
Reductive and Oxidative UV Degradation of PFAS—Status, Needs and Future Perspectives

Abstract
1. Introduction
2. Photolysis and Photochemical Decomposition Using UVC/VUV
2.1. Direct Photolysis (UV/VUV)
2.2. Photochemical Oxidation Using UVC/VUV
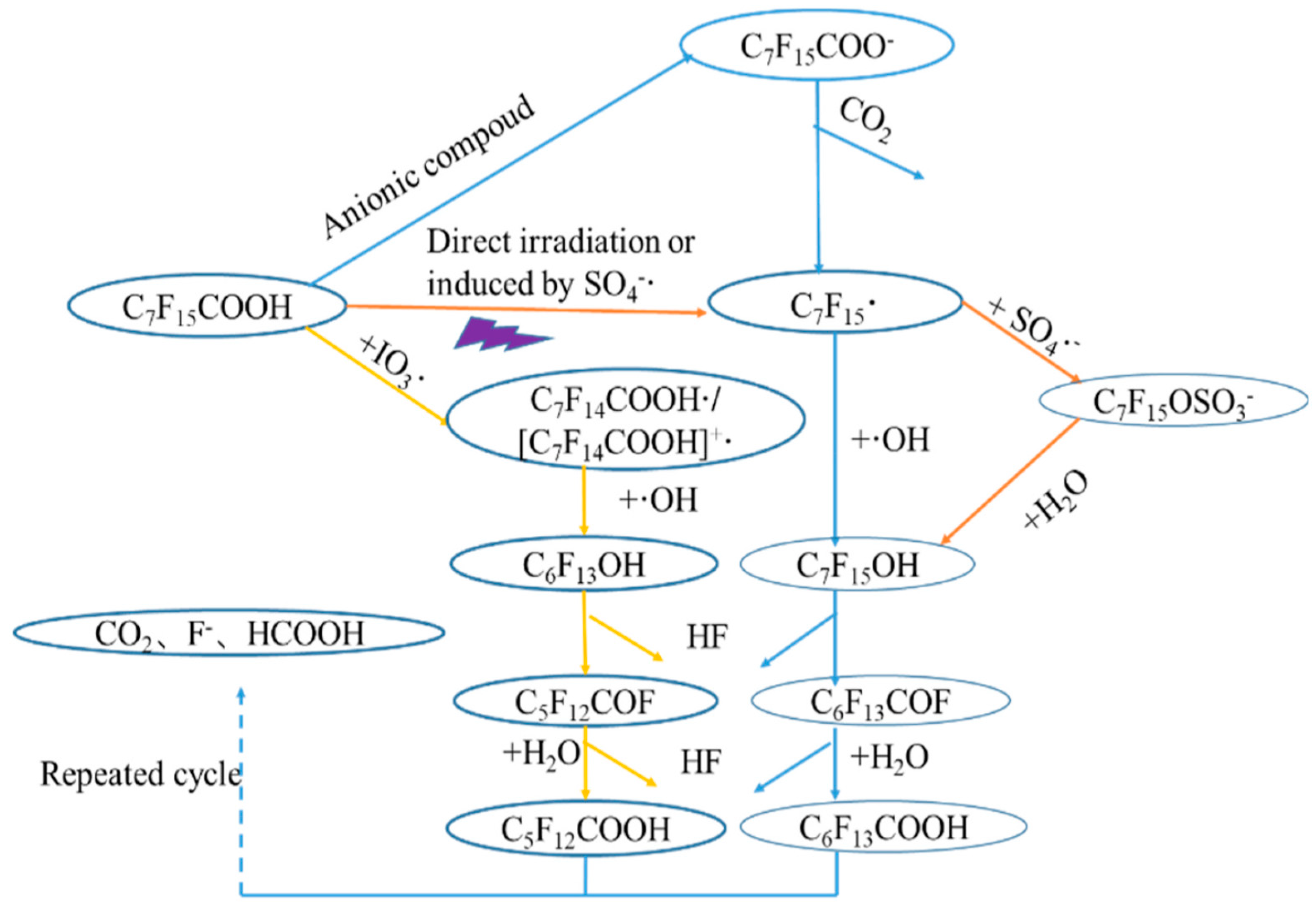
2.2.1. UV/H2O2

{kind=link}
{kind=link}
{kind=link}
| Compound | Concentrations (uM) | Matrix | pH | UV Source | Wavelength (nm) | Oxidative Agent | Degrdation (%) | Treatment Time (h) | Lamp Power (W) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PFOA | 36 | Ultrapure water | 3 to 4 | VUV | 185, 254 | Fe3+ | 100 | 72–144 | 5 | [32] |
| PFOA | 150 | Ultrapure water, surface water, WW | 2.8 | UV | 254 | SO4•− | 85.6 | 8 | [33] | |
| PFOA | 36 | milliQ | 3 to 4 | VUV | 185, 254 | Fe3+ | 51.2 | 4 | 12 | [34] |
| PFOS | 20 | milliQ | 3.6 | LP | 254 | Fe3+ | ~100 | 48 | 23 | [35] |
| PFOA | 48 | milliQ | 2 | LP | 254 | Fe3+ | 100 | 8 | 14 | [36] |
| PFOA | 20 | DI | 5 | LP | 254 | Fe3+, S2O82− | 93.9 | 5 | 9 | [37] |
| PFOA | 120 | milliQ | 4.09, 8.8, 11 | LP | 254 | CO3•− | 100 | 12 | 400 | [22] |
| PFOS | 200 | Ultrapure water | 3.0–11 | LP | 254 | S2O82− | 23.5 | 12 | 15 | [38] |
| PFOA | 48 | milliQ | 4.6 | LP | 185 | Fe3+ | 98 | 48 | 12 | [39] |
| PFDeA | 100 | DI | NG | LP | 185, 254 | S2O82− | 100 | 6 | 23 | [40] |
| PFOA | 20 | DI | 3 | LP | 254 | Fe3+ | 92.5 | 5 | 9 W | [41] |
| PFOA | 48 | DI water | 3.5 to 4 | VUV | 185, 254 | Fe3+ | 78.9 | 4 | 23 W | [42] |
