarcoding. Environ. DNA 2020, 2, 391–406. [Google Scholar] [CrossRef]
- Cawthorn, D.-M.; Steinman, H.A.; Witthuhn, R.C. Evaluation of the 16S and 12S rRNA genes as universal markers for the identification of commercial fish species in South Africa. Gene 2012, 491, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.Q.; Leasi, F.; Obertegger, U.; Kieneke, A.; Barraclough, T.G.; Fontaneto, D. The widely used small subunit 18S rDNA molecule greatly underestimates true diversity in biodiversity surveys of the meiofauna. Proc. Natl. Acad. Sci. USA 2012, 109, 16208–16212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblitckaya, A.F. Handbook of Juvenile Freshwater Fish; Lyogkaia i Pishchevaia Promyshlennost: Moscow, Russia, 1981. [Google Scholar]
- Kottelat, M.; Freyhof, J. Handbook of European Freshwater Fishes; Publications Kottelat: Cornol, Switzerland, 2007; ISBN 2839902982. [Google Scholar]
- Makeeva, A.P.; Pavlov, D.S.; Pavlov, D.A. Atlas of Larvae and Juveniles of Freshwater Fishes of Russia; KMK Scientific Press Ltd.: Moscow, Russia, 2011; ISBN 978-5-87317-714-1. [Google Scholar]
- Froese, R.; Pauly, D. (Eds.) FishBase. World Wide Web Electronic Publication. Available online: www.fishbase.org (accessed on 15 June 2021).
- Nelson, J.S.; Grande, T.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; ISBN 9781118342336. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; van Aerle, R.; Barrientos, L.; Martinez-Urtaza, J. Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Comput. Struct. Biotechnol. J. 2020, 18, 296–305. [Google Scholar] [CrossRef]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [Green Version]
- Messing, J. New M13 vectors for cloning. Methods Enzymol. 1983, 101, 20–78. [Google Scholar] [CrossRef]
- Jennings, W.B.; Ruschi, P.A.; Ferraro, G.; Quijada, C.C.; Silva-Malanski, A.C.G.; Prosdocimi, F.; Buckup, P.A. Barcoding the Neotropical freshwater fish fauna using a new pair of universal COI primers with a discussion of primer dimers and M13 primer tails. Genome 2019, 62, 77–83. [Google Scholar] [CrossRef]
- Douglas, A.M.; Georgalis, A.M.; Benton, L.R.; Canavan, K.L.; Atchison, B.A. Purification of human leucocyte DNA: Proteinase K is not necessary. Anal. Biochem. 1992, 201, 362–365. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Touchdown Polymerase Chain Reaction (PCR). Cold Spring Harb. Protoc. 2018, 2018, prot095133. [Google Scholar] [CrossRef] [PubMed]
- Makhrov, A.A.; Artamonova, V.S.; Karabanov, D.P. Finding of topmouth gudgeon Pseudorasbora parva (Temminck et Schlegel) (Actinopterygii: Cyprinidae) in the Brahmaputra River basin (Tibetan Plateau, China). Russ. J. Biol. Invasions 2013, 4, 174–179. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; ISBN 0195135857. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Ramirez-Soriano, A.; Ramos-Onsins, S.E.; Rozas, J.; Calafell, F.; Navarro, A. Statistical power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics 2008, 179, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Garrigan, D.; Lewontin, R.; Wakeley, J. Measuring the sensitivity of single-locus “neutrality tests” using a direct perturbation approach. Mol. Biol. Evol. 2010, 27, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Hurvich, C.M.; Tsai, C.-L. Regression and time series model selection in small samples. Biometrika 1989, 76, 297–307. