Adaptive Variations of Sediment Microbial Communities and Indication of Fecal-Associated Bacteria to Nutrients in a Regulated Urban River


Abstract
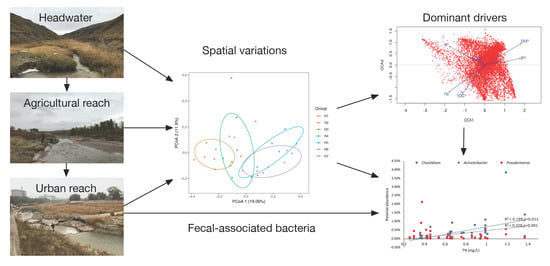
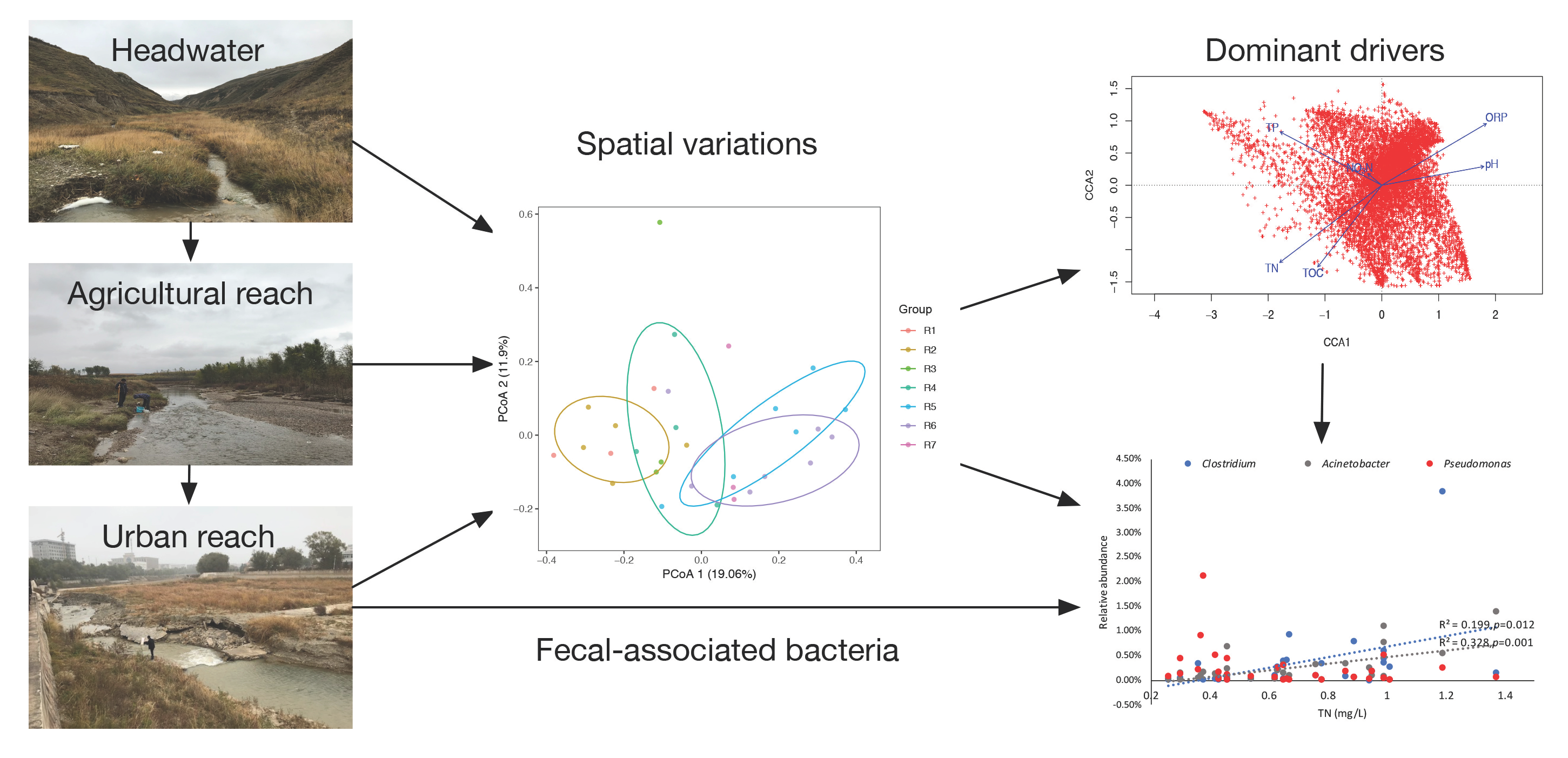{