| PFCA | 67.3 | milliQ | 1.5 | MP | 220–460 | Fe3+ | 71.2 | 24 | 200 W | [24] |
| PFOS | 40 | milliQ | LP | 254 | alkaline 2-propanol | 92 | 10 d | 32 W | [43] | |
| PFOA/PFNA | 29.6 | Aqueous solution/wax sample | 3.0–3.1 | UV | 220–460 | S2O82− | 100 | 4 | 200 W | [44] |
2.2.2. UV/VUV/Sulfite
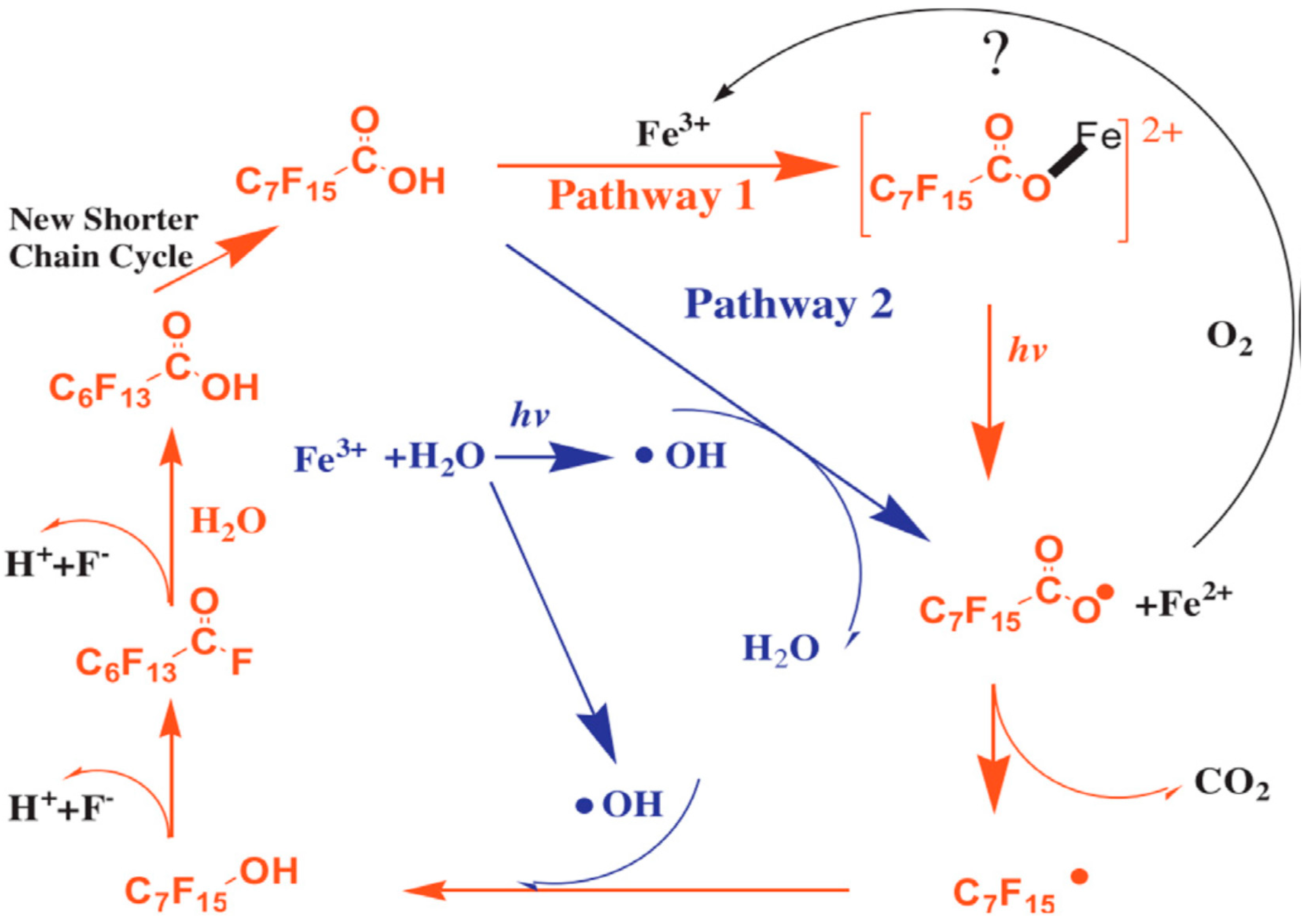
2.2.3. UV/VUV/Fe3+
2.3. Reaction by-Products during UV/VUV Photodegradation
2.4. Photochemical Oxidation Using MP UV—Impact of Experimental Conditions
3. Reductive Photodegradation
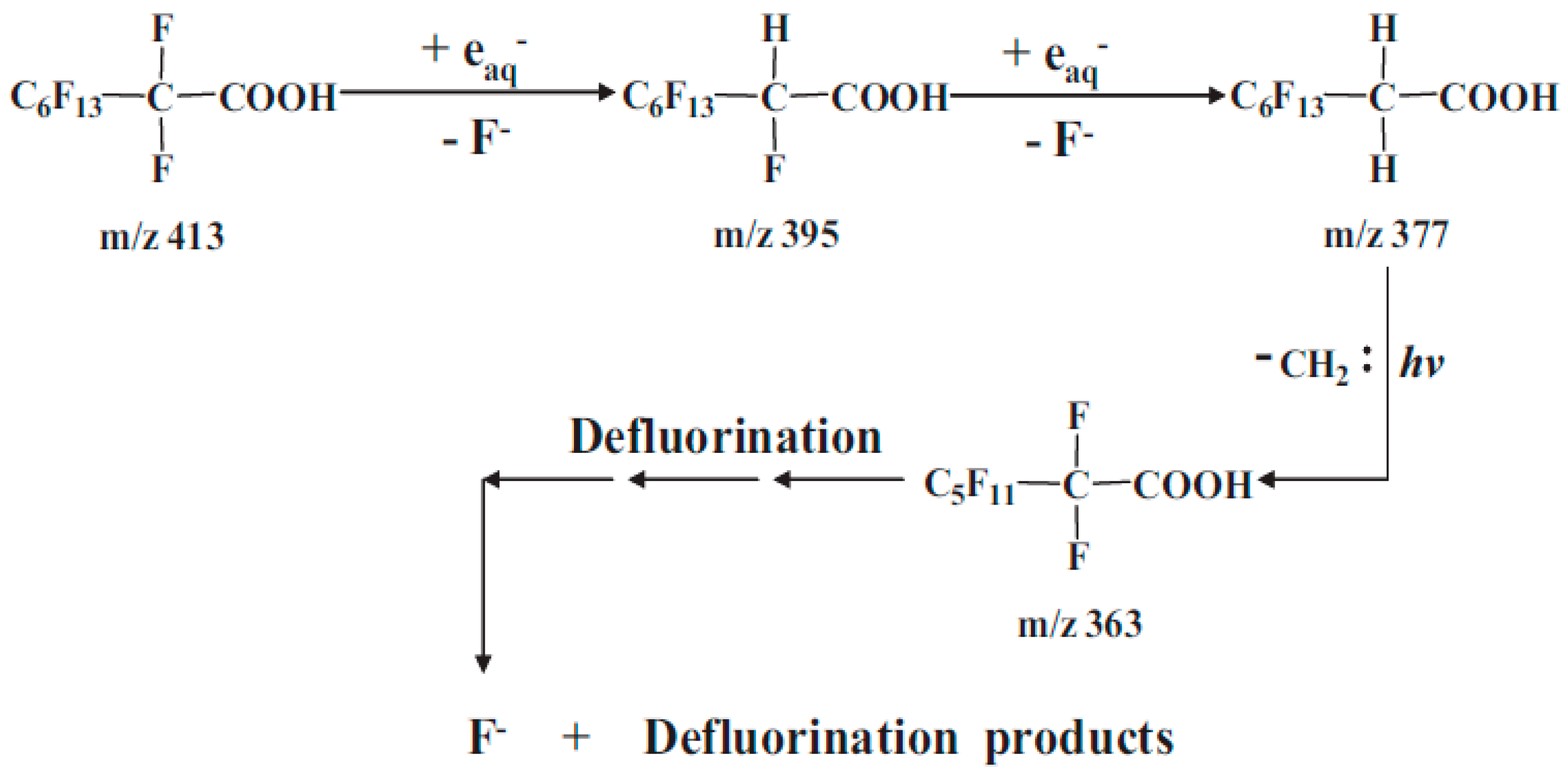
3.1. UV/VUV in Reductive Degradation of PFAS

| Compound | Concentrations (uM) | Matrix | pH | UV Source | Wavelength | Oxidative/Reducing Agent | Degrdation (%) | Treatment Time (h) | Lamp Power (W) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PFOS | 32 | Ultrapure water | 9.2 | MP UV | 200–400 | 98 | 0.5 | 250 | [70] | |
| PFOA | 37.2 | milliQ, PBS | 7–11.2 | MP UV | - | N2O, tert-butanol | 6 | 500 | [56] | |
| PFOA | 37.2 | DI, PBS, lake water, WWTP effluent | 4.3–9 | MP UV | - | none | 6 | 500 | [65] | |
| PFOA | 0.025 | 18O-water | 7 to 10 | LP UV | 254 | KI | 100 | 6 | 15 | [57] |
| PFOA | 24 | Ultrapure water | 4.5, 7.8, 10.3, 12 | LP UV | 185, 254 | none | 3 | 23 | [64] | |
| PFOA | 1.96, 2,51, 3.06 | Ultrapure water | LP UV | 185, 254 | KI | 100 | 3 | 20, 110 | [71] | |
| PFOA | 20 | DI | 10.3 | LP UV | 254 | 100 | 1–24 | 10 | [68] | |
| PFOA | 0.121, 1.21, 2.42 | Ultrapure water, tap water, river water | 3, 5.5, 7, 10 | LP UV | 185, 254 | none | 3 | 20 | [23] | |
