Fate of Diclofenac and Its Transformation and Inorganic By-Products in Different Water Matrices during Electrochemical Advanced Oxidation Process Using a Boron-Doped Diamond Electrode


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Water Matrices
2.3. Experimental Setup and Sampling
2.4. Analysis
3. Results and Discussion
3.1. Removal of Diclofenac
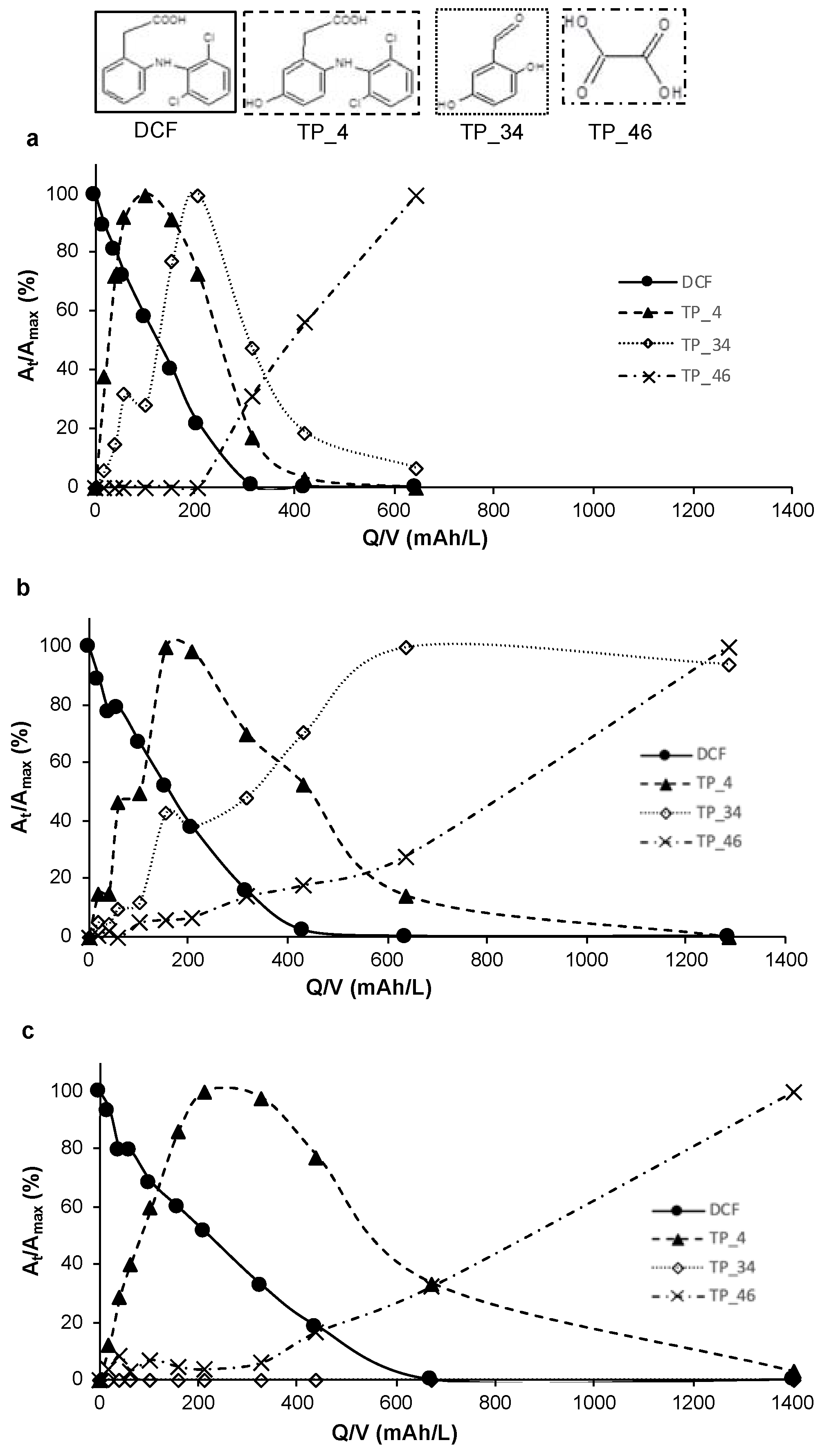
3.2. Formation and Fate of Transformation Products
3.3. Inorganic By-Product Formation
3.4. Recommended Operating Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ternes, T.A.; Meisenheimer, M.; McDowell, D.; Sacher, F.; Brauch, H.-J.; Haist-Gulde, B.; Preuss, G.; Wilme, U.; Zulei-Seibert, N. Removal of pharmaceuticals during drinking water treatment. Environ. Sci. Technol. 2002, 36, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.M.; Göbel, A.; Joss, A.; Hermann, N.; Löffler, D.; McArdell, C.S.; Ried, A.; Siegrist, H.; Ternes, T.; von Gunten, U. Oxidation of pharmaceuticals during ozonation of municipal wastewater effluents: A pilot study. Environ. Sci. Technol. 2005, 39, 8014–8022. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, E.S.; di Bernardo Dantas, A.; di Bernardo, L.; Vieira, E.M. Removal of diclofenac by conventional drinking water treatment processes and granular activated carbon filtration. Chemosphere 2013, 92, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Queloz, P.; Brovelli, A.; Margot, J.; Barry, D.A. Enhancement of micropollutant degradation at the outlet of small wastewater treatment plants. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Schröder, P.; Helmreich, B.; Škrbić, B.; Carballa, M.; Papa, M.; Pastore, C.; Emre, Z.; Oehmen, A.; Langenhoff, A.; Molinos, M.; et al. Status of hormones and pain killers in wastewater effluents across several European states—Consideration for the EU watch list considering estradiol and diclofenac. Environ. Sci. Pollut. Res. 2016, 2, 12835–12866. [Google Scholar] [CrossRef]