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Le Vinh, S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Soubrier, J.; Steel, M.; Lee, M.S.Y.; Der Sarkissian, C.; Guindon, S.; Ho, S.Y.W.; Cooper, A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012, 29, 3345–3358. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; de Maio, N.; et al. BEAST 2.5 : An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Gernhard, T. The conditioned reconstructed process. J. Theor. Biol. 2008, 253, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. A DNA-based registry for all animal species: The barcode index number (BIN) system. PLoS ONE 2013, 8, e66213. [Google Scholar] [CrossRef] [Green Version]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Collins, R.A.; Boykin, L.M.; Cruickshank, R.H.; Armstrong, K.F. Barcoding’s next top model: An evaluation of nucleotide substitution models for specimen identification. Methods Ecol. Evol. 2012, 3, 457–465. [Google Scholar] [CrossRef]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.-C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef]
- Rangel-Medrano, J.D.; Ortega-Lara, A.; Marquez, E.J. Ancient genetic divergence in bumblebee catfish of the genus Pseudopimelodus (Pseudopimelodidae: Siluriformes) from northwestern South America. PeerJ 2020, 8, e9028. [Google Scholar] [CrossRef] [PubMed]
- Microsoft R. Core Team. Microsoft R Open Application Network; Microsoft Corporsation: Redmond, WA, USA, 2021. [Google Scholar]
- Ota, R.P.; Machado, V.N.; Andrade, M.C.; Collins, R.A.; Farias, I.P.; Hrbek, T. Integrative taxonomy reveals a new species of pacu (Characiformes: Serrasalmidae: Myloplus) from the Brazilian Amazon. Neotrop. Ichthyol. 2020, 18. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Foster, E.D.; Deardorff, A. Open Science Framework (OSF). J. Med. Libr. Assoc. 2017, 105, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.P.; Barrett, M.; Hogan, G.; Flores Bueso, Y.; Claesson, M.J.; Tangney, M. Non-specific amplification of human DNA is a major challenge for 16S rRNA gene sequence analysis. Sci. Rep. 2020, 10, 16356. [Google Scholar] [CrossRef]
- Wise, C.A.; Sraml, M.; Rubinsztein, D.C.; Easteal, S. Comparative nuclear and mitochondrial genome diversity in humans and chimpanzees. Mol. Biol. Evol. 1997, 14, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.J.M.; Ip, Y.C.A.; Ng, C.S.L.; Huang, D. Takeaways from Mobile DNA Barcoding with BentoLab and MinION. Genes 2020, 11, 1121. [Google Scholar] [CrossRef]
- Longo, M.S.; O’Neill, M.J.; O’Neill, R.J. Abundant human DNA contamination identified in non-primate genome databases. PLoS ONE 2011, 6, e16410. [Google Scholar] [CrossRef] [Green Version]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Palandacic, A.; Naseka, A.; Ramler, D.; Ahnelt, H. Contrasting morphology with molecular data: An approach to revision of species complexes based on the example of European Phoxinus (Cyprinidae). BMC Evol. Biol. 2017, 17, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andujar, C.; Arribas, P.; Yu, D.W.; Vogler, A.P.; Emerson, B.C. Why the COI barcode should be the community DNA metabarcode for the metazoa. Mol. Ecol. 2018, 27, 3968–3975. [Google Scholar] [CrossRef]