| PFOS | 25 | milliQ | 9 | LP UV | 254 | KI | 6 | 15 | [67] | |
| PFDeA | 100 | DI | NG | LP UV | 185, 254 | Na2S | 100 | 6 | 23 | [40] |
| PFOA | 60.4 | aqueous solution | 3.7 | LP UV | 185, 254 | none | 2 | 15 | [20] |
3.2. Medium Pressure UV in Reductive Degradation of PFAS
3.3. Reaction By-Products during Reductive Degradation
4. Future Work and Research Needs
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baran, J.R. Fluorinated Surfactants and Repellents: 2nd Edition. Revised and Expanded Surfactant Science Series; American Chemical Society: Washington, DC, USA, 2001; Volume 123, p. 8882. [Google Scholar]
- Brooke, D.; Footitt, A.; Nwaogu, T.A. Environmental Risk Evaluation Report: Perfluorooctanesulphonate (PFOS); Environment Agency: Bristol, UK, 2004.
- Fujii, S.; Polprasert, C.; Tanaka, S.; Hong Lien, N.P.; Qiu, Y. New POPs in the water environment: Distribution, bioaccumulation and treatment of perfluorinated compounds—A review paper. J. Water Supply Res. Technol. Aqua 2007, 56, 313–326. [Google Scholar] [CrossRef]
- Du, Z.; Deng, S.; Bei, Y.; Huang, Q.; Wang, B.; Huang, J.; Yu, G. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents—A review. J. Hazard. Mater. 2014, 274, 443–454. [Google Scholar] [CrossRef]
- Ahrens, L.; Barber, J.L.; Xie, Z.; Ebinghaus, R. Longitudinal and Latitudinal Distribution of Perfluoroalkyl Compounds in the Surface Water of the Atlantic Ocean. Environ. Sci. Technol. 2009, 43, 3122–3127. [Google Scholar] [CrossRef]