- Lonappan, L.; Kaur Brar, S.; Kaur Das, R.; Verma, M.; Surampalli, R.Y. Diclofenac and its transformation products: Environmental occurence and toxicity—A review. Environ. Int. 2016, 96, 127–138. [Google Scholar] [CrossRef]
- Regnery, J.; Wing, A.D.; Alidina, M.; Drewes, J.E. Biotransformation of trace organic chemical attenuation during groundwater recharge: How useful are first-order rate constants? J. Contam. Hydrol. 2015, 17, 65–75. [Google Scholar] [CrossRef]
- Sein, M.M.; Zedda, M.; Tuerk, J.; Schmidt, T.C.; Golloch, A.; von Sonntag, C. Oxidation of diclofenac with ozone in aqueous solution. Environ. Sci. Technol. 2008, 42, 6656–6662. [Google Scholar] [CrossRef]
- Huber, C.; Preis, M.; Harvey, P.J.; Grosse, S.; Letzel, T.; Schröder, P. Emerging pollutants and plants—Metabolic activation of diclofenac by peroxidases. Chemosphere 2016, 146, 435–441. [Google Scholar] [CrossRef]
- Stülten, D.; Zühlke, S.; Lamshöft, M.; Spiteller, M. Occurrence of diclofenac and selected metabolites in sewage effluents. Sci. Tot. Environ. 2008, 405, 310–316. [Google Scholar] [CrossRef]
- Zhao, X.; Hou, Y.; Liu, H.; Qiang, Z.; Qu, J. Electro-oxidation of diclofenac at boron-doped diamond: Kinetics and mechanism. Electrochim. Acta 2009, 54, 4172–4179. [Google Scholar] [CrossRef]
- Coelho, A.D.; Sans, C.; Agüera, A.; Gómez, M.J.; Esplugas, S.; Dezotti, M. Effects of ozone pre-treatment on diclofenac: Intermediates, biodegradability and toxicity assessment. Sci. Total Environ. 2009, 407, 3572–3578. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Estrada, L.A.; Malato, S.; Gernjak, W.; Agüera, A.; Thurman, E.M.; Ferrer, I.; Fernández-Alba, A.R. Photo-Fenton Degradation of Diclofenac: Identification of main intermediates and degradation pathway. Environ. Sci. Technol. 2005, 39, 8300–8306. [Google Scholar] [CrossRef] [PubMed]
- Letzel, M.; Metzner, G.; Letzel, T. Exposure of the pharmaceutical diclofenac based on long-term measurements of the aquatic input. Environ. Int. 2009, 35, 363–368. [Google Scholar] [CrossRef] [PubMed]
- European Union Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy. Off. J. Eur. Union 2013, 226, 1–17.