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
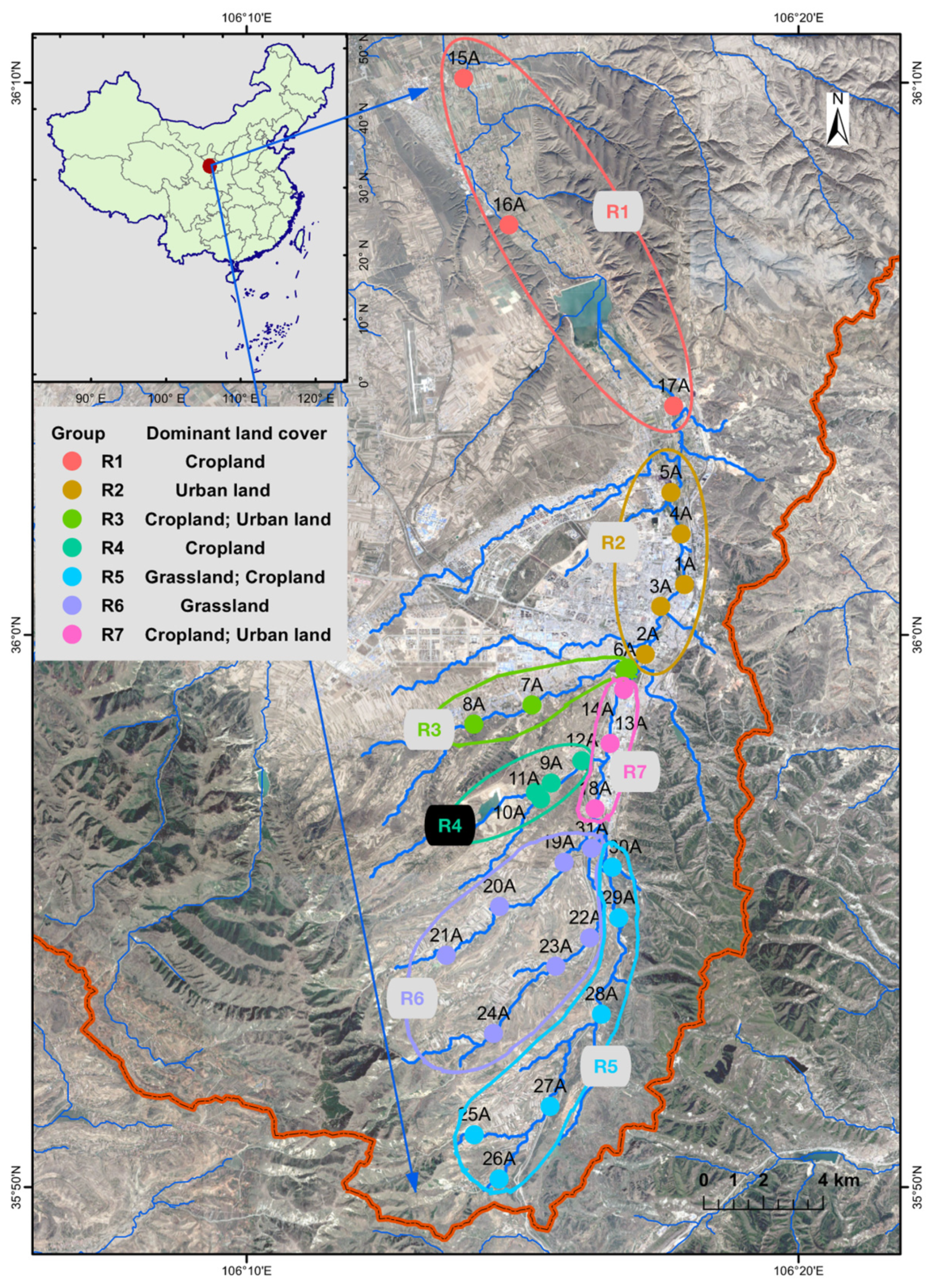
2.1. Sample Collection and Environmental Variables