- Gebbink, W.A.; van Asseldonk, L.; van Leeuwen, S.P.J. Presence of Emerging Per- and Polyfluoroalkyl Substances (PFASs) in River and Drinking Water near a Fluorochemical Production Plant in the Netherlands. Environ. Sci. Technol. 2017, 51, 11057–11065. [Google Scholar] [CrossRef]
- Gomez-Ruiz, B.; Gómez-Lavín, S.; Diban, N.; Boiteux, V.; Colin, A.; Dauchy, X.; Urtiaga, A. Efficient electrochemical degradation of poly- and perfluoroalkyl substances (PFASs) from the effluents of an industrial wastewater treatment plant. Chem. Eng. J. 2017, 322, 196–204. [Google Scholar] [CrossRef]
- Ahrens, L. Polyfluoroalkyl compounds in the aquatic environment: A review of their occurrence and fate. J. Environ. Monit. 2011, 13, 20–31. [Google Scholar] [CrossRef]
- Espana, V.A.A.; Mallavarapu, M.; Naidu, R. Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA): A critical review with an emphasis on field testing. Environ. Technol. Innov. 2015, 4, 168–181. [Google Scholar] [CrossRef]
- IISD/ENB. Summary of the meetings of the conferences of the parties to the Basel. In Proceedings of the Geneva, Switzerland. Rotterdam and Stockholm Conventions, Geneva, Switzerland, 29 April–10 May 2019. [Google Scholar]
- EPA. FACT SHEET PFOA & PFOS Drinking Water Health Advisories (EPA 800-F-16-003); EPA: Washington, DC, USA, 2016.
- EPA. EPA’s Per- and Polyfluoroalkyl Substances (PFAS) Action Plan; EPA: Washington, DC, USA, 2019.