- Capodaglio, A.G.; Bojanowska-Czajka, A.; Trojanowicz, M. Comparison of different advanced degradation processes for the removal of the pharmaceutical compounds diclofenac and carbamazepine from liquid solutions. Environ. Sci. Poll. Res. 2018, 25, 27704–27723. [Google Scholar] [CrossRef]
- Menapace, H.M.; Diaz, N.; Weiss, S. Electrochemical treatment of pharmaceutical wastewater by combining anodic oxidation with ozonation. J. Environ. Sci. Health Part A 2008, 43, 961–968. [Google Scholar] [CrossRef]
- Miklos, D.B.; Remy, C.; Jekel, M.; Linden, K.G.; Drewes, J.E.; Hübner, U. Evaluation of advanced oxidation processes for water and wastewater treatment—A critical review. Water Res. 2018, 77, 1137–1148. [Google Scholar] [CrossRef]
- Tröster, I.; Schäfer, L.; Fryda, M.; Matthée, T. Electrochemical advanced oxidation process using DiaChem® electrodes. Water Sci. Technol. 2004, 49, 207–212. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Ferro-García, M.A.; Prados-Joya, G.; Ocampo-Pérez, R. Pharmaceuticals as emerging contaminants and their removal from water. A review. Chemosphere 2013, 93, 1268–1287. [Google Scholar] [CrossRef]
- Ribeiro, A.R.; Nunes, O.C.; Pereira, M.F.R.; Silva, A.M.T. An overview on the advanced oxidation processes applied for the treatment of water pollutants defined in the recently launched Directive 2013/39/EU. Environ. Int. 2015, 7, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Oturan, M.A.; Aaron, J.-J. Advanced Oxidation Processes in Water/Wastewater Treatment: Principles and Applications. A Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Rajab, M.; Greco, G.; Heim, C.; Helmreich, B.; Letzel, T. Serial coupling of RP and zwitterionic hydrophilic intraction LC-MS: Suspects screening of diclofenac transformation products by oxidation with a boron-doped diamond electrode. J. Sep. Sci. 2013, 36, 3011–3018. [Google Scholar] [PubMed]
- Helmreich, B.; Metzger, S. Post-treatment for micropollutants removal. In Innovative Wastewater Treatment & Resource Recovery Technologies: Impacts on Energy, Economy and Environment; Lema, J.M., Suarez Martinez, S., Eds.; IWA-Publishing: London, UK, 2017; pp. 2014–2232. ISBN 13: 9781780407869. [Google Scholar]
- Brillas, E.; Garcia-Segura, S.; Skoumal, M.; Arias, C. Electrochemical incineration of diclofenac in neutral aqueous medium by anodic oxidation using Pt and boron-doped diamond anodes. Chemosphere 2010, 79, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Coria, G.; Nava, J.L.; Carreno, G. Electrooxidation of diclofenac in synthetic pharmaceutical wastewater using an electrochemical reactor equipped with a boron doped diamond electrode. J. Mex. Chem. Soc. 2014, 58, 303–308. [Google Scholar] [CrossRef]
- Faber, H.; Lutze, H.; Lareo, P.L.; Frensemeier, L.; Vogel, M.; Schmidt, T.C.; Karst, U. Liquid chromatography/mass spectrometry to study oxidative degradation of environmentally relevant pharmaceuticals by electrochemistry and ozonation. J. Chromatogr. A 2014, 1343, 152–159. [Google Scholar] [CrossRef]
- Vedenyapina, M.D.; Strel’tsova, E.D.; Davshan, N.A.; Vedenyapin, A.A. Study of the electrochemical degradation of diclofenac on a boron doped diamond electrode by UV spectroscopy. Russ. J. Appl. Chem. 2011, 84, 204–207. [Google Scholar] [CrossRef]
