Genetic Structure and Population Demography of White-Spotted Charr in the Upstream Watershed of a Large Dam



Abstract
1. Introduction
2. Materials and Methods
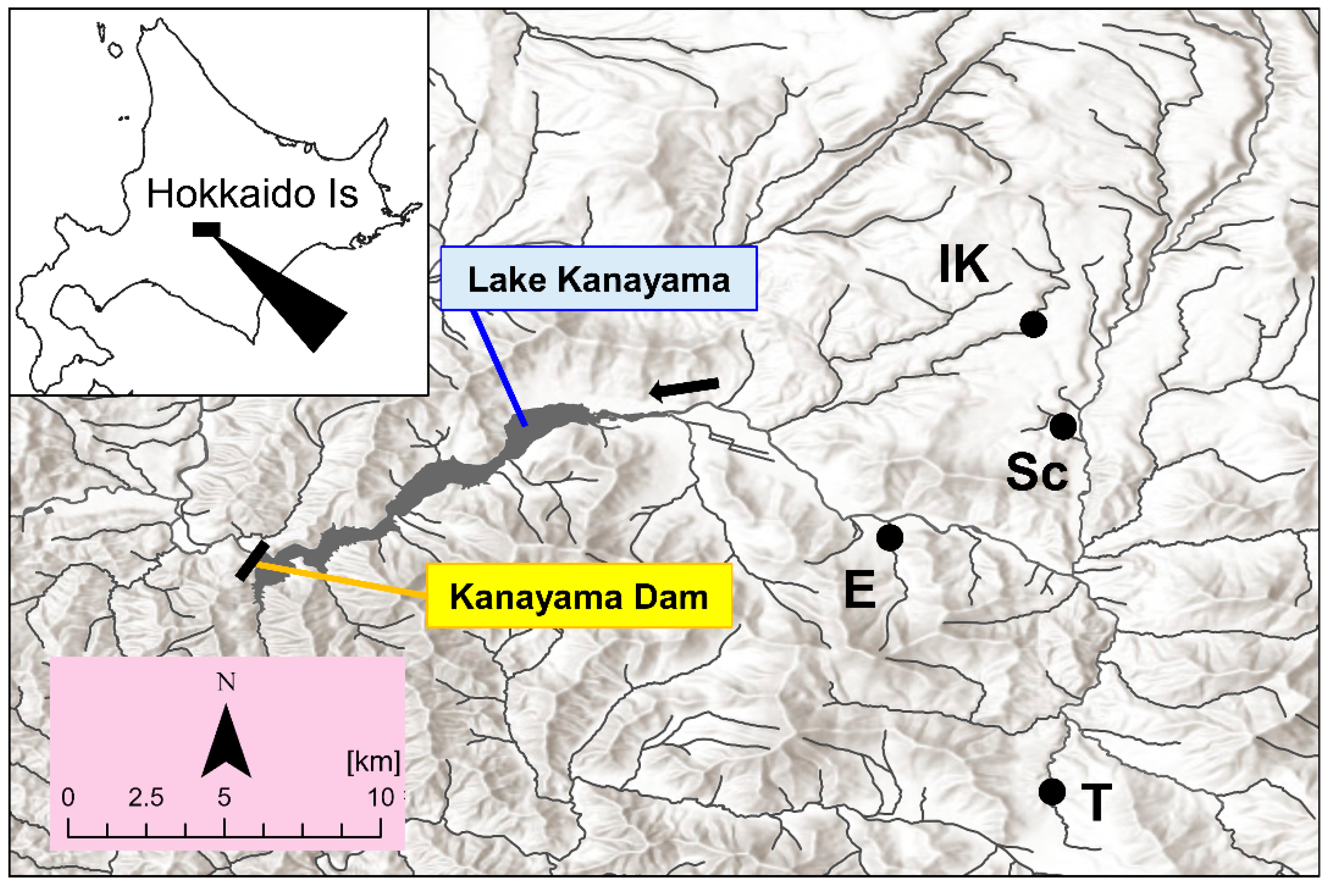
2.1. Study Area and Field Sampling
2.2. DNA Amplification and Sequencing
2.3. SNP Detection for Genetic Diversity and Population Structure Analysis
2.4. Genetic Diversity, Divergence, and Population Structure
2.5. SNP Detection for SFS
2.6. Inference of Population Demographic History
3. Results
3.1. Genetic Diversity and Divergence
3.2. Population Genetic Structure
3.3. Sibling Relationships
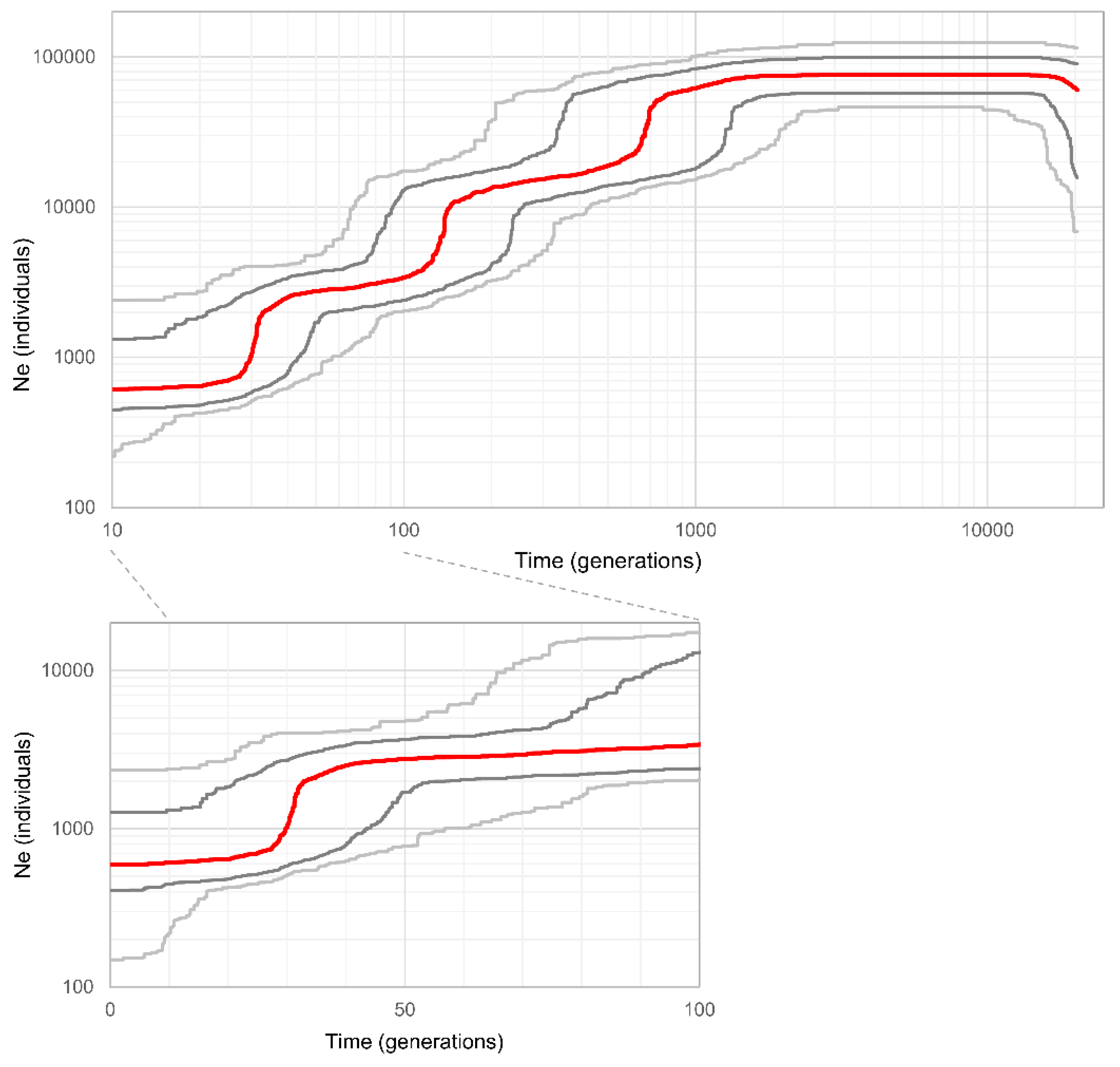
3.4. Population Demographic History
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reid, A.J.; Carlson, A.K.; Creed, I.F.; Eliason, E.J.; Gell, P.A.; Johnson, P.T.J.; Kidd, K.A.; MacCormack, T.J.; Olden, J.D.; Ormerod, S.J.; et al. Emerging threats and persistent conservation challenges for freshwater biodiversity. Biol. Rev. 2019, 94, 849–873. [Google Scholar] [CrossRef] [PubMed]
- Dodds, W.K.; Perkin, J.S.; Gerken, J.E. Human impact on freshwater ecosystem services: A global perspective. Environ. Sci. Technol. 2013, 47, 9061–9068. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Nakamura, F. Effects of fine sediment accumulation on the redd environment and the survival rate of masu salmon (Oncorhynchus masou) embryos. Landsc. Ecol. Eng. 2009, 5, 169–181. [Google Scholar] [CrossRef]
- Lande, R. Anthropogenic, Ecological and Genetic Factors in Extinction and Conservation. Res. Popul. Ecol. 1998, 40, 259–269. [Google Scholar] [CrossRef]