- Ross, I.; McDonough, J.; Miles, J.; Storch, P.; Kochunarayanan, P.T.; Kalve, E.; Hurst, J.; Dasgupta, S.S.; Burdick, J. A review of emerging technologies for remediation of PFASs. Remediat. J. 2018, 28, 101–126. [Google Scholar] [CrossRef]
- Li, Z.; Li, S.; Tang, Y.; Li, X.; Wang, J.; Li, L. Highly efficient degradation of perfluorooctanoic acid: An integrated photo-electrocatalytic ozonation and mechanism study. Chem. Eng. J. 2020, 391, 123533. [Google Scholar] [CrossRef]
- Liu, X.; Wei, W.; Xu, J.; Wang, D.; Song, L.; Ni, B.-J. Photochemical decomposition of perfluorochemicals in contaminated water. Water Res. 2020, 186, 116311. [Google Scholar] [CrossRef]
- Phong Vo, H.N.P.; Ngo, H.H.; Guo, W.; Nguyen, T.M.H.; Li, J.; Liang, H.; Deng, L.; Chen, Z.; Nguyen, T.A.H. Poly-and perfluoroalkyl substances in water and wastewater: A comprehensive review from sources to remediation. J. Water Process. Eng. 2020, 36, 101393. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Z.; Wang, Y.; Sun, W. A review on degradation of perfluorinated compounds based on ultraviolet advanced oxidation. Environ. Pollut. 2021, 291, 118014. [Google Scholar] [CrossRef] [PubMed]
- Oppenländer, T. Photooxidation and Photomineralization of Organic Matter in Water and Air. In Photochemical Purification of Water and Air; Wiley: New York, NY, USA, 2002; pp. 189–238. [Google Scholar]
- Chen, J.; Zhang, P. Photodegradation of perfluorooctanoic acid in water under irradiation of 254 nm and 185 nm light by use of persulfate. Water Sci. Technol. 2006, 54, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, P.-Y.; Liu, J. Photodegradation of perfluorooctanoic acid by 185 nm vacuum ultraviolet light. J. Environ. Sci. 2007, 19, 387–390. [Google Scholar] [CrossRef]
- Giri, R.R.; Ozaki, H.; Morigaki, T.; Taniguchi, S.; Takanami, R. UV photolysis of perfluorooctanoic acid (PFOA) in dilute aqueous solution. Water Sci. Technol. 2011, 63, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Thi, L.-A.P.; Do, H.-T.; Lee, Y.-C.; Lo, S.-L. Photochemical decomposition of perfluorooctanoic acids in aqueous carbonate solution with UV irradiation. Chem. Eng. J. 2013, 221, 258–263. [Google Scholar]
- Giri, R.R.; Ozaki, H.; Okada, T.; Taniguchi, S.; Takanami, R. Factors influencing UV photodecomposition of perfluorooctanoic acid in water. Chem. Eng. J. 2012, 180, 197–203. [Google Scholar] [CrossRef]
- Hori, H.; Yamamoto, A.; Koike, K.; Kutsuna, S.; Osaka, I.; Arakawa, R. Photochemical decomposition of environmentally persistent short-chain perfluorocarboxylic acids in water mediated by iron(II)/(III) redox reactions. Chemosphere 2007, 68, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, C.; Zhao, Z.; Cui, F.; Ou, Q.; Liu, J. Role of oxygen and superoxide radicals in promoting H2O2 production during VUV/UV radiation of water. Chem. Eng. Sci. 2021, 241, 116683. [Google Scholar] [CrossRef]
- Barki, D.; Sabach, S.; Dubowski, Y. Removal of Chlorinated Organic Pollutants from Groundwater Using a Vacuum-UV-Based Advanced Oxidation Process. ACS EST Water 2021, 1, 2076–2086. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Q.; Chen, B.; Long, L.; Zhang, G. Toward better understanding vacuum ultraviolet—iodide induced photolysis via hydrogen peroxide formation, iodine species change, and difluoroacetic acid degradation. Front. Environ. Sci. Eng. 2022, 16, 55. [Google Scholar] [CrossRef]
- Hori, H.; Hayakawa, E.; Einaga, H.; Kutsuna, S.; Koike, K.; Ibusuki, T.; Kiatagawa, H.; Arakawa, R. Decomposition of Environmentally Persistent Perfluorooctanoic Acid in Water by Photochemical Approaches. Environ. Sci. Technol. 2004, 38, 6118–6124. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅OH/⋅O− in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Schröder, H.F.; Meesters, R.J. Stability of fluorinated surfactants in advanced oxidation processes—A follow up of degradation products using flow injection–mass spectrometry, liquid chromatography–mass spectrometry and liquid chromatography–multiple stage mass spectrometry. J. Chromatogr. A 2005, 1082, 110–119. [Google Scholar] [CrossRef]