- Piper, A.M.; Batovska, J.; Cogan, N.O.I.; Weiss, J.; Cunningham, J.P.; Rodoni, B.C.; Blacket, M.J. Prospects and challenges of implementing DNA metabarcoding for high-throughput insect surveillance. Gigascience 2019, 8, giz092. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. Available online: http://www.barcodinglife.org (accessed on 15 December 2021). [CrossRef] [Green Version]
- Christopherson, C.; Sninsky, J.; Kwok, S. The effects of internal primer-template mismatches on RT-PCR: HIV-1 model studies. Nucleic Acids Res. 1997, 25, 654–658. [Google Scholar] [CrossRef] [Green Version]
- Miya, M.; Gotoh, R.O.; Sado, T. MiFish metabarcoding: A high-throughput approach for simultaneous detection of multiple fish species from environmental DNA and other samples. Fish. Sci. 2020, 86, 939–970. [Google Scholar] [CrossRef]
- Garibian, P.G.; Karabanov, D.P.; Neretina, A.N.; Taylor, D.J.; Kotov, A.A. Bosminopsis deitersi (Crustacea: Cladocera) as an ancient species group: A revision. PeerJ 2021, 9, e11310. [Google Scholar] [CrossRef]
- Betancur-R, R.; Wiley, E.O.; Arratia, G.; Acero, A.; Bailly, N.; Miya, M.; Lecointre, G.; Ortí, G. Phylogenetic classification of bony fishes. BMC Evol. Biol. 2017, 17, 162. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, A.; Swain, S.; Kar, S.K.; Hazra, R.K. Analysis of the phylogenetic relationship of Anopheles species, subgenus Cellia (Diptera: Culicidae) and using it to define the relationship of morphologically similar species. Infect. Genet. Evol. 2009, 9, 1204–1224. [Google Scholar] [CrossRef]
- Bergsten, J. A review of long-branch attraction. Cladistics 2005, 21, 163–193. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.J.; David, S.R.; Near, T.J. Gene trees, species trees, and morphology converge on a similar phylogeny of living gars (Actinopterygii: Holostei: Lepisosteidae), an ancient clade of ray-finned fishes. Mol. Phylogenet. Evol. 2012, 63, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to fail at species delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Ho, S.Y.W. The molecular clock and evolutionary timescales. Biochem. Soc. Trans. 2018, 46, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Kotov, A.A.; Garibian, P.G.; Bekker, E.I.; Taylor, D.J.; Karabanov, D.P. A new species group from the Daphnia curvirostris species complex (Cladocera: Anomopoda) from the eastern Palaearctic: Taxonomy, phylogeny and phylogeography. Zool. J. Linn. Soc. 2021, 191, 772–822. [Google Scholar] [CrossRef]
- Luzhnyak, V.A. Materials on the ichthyofauna of the Middle Don basin. J. Ichthyol. 2010, 50, 750–756. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Kodukhova, Y.V.; Kutsokon, Y.K. Expansion of stone moroko Pseudorasbora parva (Cypriniformes, Cyprinidae) into Eurasian reservoirs. Vestn. Zool. 2010, 44, 115–124. [Google Scholar]
- Karabanov, D.P.; Kodukhova, Y.V.; Slyn’ko, Y.V. New finds of topmouth gudgeon Pseudorasbora parva (Temm. et Schl., 1846) in the European part of Russia. Russ. J. Biol. Invasions 2010, 1, 156–158. [Google Scholar] [CrossRef]
- Karabanov, D.P.; Kodukhova, Y.V.; Pashkov, A.N.; Reshetnikov, A.N.; Makhrov, A.A. “Journey to the West”: Three phylogenetic lineages contributed to the invasion of Stone Moroko, Pseudorasbora parva (Actinopterygii: Cyprinidae). Russ. J. Biol. Invasions 2021, 12, 67–78. [Google Scholar] [CrossRef]