- Haddad, N.M.; Brudvig, L.A.; Clobert, J.; Davies, K.F.; Gonzalez, A.; Holt, R.D.; Lovejoy, T.E.; Sexton, J.O.; Austin, M.P.; Collins, C.D.; et al. Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci. Adv. 2015, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- WWF. Living Planet Report 2018: Aiming Higher; Grooten, M., Almond, R.E.A., Eds.; World Wildlife Fund: Gland, Switzerland, 2018. [Google Scholar]
- Morita, K.; Sahashi, G.; Miya, M.; Kamada, S.; Kanbe, T.; Araki, H. Ongoing localized extinctions of stream-dwelling white-spotted charr populations in small dammed-off habitats of Hokkaido Island, Japan. Hydrobiologia 2019, 840, 207–213. [Google Scholar] [CrossRef]
- Dunham, J.; Baxter, C.; Fausch, K.; Fredenberg, W.; Kitano, S.; Koizumi, I.; Morita, K.; Nakamura, T.; Rieman, B.; Savvaitova, K.; et al. Evolution, ecology, and conservation of Dolly Varden, white spotted char, and bull trout. Fisheries 2008, 33, 537–550. [Google Scholar] [CrossRef]
- Kikko, T.; Kai, Y.; Nakayama, K. Relationships among tributary length, census population size, and genetic variability of white-spotted charr, Salvelinus leucomaenis, in the Lake Biwa water system. Ichthyol. Res. 2009, 56, 100–104. [Google Scholar] [CrossRef]
- Nakano, S.; Koji, M.; Yamamoto, S. Change of the life cycle of Japanese charr following artificial lake construction by damming. Nippon Suisan Gakkaishi 1990, 56, 1901–1905. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nakajima, M.; Taniguchi, N. Loss of genetic variation and increased population differentiation in geographically peripheral populations of Japanese char Salvelinus leucomaenis. Aquaculture 2010, 308, S20–S27. [Google Scholar] [CrossRef]
- Fausch, K.D.; Nakano, S.; Ishigaki, K. Distribution of two congeneric charrs in streams of Hokkaido Island, Japan: Considering multiple factors across scales. Oecologia 1994, 100, 1–12. [Google Scholar] [CrossRef]