- Liang, X.; Cheng, J.; Yang, C.; Yang, S. Factors influencing aqueous perfluorooctanoic acid (PFOA) photodecomposition by VUV irradiation in the presence of ferric ions. Chem. Eng. J. 2016, 298, 291–299. [Google Scholar] [CrossRef]
- Qian, Y.; Guo, X.; Zhang, Y.; Peng, Y.; Sun, P.; Huang, C.-H.; Niu, J.; Zhou, X.; Crittenden, J.C. Perfluorooctanoic Acid Degradation Using UV–Persulfate Process: Modeling of the Degradation and Chlorate Formation. Environ. Sci. Technol. 2016, 50, 772–781. [Google Scholar] [CrossRef]
- Cheng, J.-H.; Liang, X.-Y.; Yang, S.-W.; Hu, Y.-Y. Photochemical defluorination of aqueous perfluorooctanoic acid (PFOA) by VUV/Fe3+ system. Chem. Eng. J. 2014, 239, 242–249. [Google Scholar] [CrossRef]
- Jin, L.; Zhang, P.; Shao, T.; Zhao, S. Ferric ion mediated photodecomposition of aqueous perfluorooctane sulfonate (PFOS) under UV irradiation and its mechanism. J. Hazard. Mater. 2014, 271, 9–15. [Google Scholar] [CrossRef]
- Ohno, M.; Ito, M.; Ohkura, R.; Mino, E.; Kose, T.; Okuda, T.; Nakai, S.; Kawata, K.; Nishijima, W. Photochemical decomposition of perfluorooctanoic acid mediated by iron in strongly acidic conditions. J. Hazard. Mater. 2014, 268, 150–155. [Google Scholar] [CrossRef]
- Song, Z.; Tang, H.; Wang, N.; Wang, X.; Zhu, L. Activation of persulfate by UV and Fe2+for the defluorination of perfluorooctanoic acid. Adv. Environ. Res. 2014, 3, 185–197. [Google Scholar] [CrossRef]
- Yang, S.; Cheng, J.; Sun, J.; Hu, Y.; Liang, X. Defluorination of Aqueous Perfluorooctanesulfonate by Activated Persulfate Oxidation. PLoS ONE 2013, 8, e74877. [Google Scholar] [CrossRef]
- Ohno, M.; Kubo, Y.; Mino, E.; Kose, T.; Nakai, S.; Nishijima, W.; Kawata, K. Effect of pH and Coexisting Species on the Photochemical Decomposition of Perfluorooctanoic Acid by Iron (III) Sulphate. J. Water Environ. Technol. 2012, 10, 129–140. [Google Scholar] [CrossRef][Green Version]
- Wang, B.; Cao, M.; Tan, Z.; Wang, L.; Yuan, S.; Chen, J. Photochemical decomposition of perfluorodecanoic acid in aqueous solution with VUV light irradiation. J. Hazard. Mater. 2010, 181, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xiang, Q.; Lei, M.; Yan, J.; Zhu, L.; Zou, J. Efficient degradation of perfluorooctanoic acid by UV–Fenton process. Chem. Eng. J. 2012, 184, 156–162. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Pan, G.; Chen, H. Ferric ion mediated photochemical decomposition of perfluorooctanoic acid (PFOA) by 254 nm UV light. J. Hazard. Mater. 2008, 160, 181–186. [Google Scholar] [CrossRef]
- Yamamoto, T.; Noma, Y.; Sakai, S.-I.; Shibata, Y. Photodegradation of Perfluorooctane Sulfonate by UV Irradiation in Water and Alkaline 2-Propanol. Environ. Sci. Technol. 2007, 41, 5660–5665. [Google Scholar] [CrossRef]
- Hori, H.; Yamamoto, A.; Hayakawa, E.; Taniyasu, S.; Yamashita, N.; Kutsuna, S.; Kiatagawa, H.; Arakawa, R. Efficient Decomposition of Environmentally Persistent Perfluorocarboxylic Acids by Use of Persulfate as a Photochemical Oxidant. Environ. Sci. Technol. 2005, 39, 2383–2388. [Google Scholar] [CrossRef]
- Barisci, S.; Suri, R. Removal of polyfluorinated telomer alcohol by Advanced Oxidation Processes (AOPs) in different water matrices and evaluation of degradation mechanisms. J. Water Process. Eng. 2021, 39, 101745. [Google Scholar] [CrossRef]
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Bahnemann, D.C.J.; Fox, M.A.; Pelizzetti, E.; Pichat, P.; Serpone, N. Photocatalytic treatment of waters. In Aquatic and Surface Photochemistry; Helz, G.R., Zepp, R.G., Crosby, D.G., Eds.; Lewis Publishers: Boca Raton, FL, USA, 1994; pp. 261–316. [Google Scholar]
- Guan, Y.-H.; Ma, J.; Li, X.-C.; Fang, J.-Y.; Chen, L.-W. Influence of pH on the Formation of Sulfate and Hydroxyl Radicals in the UV/Peroxymonosulfate System. Environ. Sci. Technol. 2011, 45, 9308–9314. [Google Scholar] [CrossRef]
- Yuan, R.; Wang, Z.; Hu, Y.; Wang, B.; Gao, S. Probing the radical chemistry in UV/persulfate-based saline wastewater treatment: Kinetics modeling and byproducts identification. Chemosphere 2014, 109, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Anipsitakis, G.P.; Dionysiou, D.D.; Gonzalez, M.A. Cobalt-Mediated Activation of Peroxymonosulfate and Sulfate Radical Attack on Phenolic Compounds. Implications of Chloride Ions. Environ. Sci. Technol. 2006, 40, 1000–1007. [Google Scholar] [PubMed]