- Hirsch, P.E.; N’Guyen, A.; Adrian-Kalchhauser, I.; Burkhardt-Holm, P. What do we really know about the impacts of one of the 100 worst invaders in Europe? A reality check. Ambio 2016, 45, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, D.A.; Sorokin, P.A.; Vasil’ev, V.P.; Chernova, N.V.; Vasil’eva, E.D. Reconstruction of phylogenetic relations of Ponto-Caspian gobies (Gobiidae, Perciformes) based on mitochondrial genome variation and some problems of their taxonomy. J. Ichthyol. 2013, 53, 702–712. [Google Scholar] [CrossRef]
- Freyhof, J. Diversity and distribution of freshwater Gobies from the Mediterranean, the Black and Caspian Seas. In The Biology of Gobies; Patzner, R., Van Tassell, J.L., Kovacic, M., Kapoor, B.G., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 279–288. ISBN 9780429062872. [Google Scholar]
- Kodukhova, Y.V.; Karabanov, D.P. Finding of Longtail Dwarf Goby Knipowitschia longecaudata (Actinopterygii: Gobiidae) in the upper part of unregulated section of the Volga River. Inland Water Biol. 2021, 14, 620–625. [Google Scholar] [CrossRef]
- Kodukhova, Y.V.; Borovikova, E.A.; Karabanov, D.P. First record of stellate tadpole goby Benthophilus stellatus (Sauvage, 1874) (Actinopterygii: Gobiidae) in the Rybinsk Reservoir. Inland Water Biol. 2016, 9, 428–430. [Google Scholar] [CrossRef]
- Slynko, Y.V.; Borovikova, E.A.; Gurovskii, A.N. Phylogeography and origin of freshwater populations of tubenose gobies of genus Proterorhinus (Gobiidae: Pisces) in Ponto-Caspian Basin. Russ. J. Genet. 2013, 49, 1144–1154. [Google Scholar] [CrossRef]
- Neilson, M.E.; Stepien, C.A. Evolution and phylogeography of the tubenose goby genus Proterorhinus (Gobiidae: Teleostei): Evidence for new cryptic species. Biol. J. Linn. Soc. 2009, 96, 664–684. [Google Scholar] [CrossRef] [Green Version]
- Reshetnikov, A.N. The current range of Amur sleeper Perccottus glenii Dybowski, 1877 (Odontobutidae, Pisces) in Eurasia. Russ. J. Biol. Invasions 2010, 1, 119–126. [Google Scholar] [CrossRef]
- Kiryukhina, N.A. Molecular and genetic variability in populations of Syngnathus nigrolineatus Eichwald 1831 and ways of expansion in the Volga River basins on the basis of mitochondrial DNA sequence analysis. Russ. J. Biol. Invasions 2013, 4, 249–254. [Google Scholar] [CrossRef]
- Borovikova, E.A.; Artamonova, V.S. Vendace (Coregonus albula) and least cisco (Coregonus sardinella) are a single species: Evidence from revised data on mitochondrial and nuclear DNA polymorphism. Hydrobiologia 2021, 848, 4241–4262. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence 5′-3′ | Annealing Temperature (°C) | Full Amplicon Length (Kb) |
|---|---|---|---|---|
| COI (long) | MifCOI-F | tgt aaa acg acg gcc agt tCA CAA AGA VAT TGG YAC CCT ITA | 52 | 0.7 |
| MifCOI-R | cag gaa aca gct atg acta CIG GGT GIC CRA ARA AYC ARA AIA | |||
| COI (short) | ifCOImb-F | GGD ACH GGN TGA ACD GTH TAY CCB CC | 56 | 0.4 |
| ifCOImb-R | TGD CCA AAR AAY CAG AAB ARG TGA TG | |||
| 16S | if16Sa | CTG CCC TGT GAC CAA AAG TT | 52 | 0.6 |
| if16Sb | GGT CCT TTC GTA CTA GGA AG | |||
| 18S | if18S1 | TAA CAT ATG CTT GTC TCA AAG | 50 | 0.5 |
| if18S2 | CCT GTA TTG TTA TTT TTC GTC AC | |||
| M13s | M13seq-F | tgt aaa acg acg gcc agt t | 55 | Seq. |
| M13seq-R | cag gaa aca gct atg act a |
| Locus | Position | Best Model (BIC) |
|---|---|---|
| 1st | TIM3e {0.1698, 2.1566, 12.5089} + FQ + G4 {0.1709} | |
| COI | 2nd | F81 + F |
| 3rd | TIM3 {0.7556, 9.7639, 4.9699} + F {0.2885, 0.3166, 0.1141, 0.2805} + R3 {0.0078, 0.0093, 0.6312, 0.5571, 0.3608, 1.7961} | |
| 16S | TPM2 {3.2961, 8.5094} + F {0.2888, 0.2442, 0.2371, 0.2298} + G4 {0.2244} | |