2.2. Extraction of DNA and Sequencing of 16S rRNA Amplicon
2.3. Bioinformatics Analysis
2.4. Diversity and Statistical Analysis
3. Results
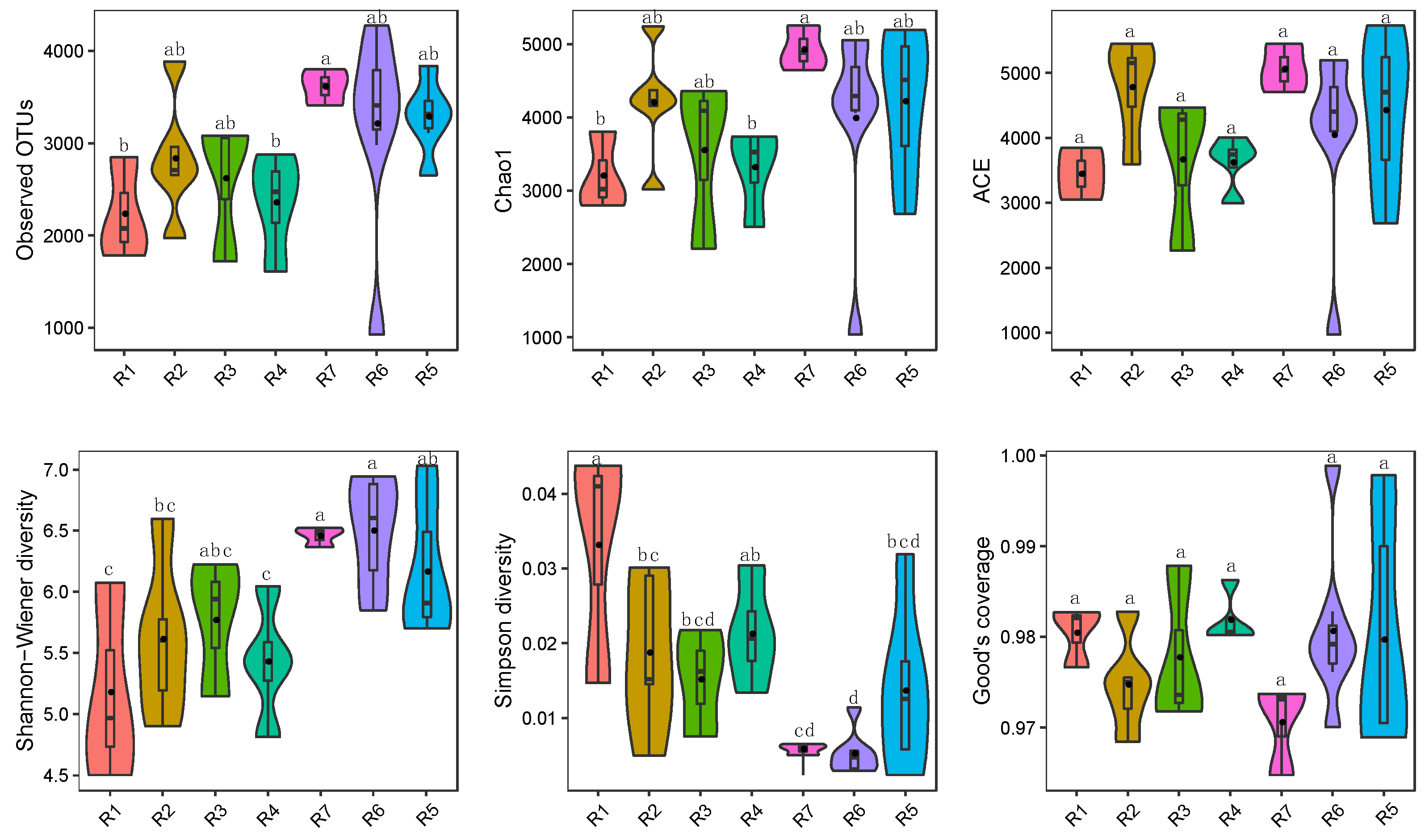
3.1. Microbial Alpha Diversity in Qingshui River