- He, X.; Mezyk, S.P.; Michael, I.; Fatta-Kassinos, D.; Dionysiou, D.D. Degradation kinetics and mechanism of β-lactam antibiotics by the activation of H2O2 and Na2S2O8 under UV-254nm irradiation. J. Hazard. Mater. 2014, 279, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Lutze, H.V.; Kerlin, N.; Schmidt, T.C. Sulfate radical-based water treatment in presence of chloride: Formation of chlorate, inter-conversion of sulfate radicals into hydroxyl radicals and influence of bicarbonate. Water Res. 2015, 72, 349–360. [Google Scholar] [CrossRef]
- Heidt, L.J. The Photolysis of Persulfate. J. Chem. Phys. 1942, 10, 297–302. [Google Scholar] [CrossRef]
- Liu, D.; Xiu, Z.; Liu, F.; Wu, G.; Adamson, D.; Newell, C.; Vikesland, P.; Tsai, A.-L.; Alvarez, P.J. Perfluorooctanoic acid degradation in the presence of Fe(III) under natural sunlight. J. Hazard. Mater. 2013, 262, 456–463. [Google Scholar] [CrossRef]
- Huang, J.; Wang, X.; Pan, Z.; Li, X.; Ling, Y.; Li, L. Efficient degradation of perfluorooctanoic acid (PFOA) by photocatalytic ozonation. Chem. Eng. J. 2016, 296, 329–334. [Google Scholar] [CrossRef]
- Lyu, X.-J.; Li, W.-W.; Lam, K.S.P.; Yu, H.-Q. Insights into perfluorooctane sulfonate photodegradation in a catalyst-free aqueous solution. Sci. Rep. 2015, 5, 9353. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, C.-J.; Chen, P.; Zhou, Q.; Zhang, W.-X. Effect of initial solution pH on photo-induced reductive decomposition of perfluorooctanoic acid. Chemosphere 2014, 107, 218–223. [Google Scholar] [CrossRef]
- Sima, J.; Makanova, J. Photochemistry of iron (III) complexes. Coord. Chem. Rev. 1997, 160, 161–189. [Google Scholar] [CrossRef]
- King, M.; France, J.; Fisher, F.; Beine, H. Measurement and modelling of UV radiation penetration and photolysis rates of nitrate and hydrogen peroxide in Antarctic sea ice: An estimate of the production rate of hydroxyl radicals in first-year sea ice. J. Photochem. Photobiol. A Chem. 2005, 176, 39–49. [Google Scholar] [CrossRef]
- Gonzalez, M.; Oliveros, E.; Worner, M.; Braun, A. Vacuum-ultraviolet photolysis of aqueous reaction systems. J. Photochem. Photobiol. C Photochem. Rev. 2004, 5, 225–246. [Google Scholar] [CrossRef]
- Sörensen, M.; Frimmel, F.H. Photochemical Degradation of Hydrophilic Xenobiotics in the UV/H2O2-process. Influence of Bicarbonate on the Degradation Rate of EDTA, 2-Amino-1-naphthalenesulfonate, Diphenyl-4-sulfonate, and 4,4′-Diaminostilbene-2,2′-disulfonate. Acta Hydrochim. Hydrobiol. 1996, 24, 185–188. [Google Scholar] [CrossRef]
- Hogfeldt, E. Stability Constants of Metal–Iron Complexes Part A: Inorganic Ligands, 2nd ed.; Pergamon Press: London, UK, 1982. [Google Scholar]
- Goss, K.-U. The pKa Values of PFOA and Other Highly Fluorinated Carboxylic Acids. Environ. Sci. Technol. 2008, 42, 456–458. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P. Effects of pH on photochemical decomposition of perfluorooctanoic acid in different atmospheres by 185nm vacuum ultraviolet. J. Environ. Sci. 2014, 26, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.-J.; Li, W.-W.; Lam, P.K.; Yu, H.-Q. Photodegradation of perfluorooctane sulfonate in environmental matrices. Sep. Purif. Technol. 2015, 151, 172–176. [Google Scholar] [CrossRef]
- Sauer, M.C.; Crowell, R.A.; Shkrob, I.A. Electron Photodetachment from Aqueous Anions. 1. Quantum Yields for Generation of Hydrated Electron by 193 and 248 nm Laser Photoexcitation of Miscellaneous Inorganic Anions. J. Phys. Chem. A 2004, 108, 5490–5502. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, C.; Li, F.; Chen, J.; Zhou, Q. Photo-reductive defluorination of perfluorooctanoic acid in water. Water Res. 2010, 44, 2939–2947. [Google Scholar] [CrossRef]
- Song, Z.; Tang, H.; Wang, N.; Zhu, L. Reductive defluorination of perfluorooctanoic acid by hydrated electrons in a sulfite-mediated UV photochemical system. J. Hazard. Mater. 2013, 262, 332–338. [Google Scholar] [CrossRef]