| 18S | TIM2e {0.4296, 0.2174, 3.6492} + FQ + G4 {0.1817} |
| Primers Set | Percentage of PCR and Sequencings Success |
|---|---|
| LCO\HCO [8] | 78 |
| Fish-1 [12] | 84 |
| Fish-2 [12] | 78 |
| COI-1 [13] | 87 |
| MifCOI | 91 |
| ifCOImb | 98 |
| if16S | 98 |
| if18S | 97 |
| Loci/Orders | N | ns | G + C | S | Eta | h | Hd | Pi | k | Fs | D |
|---|---|---|---|---|---|---|---|---|---|---|---|
| COI (mitochondrial) | |||||||||||
| Clupeiformes | 55 | 669 | 0.486 | 173 | 206 | 36 | 0.97 | 0.087 | 58.2 | 1.81 | 1.04 |
| Cypriniformes | 17 | 669 | 0.451 | 172 | 220 | 16 | 0.99 | 0.079 | 53.2 | −0.63 | −0.78 |
| Gobiiformes | 49 | 669 | 0.478 | 233 | 342 | 24 | 0.94 | 0.129 | 86.7 | 15.9 | 0.47 |
| Osmeriformes | 4 | 669 | 0.486 | 8 | 8 | 3 | 0.83 | 0.006 | 4.00 | 1.16 | −0.82 |
| Perciformes | 1 | 669 | 0.461 | 0 | 0 | 1 | - | 0 | - | - | - |
| Salmoniformes | 5 | 669 | 0.495 | 4 | 4 | 3 | 0.70 | 0.003 | 1.80 | 0.46 | −0.41 |
| Syngnathiformes | 15 | 669 | 0.476 | 21 | 21 | 9 | 0.84 | 0.008 | 5.10 | −1.04 | −0.86 |
| COI (total) | 146 | 669 | 0.478 | 277 | 515 | 92 | 0.98 | 0.177 | 118.8 | 1.21 | 0.92 |
| 16S (mitochondrial) | |||||||||||
| Clupeiformes | 39 | 577 | 0.502 | 70 | 82 | 12 | 0.73 | 0.028 | 16.22 | 6.93 | −0.59 |
| Cypriniformes | 19 | 576 | 0.450 | 32 | 33 | 11 | 0.78 | 0.016 | 9.43 | 0.21 | −0.01 |
| Gobiiformes | 19 | 581 | 0.486 | 121 | 154 | 11 | 0.93 | 0.070 | 39.78 | 6.40 | −0.40 |
| 16S (total) | 77 | 581 | 0.483 | 176 | 261 | 32 | 0.91 | 0.119 | 66.24 | 14.8 | 0.85 |
| 18S (nuclear) | |||||||||||
| Clupeiformes | 43 | 458 | 0.559 | 15 | 16 | 7 | 0.70 | 0.010 | 4.70 | 3.75 | 0.86 |
| Cypriniformes | 7 | 486 | 0.573 | 11 | 11 | 4 | 0.71 | 0.012 | 5.81 | 2.07 | 1.58 |
| Gobiiformes | 27 | 445 | 0.546 | 11 | 12 | 7 | 0.63 | 0.004 | 1.80 | −0.95 | −1.41 |
| Osmeriformes | 4 | 463 | 0.555 | 0 | 0 | 1 | - | 0 | - | - | - |
| Perciformes | 1 | 442 | 0.545 | 0 | 0 | 1 | - | 0 | - | - | - |
| Salmoniformes | 4 | 441 | 0.540 | 2 | 2 | 3 | 0.83 | 0.002 | 1.00 | −0.88 | −0.71 |
| Syngnathiformes | 5 | 417 | 0.581 | 1 | 1 | 2 | 0.60 | 0.001 | 0.60 | 0.62 | 1.22 |
| 18S (total) | 91 | 486 | 0.546 | 51 | 59 | 19 | 0.87 | 0.041 | 15.60 | 6.80 | 1.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karabanov, D.P.; Bekker, E.I.; Pavlov, D.D.; Borovikova, E.A.; Kodukhova, Y.V.; Kotov, A.A. New Sets of Primers for DNA Identification of Non-Indigenous Fish Species in the Volga-Kama Basin (European Russia). Water 2022, 14, 437. https://doi.org/10.3390/w14030437
Karabanov DP, Bekker EI, Pavlov DD, Borovikova EA, Kodukhova YV, Kotov AA. New Sets of Primers for DNA Identification of Non-Indigenous Fish Species in the Volga-Kama Basin (European Russia). Water. 2022; 14(3):437. https://doi.org/10.3390/w14030437
Chicago/Turabian StyleKarabanov, Dmitry P., Eugeniya I. Bekker, Dmitry D. Pavlov, Elena A. Borovikova, Yulia V. Kodukhova, and Alexey A. Kotov. 2022. "New Sets of Primers for DNA Identification of Non-Indigenous Fish Species in the Volga-Kama Basin (European Russia)" Water 14, no. 3: 437. https://doi.org/10.3390/w14030437
APA StyleKarabanov, D. P., Bekker, E. I., Pavlov, D. D., Borovikova, E. A., Kodukhova, Y. V., & Kotov, A. A. (2022). New Sets of Primers for DNA Identification of Non-Indigenous Fish Species in the Volga-Kama Basin (European Russia). Water, 14(3), 437. https://doi.org/10.3390/w14030437