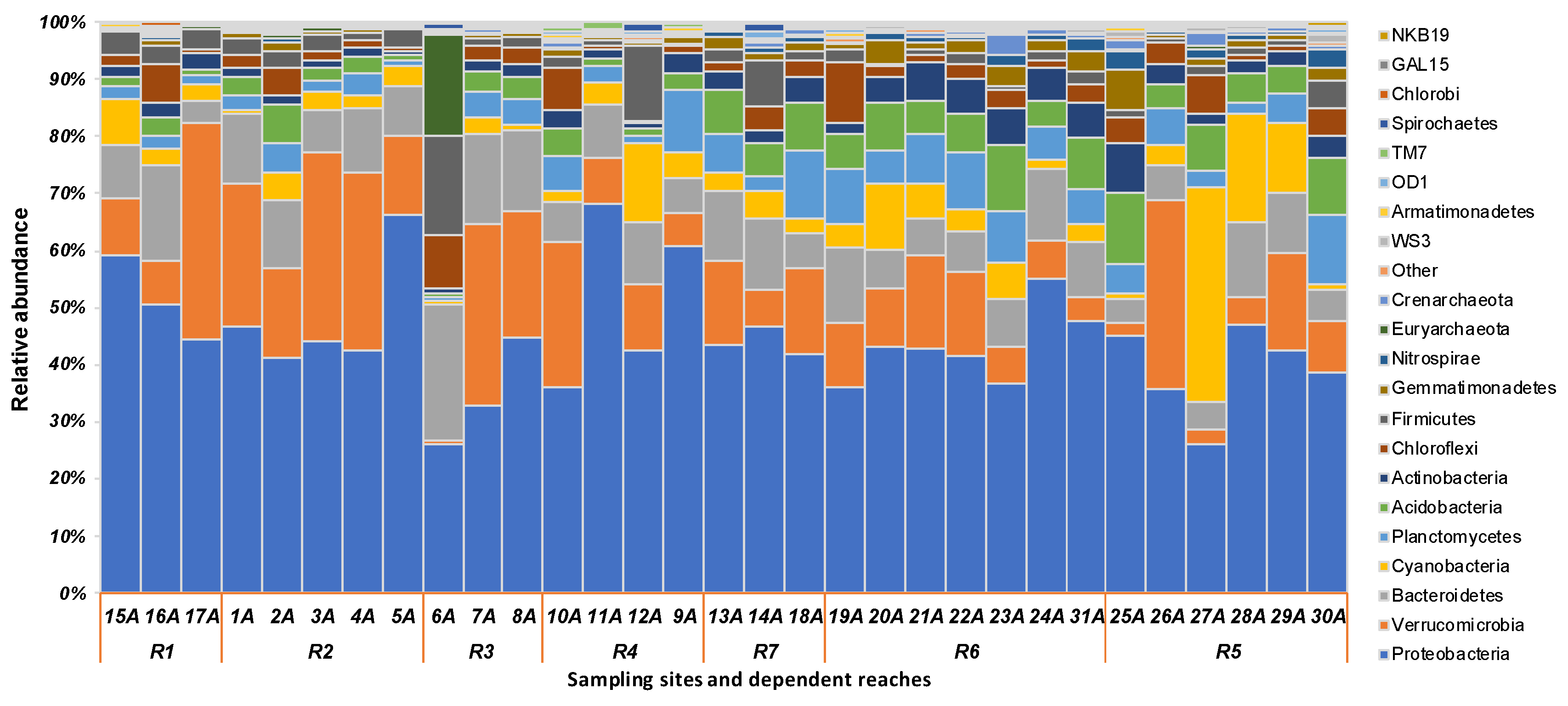
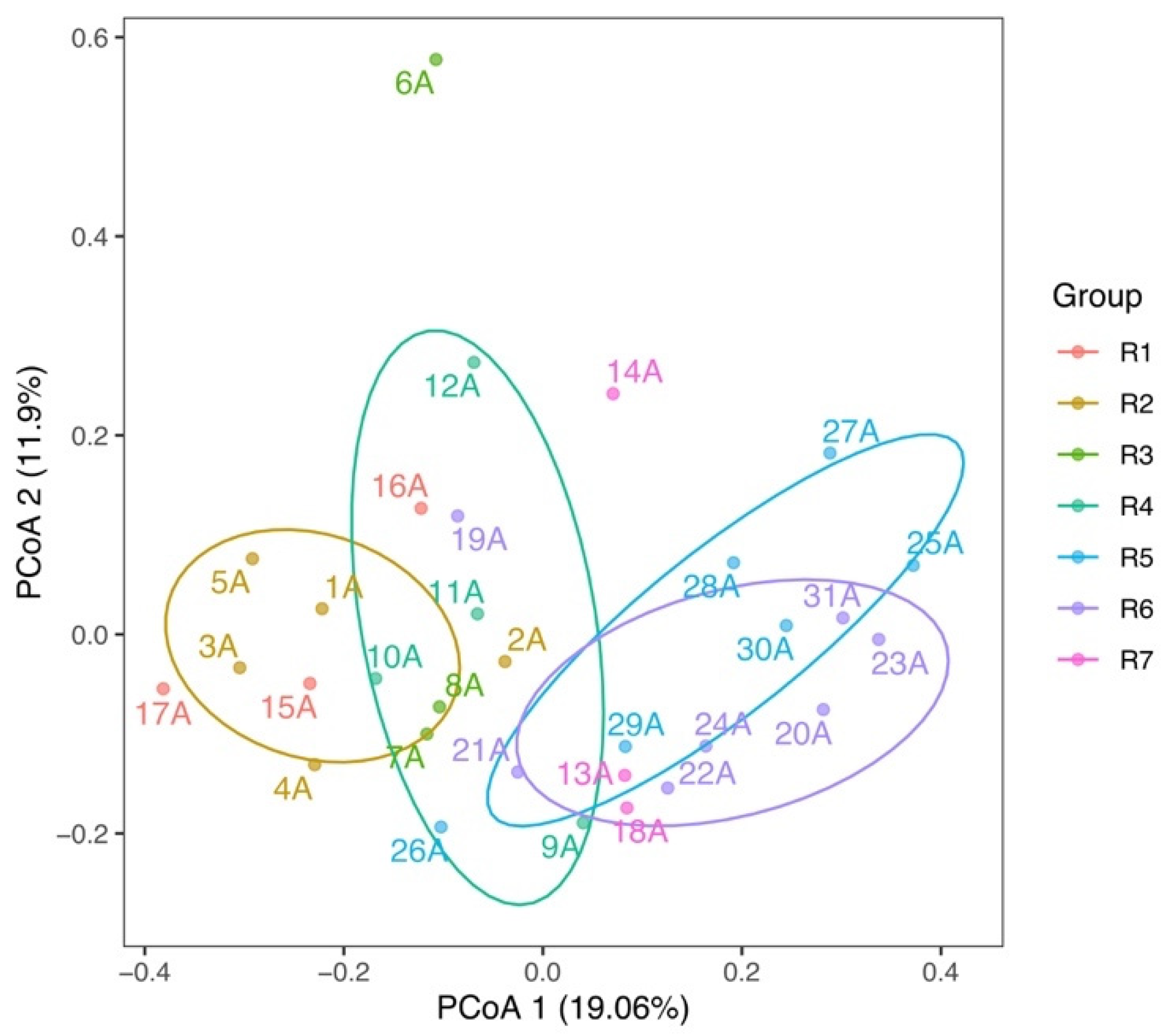
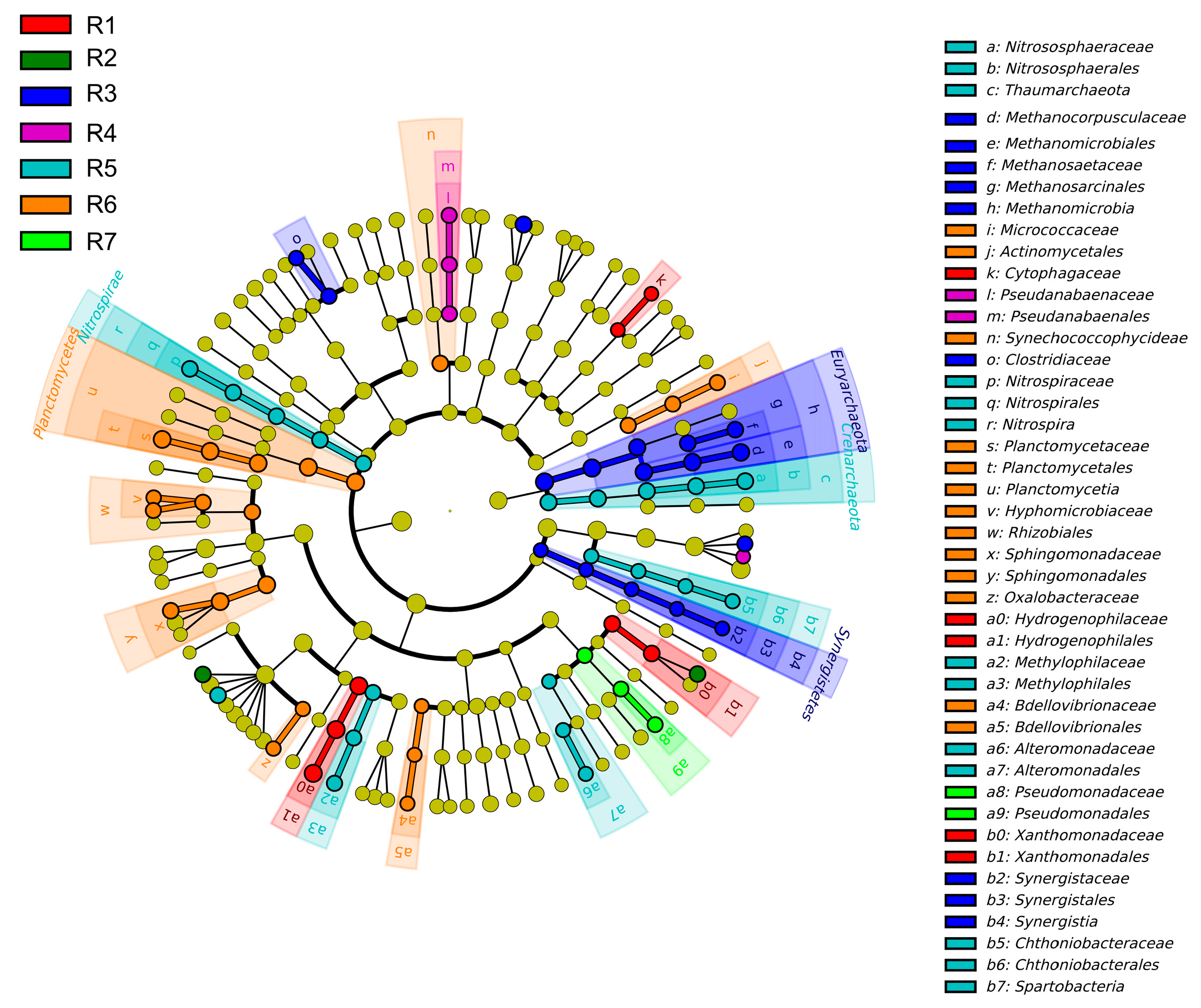