- Kikko, T.; Kai, Y.; Kuwahara, M.; Nakayama, K. Genetic diversity and population structure of white-spotted charr, Salvelinus leucomaenis, in the Lake Biwa water system inferred from AFLP analysis. Ichthyol. Res. 2008, 55, 141–147. [Google Scholar] [CrossRef]
- Morita, K.; Yamamoto, S.; Hoshino, N. Extreme life history change of white-spotted char (Salvelinus ieucomaenis) after damming. Can. J. Fish. Aquat. Sci. 2000, 57, 1300–1306. [Google Scholar] [CrossRef]
- Yamada, T.; Koizumi, I.; Urabe, H.; Nakamura, F. Temperature-dependent swimming performance differs by species: Implications for condition-specific competition between stream salmonids. Zoolog. Sci. 2020, 37, 1–5. [Google Scholar] [CrossRef]
- Woodward, G.; Perkins, D.M.; Brown, L.E. Climate change and freshwater ecosystems: Impacts across multiple levels of organization. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2093–2106. [Google Scholar] [CrossRef] [PubMed]
- Lamphere, B.A.; Blum, M.J. Genetic estimates of population structure and dispersal in a benthic stream fish. Ecol. Freshw. Fish 2012, 21, 75–86. [Google Scholar] [CrossRef]
- Jiang, S.; Luo, M.X.; Gao, R.H.; Zhang, W.; Yang, Y.Z.; Li, Y.J.; Liao, P.C. Isolation-by-environment as a driver of genetic differentiation among populations of the only broad-leaved evergreen shrub Ammopiptanthus mongolicus in Asian temperate deserts. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Palstra, F.P.; Ruzzante, D.E. Genetic estimates of contemporary effective population size: What can they tell us about the importance of genetic stochasticity for wild population persistence? Mol. Ecol. 2008, 17, 3428–3447. [Google Scholar] [CrossRef]
- Yamamoto, S.; Morita, K.; Kitano, S.; Watanabe, K.; Koizumi, I.; Maekawa, K.; Takamura, K. Phylogeography of white-spotted charr (Salvelinus leucomaenis) inferred from mitochondrial DNA sequences. Zoolog. Sci. 2004, 21, 229–240. [Google Scholar] [CrossRef]