- Frontistis, Z.; Brebou, C.; Venieri, D.; Mantzavinos, D.; Katsaounis, A. BDD anodic oxidation as tertiary wastewater treatment for the removal of emerging micro-pollutants, pathogens and organic matter. J. Chem. Technol. Biot. 2011, 86, 1233–1236. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Quiroz Alfaro, M.A. Recent environmental applications of diamond electrode: Critical review. J. Environ. Eng. Manag. 2008, 18, 155–172. [Google Scholar]
- Rajab, M.; Heim, C.; Letzel, T.; Drewes, J.E.; Helmreich, B. Oxidation of bisphenol A by boron-doped diamond electrode in different water matrices: Transformation products and inorganic by-products. Int. J. Environ. Sci. Technol. 2016, 13, 2539–2548. [Google Scholar] [CrossRef]
- Schmalz, V.; Dittmar, T.; Fischer, D.; Worch, E. Diamond electrodes in decentralized wastewater treatment: Electrochemical degradation of the chemical oxygen demand (COD) in wastewater with high organic loads from hardening shops. CIT 2008, 80, 1545–1550. [Google Scholar]
- Honda, Y.; Ivandini, T.A.; Watanabe, T.; Murata, K.; Einaga, Y. An electrolyte-free system for ozone generation using heavily boron-doped diamond electrodes. Diam. Relat. Mater. 2013, 40, 7–11. [Google Scholar] [CrossRef]
- Michaud, P.-A.; Panizza, M.; Ouattara, L.; Diaco, T.; Foti, G.; Comninellis, C. Electrochemical oxidation of water on synthetic boron-doped diamond thin film anodes. J. Appl. Electrochem. 2003, 33, 151–154. [Google Scholar] [CrossRef]
- Panizza, M.; Brillas, E.; Comninellis, C. Application of boron-doped diamond electrodes for wastewater treatment. J. Environ. Eng. Manag. 2008, 18, 139–153. [Google Scholar]
- Lara-Ramos, J.A.; Saez, C.; Machuca-Martínez, F.; Rodrigo, M.A. Electro-ozonizers: A new approach for an old problem. Sep. Purif. Technol. 2020, 241. [Google Scholar] [CrossRef]
- Von Gunten, U. Ozonation of drinking water: Part I. Oxidation kinetics and product formation. Water Res. 2003, 37, 1443–1467. [Google Scholar] [CrossRef]
- Calza, P.; Sakkas, V.A.; Medana, C.; Baiocchi, C.; Dimou, A.; Pelizzetti, E.; Albanis, T. Photocatalytic degradation study of diclofenac over aqueous TiO2 suspension. Appl. Catal. B-Environ. 2006, 67, 197–205. [Google Scholar] [CrossRef]
- Merel, S.; Lege, S.; Yanez Heras, J.E.; Zwiener, C. Assessment of n-oxide formation during wastewater ozonation. Environ. Sci. Technol. 2017, 51, 410–417. [Google Scholar] [CrossRef]
- Ternes, T.A.; Prasse, C.; Eversloh, C.L.; Knopp, G.; Cornel, P.; Schulte-Oehlmann, U.; Schwartz, T.; Alexander, J.; Seitz, W.; Coors, A.; et al. Integrated evaluation concept to assess the efficiacy of advanced wastewater treatment processes for the elimination of micropollutants and pathogens. Environ. Sci. Technol. 2017, 51, 308–319. [Google Scholar] [CrossRef]
- Wert, E.C.; Gonzales, S.; Dong, M.M.; Rosario-Ortiz, F.L. Evaluation of enhanced coagulation pretreatment to improve ozone oxidation efficiency in wastewater. Water Res. 2009, 45, 5191–5199. [Google Scholar] [CrossRef]
- Heim, C.; Ureña de Vivanco, M.; Rajab, M.; Müller, E.; Letzel, T.; Helmreich, B. Rapid inactivation of waterborne bacteria using boron-doped diamond electrodes. Int. J. Environ. Sci. Technol. 2015, 12, 3061–3070. [Google Scholar] [CrossRef]