- Li, X.; Ma, J.; Liu, G.; Fang, J.; Yue, S.; Guan, Y.; Chen, L.; Liu, X. Efficient Reductive Dechlorination of Monochloroacetic Acid by Sulfite/UV Process. Environ. Sci. Technol. 2012, 46, 7342–7349. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Dong, W.; Luo, C.; Liu, T. Efficient Reductive Decomposition of Perfluorooctanesulfonate in a High Photon Flux UV/Sulfite System. Environ. Sci. Technol. 2016, 50, 10554–10561. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.R.; Ozaki, H.; Guo, X.W.; Takanami, R.; Taniguchi, S. Oxidativ–reductive photodecomposition of perfluorooctanoic acid in water. Int. J. Environ. Sci. Technol. 2013, 11, 1277–1284. [Google Scholar] [CrossRef]
- Park, H.; Vecitis, C.D.; Cheng, J.; Choi, W.; Mader, B.T.; Hoffmann, M.R. Reductive Defluorination of Aqueous Perfluorinated Alkyl Surfactants: Effects of Ionic Headgroup and Chain Length. J. Phys. Chem. A 2009, 113, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry, 5th ed.; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Fang, G.; Gao, J.; Dionysiou, D.D.; Liu, C.; Zhou, D. Activation of Persulfate by Quinones: Free Radical Reactions and Implication for the Degradation of PCBs. Environ. Sci. Technol. 2013, 47, 4605–4611. [Google Scholar] [CrossRef]
- Weeks, J.L.; Meaburn, G.M.A.C.; Gordon, S. Absorption Coefficients of Liquid Water and Aqueous Solutions in the Far Ultraviolet. Radiat. Res. 1963, 19, 559. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, J.; Xiao, Y.; Du, J. Efficient Removal of Polycyclic Aromatic Hydrocarbons, Dyes, and Heavy Metal Ions by a Homopolymer Vesicle. ACS Appl. Mater. Interfaces 2018, 10, 713–722. [Google Scholar] [CrossRef]
- Moriwaki, H.; Takagi, Y.; Tanaka, M.; Tsuruho, K.; Okitsu, K.; Maeda, Y. Sonochemical Decomposition of Perfluorooctane Sulfonate and Perfluorooctanoic Acid. Environ. Sci. Technol. 2005, 39, 3388–3392. [Google Scholar] [CrossRef]
- Grossweiner, L.I.; Swenson, G.W.; Zwicker, E.F. Photochemical Generation of the Hydrated Electron. Science 1963, 141, 805–806. [Google Scholar] [CrossRef]
- Dillert, R.; Bahnemann, D.B.D.; Hidaka, H. Light-induced degradation of perfluorocarboxylic acids in the presence of titanium dioxide. Chemosphere 2007, 67, 785–792. [Google Scholar] [CrossRef]
- Colosi, L.M.; Pinto, R.A.; Huang, Q.; Weber, W.J.J. Peroxidase-mediated degradation of perfluorooctanoic acid. Environ. Toxicol. Chem. 2009, 28, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.E.F.; Quina, F.H.; Nascimento, C.A.O.; Silva, D.N.; Chiavone-Filho, O. Treatment of Saline Wastewater Contaminated with Hydrocarbons by the Photo-Fenton Process. Environ. Sci. Technol. 2004, 38, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Nohara, K.; Toma, M.; Kutsuna, S.; Takeuchi, K.; Ibusuki, T. Cl Atom-Initiated Oxidation of Three Homologous Methyl Perfluoroalkyl Ethers. Environ. Sci. Technol. 2001, 35, 114–120. [Google Scholar] [CrossRef] [PubMed]


| Property | PFOA (Free Acid) | PFOS (Potassium Salt) |
|---|---|---|
| * Physical description | White powder/waxy white solid | White powder PFOA |
| Molecular formula | C8HF15O2 | C8HF17O3S |
| Molecular weight (g mol−1) | 414 | 538 |
| Water solubility at 25 °C (mg L−1) | 9.5 × 103 | 680 |
| Melting Point (°C) | 45–50 | >400 |
| Boiling point (°C) | 189–192 | Not measurable |
| Vapour pressure at 25 °C (Pa) | 4.2 | 2.48 × 10–8 |
| Organic–carbon partition coefficient (log Koc) | 2.06 | ** 2.57 |
| Henry’s law constant (atm-m3 mol−1) | Not measurable | 3.05 × 10–9 |
| Half-Life | *** 90 days, **** >92 years (at 25 °C) | *** 114 days, **** >41 years (at 25 °C) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umar, M. Reductive and Oxidative UV Degradation of PFAS—Status, Needs and Future Perspectives. Water 2021, 13, 3185. https://doi.org/10.3390/w13223185
Umar M. Reductive and Oxidative UV Degradation of PFAS—Status, Needs and Future Perspectives. Water. 2021; 13(22):3185. https://doi.org/10.3390/w13223185
Chicago/Turabian StyleUmar, Muhammad. 2021. "Reductive and Oxidative UV Degradation of PFAS—Status, Needs and Future Perspectives" Water 13, no. 22: 3185. https://doi.org/10.3390/w13223185
APA StyleUmar, M. (2021). Reductive and Oxidative UV Degradation of PFAS—Status, Needs and Future Perspectives. Water, 13(22), 3185. https://doi.org/10.3390/w13223185