- Yamamoto, S.; Morita, K.; Koizumi, I.; Maekawa, K. Genetic differentiation of white-spotted charr (Salvelinus leucomaenis) populations after habitat fragmentation: Spatial-temporal changes in gene frequencies. Conserv. Genet. 2004, 5, 529–538. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kubota, H.; Hasegawa, K.; Nakamura, T. Census and effective population sizes of white-spotted charr (Salvelinus leucomaenis) in a fragmented landscape. Ecol. Freshw. Fish 2016, 25, 612–621. [Google Scholar] [CrossRef]
- Yamamoto, T.; Tsuda, Y.; Takayama, K.; Nagashima, R.; Tateishi, Y.; Kajita, T. The presence of a cryptic barrier in the West Pacific Ocean suggests the effect of glacial climate changes on a widespread sea-dispersed plant, Vigna marina (Fabaceae). Ecol. Evol. 2019, 9, 8429–8440. [Google Scholar] [CrossRef]
- Nunziata, S.O.; Lance, S.L.; Scott, D.E.; Lemmon, E.M.; Weisrock, D.W. Genomic data detect corresponding signatures of population size change on an ecological time scale in two salamander species. Mol. Ecol. 2017, 26, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Shafer, A.B.A.; Wolf, J.B.W.; Alves, P.C.; Bergström, L.; Bruford, M.W.; Brännström, I.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R.; et al. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015, 30, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Minami-Furano-cho Historical Record Compilation committee. Minami-Furano-cho Historical Record; Minami-Furano Town Office: Minami-Furano, Japan, 1991; Book 2. (In Japanese) [Google Scholar]
- Kanayama dam management branch, Hokkaido Regional Development Bureau, MLIT, Japan. Available online: https://www.hkd.mlit.go.jp/sp/sorati_kasen/kluhh400000062t2.html (accessed on 6 June 2020). (In Japanese).