- Greco, G.; Grosse, S.; Letzel, T. Serial coupling of reversed-phase and zwitterionic hydrophilic interaction LC/MS for the analysis of polar and nonpolar phenols in wine. J. Sep. Sci. 2013, 36, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Stadlmair, L.F.; Grosse, S.; Letzel, T.; Drewes, J.E.; Grassmann, J. Comprehensive MS-based screening and identification of pharmaceutical transformation products formed during enzymatic conversion. Anal. Bioanal. Chem. 2019, 411, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Baird, R.B.; Eaton, A.D.; Rice, E.W. Standard Methods for the Examination of Water and Wastewater, 23rd ed.; American Public Health Association: Washington, DC, USA, 2017. [Google Scholar]
- Bader, H.; Hoigné, J. Determination of ozone in water by the indigo method. Water Res. 1981, 15, 449–456. [Google Scholar] [CrossRef]
- Heim, C.; Ureña de Vivanco, M.; Rajab, M.; Glas, K.; Horn, H.; Helmreich, B.; Letzel, T. Ozone II: Characterization of in situ ozone generation using diamond electrodes. BrewingScience 2011, 64, 83–88. [Google Scholar]
- Bergmann, H.M.E.; Rollin, J.; Iourtchouk, T. The occurence of perchlorate during drinking water electrolysis using BDD anodes. Electrochim. Acta 2009, 54, 2102–2107. [Google Scholar] [CrossRef]
- Bergmann, H.M.E.; Rollin, J. Product and by-product formation in laboratory studies on disinfection electrolysis of water using boron-doped diamond electrodes. Catal. Today 2007, 124, 198–203. [Google Scholar] [CrossRef]
- Polcaro, A.M.; Vacca, A.; Mascia, M.; Ferrara, F. Product and by-product formation in electrolysis of dilute chloride solutions. J. Appl. Electrochem. 2008, 38, 979–984. [Google Scholar] [CrossRef]
- Lee, Y.; von Gunten, U. Oxidative transformation of micropollutants during municipal wastewater treatment: Comparison of kinetic aspects of selective (chlorine, chlorine dioxide, ferrate VI, and ozone) and non-selective oxidants (hydroxyl radical). Water Res. 2010, 44, 555–566. [Google Scholar] [CrossRef]
- Polcaro, A.M.; Vacca, A.; Mascia, M.; Palmas, S.; Rodriguez Ruiz, J. Electrochemical treatment of waters with BDD anodes: Kinetics of the reactions involving chlorides. J. Appl. Electrochem. 2009, 39, 2083–2092. [Google Scholar] [CrossRef]
- Soufan, M.; Deborde, M.; Legube, B. Aqueous chlorination of diclofenac: Kinetic study and transformation products identification. Water Res. 2012, 46, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Vogna, D.; Marotta, R.; Napolitano, A.; Andreozzi, R.; d’Ischia, M. Advanced oxidation of the pharmaceutical drug diclofenac with UV/H2O2 and ozone. Water Res. 2004, 38, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Faber, H.; Melles, D.; Brauckmann, C.; Wehe, C.A.; Wentker, K.; Karst, U. Simulation of the oxidative metabolism of diclofenac by electrochemistry/(liquid chromatography/)mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Gandini, D.; Mahé, E.; Michaud, P.A.; Haenni, W.; Perret, A.; Comninellis, C. Oxidation of carboxylic acids at boron-doped diamond electrodes for wastewater treatment. J. Appl. Electrochem. 2000, 30, 1345–1350. [Google Scholar] [CrossRef]
- Bergmann, H.M.E.; Iourtchouk, T.; Rollin, J. The occurence of bromate and perbromate on BDD anodes during electrolysis of aqueous systems containing bromide: First systematic experimental studies. J. Appl. Electrochem. 2011, 41, 1109–1123. [Google Scholar] [CrossRef]