- Suyama, Y.; Matsuki, Y. MIG-seq: An effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Pritchord, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Wagner, A.P.; Taper, M.L. ML-RELATE: A computer program for maximum likelihood estimation of relatedness and relationship. Mol. Ecol. Notes 2006, 6, 576–579. [Google Scholar] [CrossRef]
- Faulks, L.; Östman, Ö. Genetic diversity and hybridisation between native and introduced salmonidae fishes in a Swedish alpine lake. PLoS ONE 2016, 11, e0152732. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, Y.X. Exploring population size changes using SNP frequency spectra. Nat. Genet. 2015, 47, 555–559. [Google Scholar] [CrossRef]
- easySFS. Available online: https://github.com/isaacovercast/easySFS (accessed on 6 June 2020).
- Jacobs, A.; Hughes, M.R.; Robinson, P.C.; Adams, C.E.; Elmer, K.R. The genetic architecture underlying the evolution of a rare piscivorous life history form in brown trout after secondary contact and strong introgression. Genes 2018, 9, 280. [Google Scholar] [CrossRef]
- Delrieu-Trottin, E.; Hubert, N.; Giles, E.C.; Chifflet-Belle, P.; Suwalski, A.; Neglia, V.; Rapu-Edmunds, C.; Mona, S.; Saenz-Agudelo, P. Coping with Pleistocene climatic fluctuations: Demographic responses in remote endemic reef fishes. Mol. Ecol. 2020, 29, 2218–2233. [Google Scholar] [CrossRef]
- Souissi, A.; Bonhomme, F.; Manchado, M.; Bahri-Sfar, L.; Gagnaire, P.A. Genomic and geographic footprints of differential introgression between two divergent fish species (Solea spp.). Heredity 2018, 121, 579–593. [Google Scholar] [CrossRef]
- Tine, M.; Kuhl, H.; Gagnaire, P.A.; Louro, B.; Desmarais, E.; Martins, R.S.T.; Hecht, J.; Knaust, F.; Belkhir, K.; Klages, S.; et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun. 2014, 5, 5770. [Google Scholar] [CrossRef]
- Le Moan, A.; Gagnaire, P.A.; Bonhomme, F. Parallel genetic divergence among coastal-marine ecotype pairs of European anchovy explained by differential introgression after secondary contact. Mol. Ecol. 2016, 25, 3187–3202. [Google Scholar] [CrossRef]
- Fischer, M.C.; Rellstab, C.; Leuzinger, M.; Roumet, M.; Gugerli, F.; Shimizu, K.K.; Holderegger, R.; Widmer, A. Estimating genomic diversity and population differentiation-an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genomics 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Lemopoulos, A.; Prokkola, J.M.; Uusi-Heikkilä, S.; Vasemägi, A.; Huusko, A.; Hyvärinen, P.; Koljonen, M.L.; Koskiniemi, J.; Vainikka, A. Comparing RADseq and microsatellites for estimating genetic diversity and relatedness-Implications for brown trout conservation. Ecol. Evol. 2019, 9, 2106–2120. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, S.; Laporte, M.; Perrier, C.; Sirois, P.; Bernatchez, L. Investigating genomic and phenotypic parallelism between piscivorous and planktivorous lake trout (Salvelinus namaycush) ecotypes by means of RADseq and morphometrics analyses. Mol. Ecol. 2016, 25, 4773–4792. [Google Scholar] [CrossRef] [PubMed]
- Ferchaud, A.L.; Laporte, M.; Perrier, C.; Bernatchez, L. Impact of supplementation on deleterious mutation distribution in an exploited salmonid. Evol. Appl. 2018, 11, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Narum, S.R.; Gallardo, P.; Correa, C.; Matala, A.; Hasselman, D.; Sutherland, B.J.G.; Bernatchez, L. Genomic patterns of diversity and divergence of two introduced salmonid species in Patagonia, South America. Evol. Appl. 2017, 10, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, F.C.; Sauvage, C.; Marie, A.; Garant, D.; Bernatchez, L. Dynamics of introgressive hybridization assessed by SNP population genomics of coding genes in stocked brook charr (Salvelinus fontinalis). Mol. Ecol. 2012, 21, 2877–2895. [Google Scholar] [CrossRef] [PubMed]
- Bourret, V.; Kent, M.P.; Primmer, C.R.; Vasemägi, A.; Karlsson, S.; Hindar, K.; McGinnity, P.; Verspoor, E.; Bernatchez, L.; Lien, S. SNP-array reveals genome-wide patterns of geographical and potential adaptive divergence across the natural range of Atlantic salmon (Salmo salar). Mol. Ecol. 2013, 22, 532–551. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nakajima, M.; Taniguchi, N. Population structure and conservation genetics of anadromous white-spotted char (Salvelinus leucomaenis) on Hokkaido Island: Detection of isolation-by-distance. Environ. Biol. Fishes 2016, 99, 513–525. [Google Scholar] [CrossRef]
- Yamamoto, S.; Morita, K.; Sahashi, G. Spatial and temporal changes in genetic structure and diversity of isolated white-spotted charr (Salvelinus leucomaenis) populations. Hydrobiologia 2019, 840, 35–48. [Google Scholar] [CrossRef]
- Morita, K.; Morita, S.H.; Nagasawa, T.; Kuroki, M. Migratory patterns of anadromous white-spotted charr Salvelinus leucomaenis in Eastern Hokkaido, Japan: The solution to a mystery? J. Ichthyol. 2013, 53, 809–819. [Google Scholar] [CrossRef]
- Kanno, Y.; Vokoun, J.C.; Letcher, B.H. Fine-scale population structure and riverscape genetics of brook trout (Salvelinus fontinalis) distributed continuously along headwater channel networks. Mol. Ecol. 2011, 20, 3711–3729. [Google Scholar] [CrossRef]
- Sandlund, O.T.; Karlsson, S.; Thorstad, E.B.; Berg, O.K.; Kent, M.P.; Norum, I.C.J.; Hindar, K. Spatial and temporal genetic structure of a river-resident Atlantic salmon (Salmo salar) after millennia of isolation. Ecol. Evol. 2014, 4, 1538–1554. [Google Scholar] [CrossRef]
- Kitanishi, S.; Ikeda, T.; Yamamoto, T. Short-term temporal instability in fine-scale genetic structure of masu salmon. Freshw. Biol. 2017, 62, 1655–1664. [Google Scholar] [CrossRef]
- Tsuda, Y.; Nakao, K.; Ide, Y.; Tsumura, Y. The population demography of Betula maximowicziana, a cool-temperate tree species in Japan, in relation to the last glacial period: Its admixture-like genetic structure is the result of simple population splitting not admixing. Mol. Ecol. 2015, 24, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Yokota, A. Population viability of stream-resident salmonids after habitat fragmentation: A case study with white-spotted charr (Salvelinus leucomaenis) by an individual based model. Ecol. Modell. 2002, 155, 85–94. [Google Scholar] [CrossRef]
- Otsuka, S. Gold mines in Yubari and Sorachi, Hokkaido. Jour. Tokyo Geogr. Soc. 1898, 10, 324–333. (In Japanese) [Google Scholar] [CrossRef]
- Minami-Furano-cho Historical Record Compilation Committee. Minami-Furano-cho Historical Record; Minami-Furano Town Office: Minami-Furano, Japan, 1991; Book 1. (In Japanese) [Google Scholar]
- Nagasaka, A.; Nakamura, F. The influences of land-use changes on hydrology and riparian environment in a northern Japanese landscape. Landsc. Ecol. 1999, 14, 543–556. [Google Scholar] [CrossRef]