- Palmas, S.; Polcaro, A.M.; Vacca, A.; Mascia, M.; Ferrara, F. Influence of the operating conditions on the electrochemical disinfection process of natural waters at BDD electrodes. J. Appl. Electrochem. 2007, 37, 1357–1365. [Google Scholar] [CrossRef]
- Von Gunten, U. Ozonation of drinking water: Part II. Disinfection and by-product formation in presence of bromide, iodide or chlorine. Water Res. 2003, 37, 1469–1487. [Google Scholar] [CrossRef]
- Sánchez-Carretero, A.; Sáez, C.; Cañizares, P.; Rodrigo, M.A. Electrochemical production of perchlorates using conductive diamond electrolyses. Chem. Eng. J. 2011, 166, 710–714. [Google Scholar] [CrossRef]
- Isidro, J.; Brackemeyer, D.; Sáez, C.; Llanos, J.; Lobato, J.; Cañizares, P.; Matthée, T.; Rodrigo, M.A. Testing the use of cells equipped with solid polymer electrolytes for electro-disinfection. Sci. Total Environ. 2020, 725. [Google Scholar] [CrossRef]
- Rajab, M.; Heim, C.; Letzel, T.; Drewes, J.E.; Helmreich, B. Electrochemical disinfection using boron-doped diamond electrode—The synergetic effects of in situ ozone and free chlorine generation. Chemosphere 2015, 121, 47–53. [Google Scholar] [CrossRef]
- Heeb, M.B.; Criquet, J.; Zimmermann-Steffens, S.G.; von Gunten, U. Oxidative treatment of bromide-containing waters: Formation of bromine and its reactions with inorganic and organic compounds—A critical review. Water Res. 2014, 48, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Mascia, M.; Palmas, S.; Mais, L.; Rizzardini, S. On the formation of bromate and chlorate ions during electrolysis with boron-doped diamond anode for seawater treatment. J. Chem. Technol. Biotechnol. 2013, 88, 2244–2251. [Google Scholar] [CrossRef]
- Bergmann, M.E.H.; Korparal, A.S.; Iourtchouk, T. Electrochemical advanced oxidation processes, formation of halogenate and perhalogenate species—A critical review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 348–390. [Google Scholar] [CrossRef]
- Bojanowska-Czajka, A.; Kciuk, G.; Gumiela, M.; Borowiecka, S.; Nalecz-Jawecki, G.; Koc, A.; Garcia-Reyes, J.F.; Solpan Ozbay, D.; Trojanowicz, M. Analytical, toxicological and kinetic investigation of decomposition of the drug diclofenac in waters and wastes using gamma radiation. Environ. Sci. Pollut. Res. 2015, 22, 20255–20270. [Google Scholar] [CrossRef] [PubMed]
- Diniz, M.S.; Salgado, R.; Pereira, V.J.; Carvalho, G.; Oehmen, A.; Reis, M.A.M.; Noronha, J.P. Ecotoxicity of ketoprofen, diclofenac, atenolol and their photolysis byproducts in zebrafish (Danio rerio). Sci. Total Environ. 2015, 505, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Nie, E.; Xu, J.; Yan, S.; Cooper, W.J.; Song, W. Degradation of Diclofenac by Advanced Oxidation and Reduction Processes: Kinetic Studied, Degradation Pathways and Toxicity Assessments. Water Res. 2013, 47, 1909–1918. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Ferro, S.; de Battisti, A. Electrochemical incineration of oxalic acid: Reactivity and engineering parameters. J. Appl. Electrochem. 2005, 35, 1087–1093. [Google Scholar] [CrossRef]
- Hübner, U.; von Gunten, U.; Jekel, M. Evaluation of the persistence of transformation products from ozonation of trace organic compounds—A critical review. Water Res. 2015, 68, 150–170. [Google Scholar] [CrossRef]
- Liwarska-Bizukojc, E.; Galamon, M.; Bernat, P. Kinetics of Biological Removal of the Selected Micropollutants and Their Effect on Activated Sludge Biomass. Water Air Soil Pollut. 2018, 229, 356. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Deionized Water | Hard Drinking Water | Wastewater Effluent |