- Buhl, K.J.; Hamilton, S.J. Comparative toxicity of inorganic contaminants released by placer mining to early life stages of salmonids. Ecotoxicol. Environ. Saf. 1990, 20, 325–342. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nakajima, M.; Taniguchi, N. Mitochondrial genetic evidence for recent population expansion of the white-spotted char (Salvelinus leucomaenis) without geographic patterns from Northern Japan to Central Honshu. Fish Genet. Breed. Sci. 2015, 44, 5–16. [Google Scholar]
- Niizato, T.; Shigeno, K.; Takashimizu, Y. Ainu oral traditions and historical records on earthquakes in Hokkaido, Japan. Hist. Earthq. 2006, 20, 121–136, (In Japanese with English abstract). [Google Scholar]
- Lapierre, M.; Lambert, A.; Achaz, G. Accuracy of demographic inferences from the site frequency spectrum: The case of the yoruba population. Genetics 2017, 206, 139–449. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. A Primer of Conservation Genetics; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Kitano, S.; Ohdachi, S.; Koizumi, I.; Hasegawa, K. Hybridization between native white-spotted charr and nonnative brook trout in the upper Sorachi River, Hokkaido, Japan. Ichthyol. Res. 2014, 61, 1–8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Name | N | HO (SE) | HE (SE) | FIS (SE) |
|---|---|---|---|---|---|
| T | Tomamu River | 13 | 0.213 (0.010) | 0.211 (0.009) | 0.025 (0.113) |
| Sc | A tributary of the Seesorapuchi River | 12 | 0.209 (0.011) | 0.209 (0.009) | 0.043 (0.100) |
| E | Ecchudantai-no-sawa River | 11 | 0.215 (0.010) | 0.206 (0.008) | 0.018 (0.094) |
| IK | Ikutora River | 16 | 0.203 (0.009) | 0.204 (0.008) | 0.031 (0.128) |
| T | Sc | E | IK | |
|---|---|---|---|---|
| T | 0.000 | |||
| Sc | 0.017 * | 0.000 | ||
| E | 0.011 | 0.004 | 0.000 | |
| IK | 0.022 * | 0.016 * | 0.008 | 0.000 |
| Pairs | FS | HS | U |
|---|---|---|---|
| Within | |||
| T | 0.051 | 0.077 | 0.872 |
| Sc | 0.015 | 0.106 | 0.879 |
| E | 0.000 | 0.036 | 0.964 |
| IK | 0.008 | 0.092 | 0.900 |
| Between | |||
| T-Sc | 0.000 | 0.026 | 0.974 |
| T-E | 0.000 | 0.014 | 0.986 |
| T-IK | 0.000 | 0.014 | 0.986 |
| Sc-E | 0.000 | 0.015 | 0.985 |
| Sc-IK | 0.000 | 0.016 | 0.984 |
| E-IK | 0.000 | 0.034 | 0.966 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakajima, S.; Hirota, S.K.; Matsuo, A.; Suyama, Y.; Nakamura, F. Genetic Structure and Population Demography of White-Spotted Charr in the Upstream Watershed of a Large Dam. Water 2020, 12, 2406. https://doi.org/10.3390/w12092406
Nakajima S, Hirota SK, Matsuo A, Suyama Y, Nakamura F. Genetic Structure and Population Demography of White-Spotted Charr in the Upstream Watershed of a Large Dam. Water. 2020; 12(9):2406. https://doi.org/10.3390/w12092406
Chicago/Turabian StyleNakajima, Souta, Shun K. Hirota, Ayumi Matsuo, Yoshihisa Suyama, and Futoshi Nakamura. 2020. "Genetic Structure and Population Demography of White-Spotted Charr in the Upstream Watershed of a Large Dam" Water 12, no. 9: 2406. https://doi.org/10.3390/w12092406
APA StyleNakajima, S., Hirota, S. K., Matsuo, A., Suyama, Y., & Nakamura, F. (2020). Genetic Structure and Population Demography of White-Spotted Charr in the Upstream Watershed of a Large Dam. Water, 12(9), 2406. https://doi.org/10.3390/w12092406