|---|---|---|---|---|
| chloride | mg/L | n.d. | 250 | 180 ± 10 |
| bromide | mg/L | n.d. | 1.0 | <0.5 |
| nitrate | mg/L | n.d. | 50.0 | 48.5 |
| sulfate | mg/L | n.d. | 200 | 45.4 |
| iodide | mg/L | n.d. | 0.1 | n.d. |
| DOC | mmol/L | n.d. | 3.6 | 12.0 |
| pH | - | 5.5 | 8.9 | 8.0 |
| el. conductivity | µS/cm | 0.06 | 1230 | 1170 |
| Deionized Water | Drinking Water | Wastewater Effluent | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| j | Q/V | Br− | Cl− | ClO3− | ClO4− | Q/V | Br− | Cl− | ClO3− | ClO4− | Q/V | Br− | Cl− | ClO3− | ClO4− | ||
| (mA/cm2) | (mAh/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mAh/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mAh/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | ||
| 42 | 0 | 0.989 | 233 | <0.5 | <1 | 0 | <0.5 | 186 | <0.5 | <1 | |||||||
| 14.6 | 0.898 | 219 | 0.709 | <1 | 29.9 | <0.5 | 184 | 0.913 | <1 | ||||||||
| 29.9 | 0.841 | 215 | 1.12 | 1.26 | 95.3 | <0.5 | 176 | 2.61 | 2.34 | ||||||||
| 195 | 0.406 | 214 | 8.63 | 8.15 | 199 | <0.5 | 175 | 5.95 | 4.96 | ||||||||
| 167 | 0 | 1.05 | 234 | <0.5 | <1 | 0 | <0.5 | 186 | <0.5 | <1 | |||||||
| 58.8 | 0.911 | 219 | 0.987 | 2.35 | 121 | <0.5 | 176 | 1.25 | 2.50 | ||||||||
| 120 | 0.825 | 221 | 1.79 | 4.59 | 380 | <0.5 | 170 | 4.07 | 8.18 | ||||||||
| 375 | 0.616 | 218 | 5.55 | 17.7 | 791 | <0.5 | 167 | 9.40 | 19.6 | ||||||||
| 292 | 0 | <0.05 | 0.096 | <0.05 | <0.1 | 0 | 1.06 | 239 | <0.5 | <1 | 0 | <0.5 | 186 | <0.5 | <1 | ||
| 101 | <0.05 | 0.794 | <0.05 | 0.115 | 102 | 0.818 | 222 | 1.08 | 4.07 | 214 | <0.5 | 185 | 1.82 | 4.55 | |||
| 207 | <0.05 | 1.50 | <0.05 | 0.218 | 208 | 0.703 | 222 | 1.86 | 11.7 | 674 | <0.5 | 177 | 5.59 | 16.9 | |||
| 644 | <0.05 | 3.10 | 0.108 | 0.921 | 637 | 0.541 | 215 | 6.55 | 26.3 | 1403 | <0.5 | 169 | 11.9 | 30.0 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heim, C.; Rajab, M.; Greco, G.; Grosse, S.; Drewes, J.E.; Letzel, T.; Helmreich, B. Fate of Diclofenac and Its Transformation and Inorganic By-Products in Different Water Matrices during Electrochemical Advanced Oxidation Process Using a Boron-Doped Diamond Electrode. Water 2020, 12, 1686. https://doi.org/10.3390/w12061686
Heim C, Rajab M, Greco G, Grosse S, Drewes JE, Letzel T, Helmreich B. Fate of Diclofenac and Its Transformation and Inorganic By-Products in Different Water Matrices during Electrochemical Advanced Oxidation Process Using a Boron-Doped Diamond Electrode. Water. 2020; 12(6):1686. https://doi.org/10.3390/w12061686
Chicago/Turabian StyleHeim, Carolin, Mohamad Rajab, Giorgia Greco, Sylvia Grosse, Jörg E. Drewes, Thomas Letzel, and Brigitte Helmreich. 2020. "Fate of Diclofenac and Its Transformation and Inorganic By-Products in Different Water Matrices during Electrochemical Advanced Oxidation Process Using a Boron-Doped Diamond Electrode" Water 12, no. 6: 1686. https://doi.org/10.3390/w12061686
APA StyleHeim, C., Rajab, M., Greco, G., Grosse, S., Drewes, J. E., Letzel, T., & Helmreich, B. (2020). Fate of Diclofenac and Its Transformation and Inorganic By-Products in Different Water Matrices during Electrochemical Advanced Oxidation Process Using a Boron-Doped Diamond Electrode. Water, 12(6), 1686. https://doi.org/10.3390/w12061686