3.2. Spatial Variations of Microbial Composition and Beta Diversity
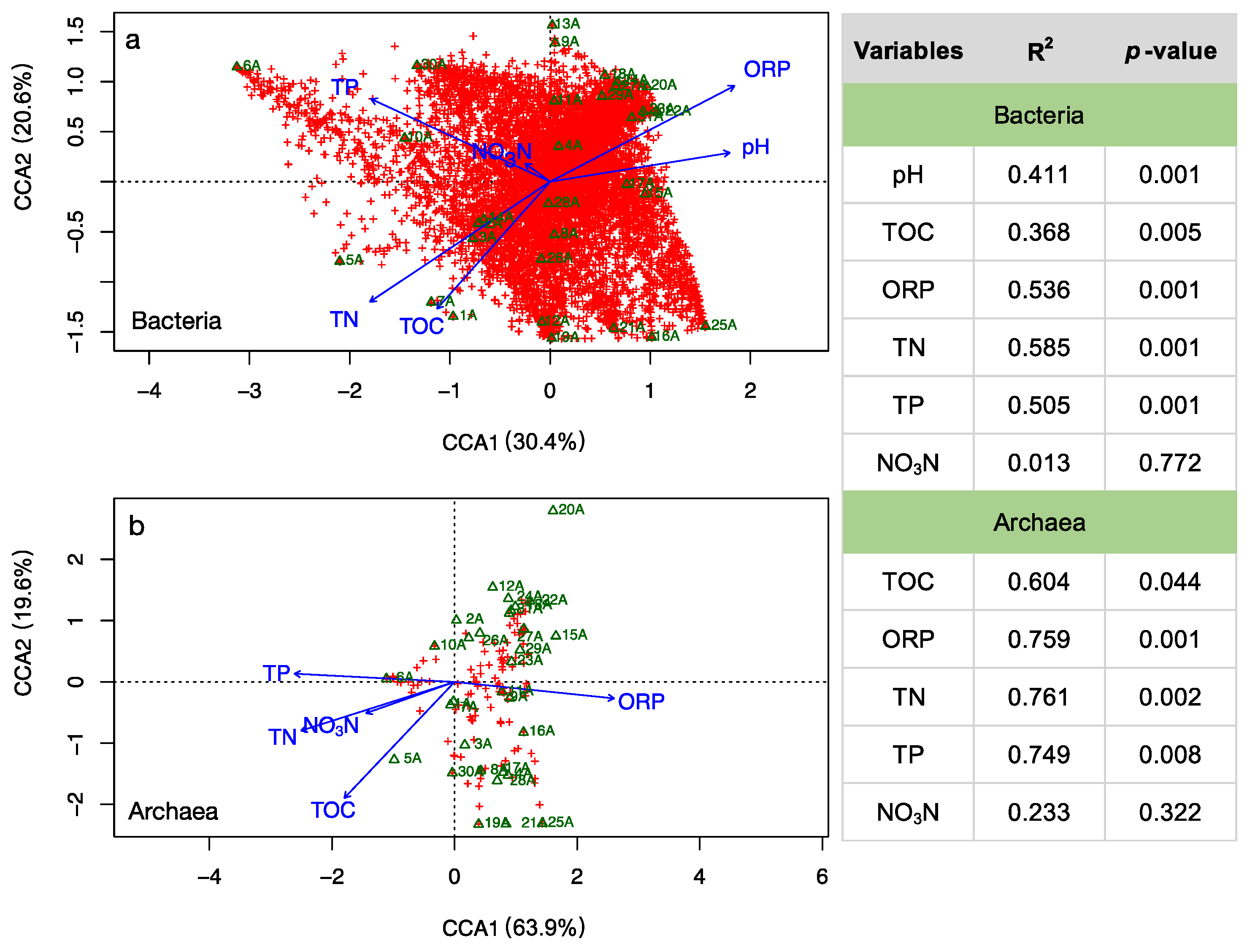
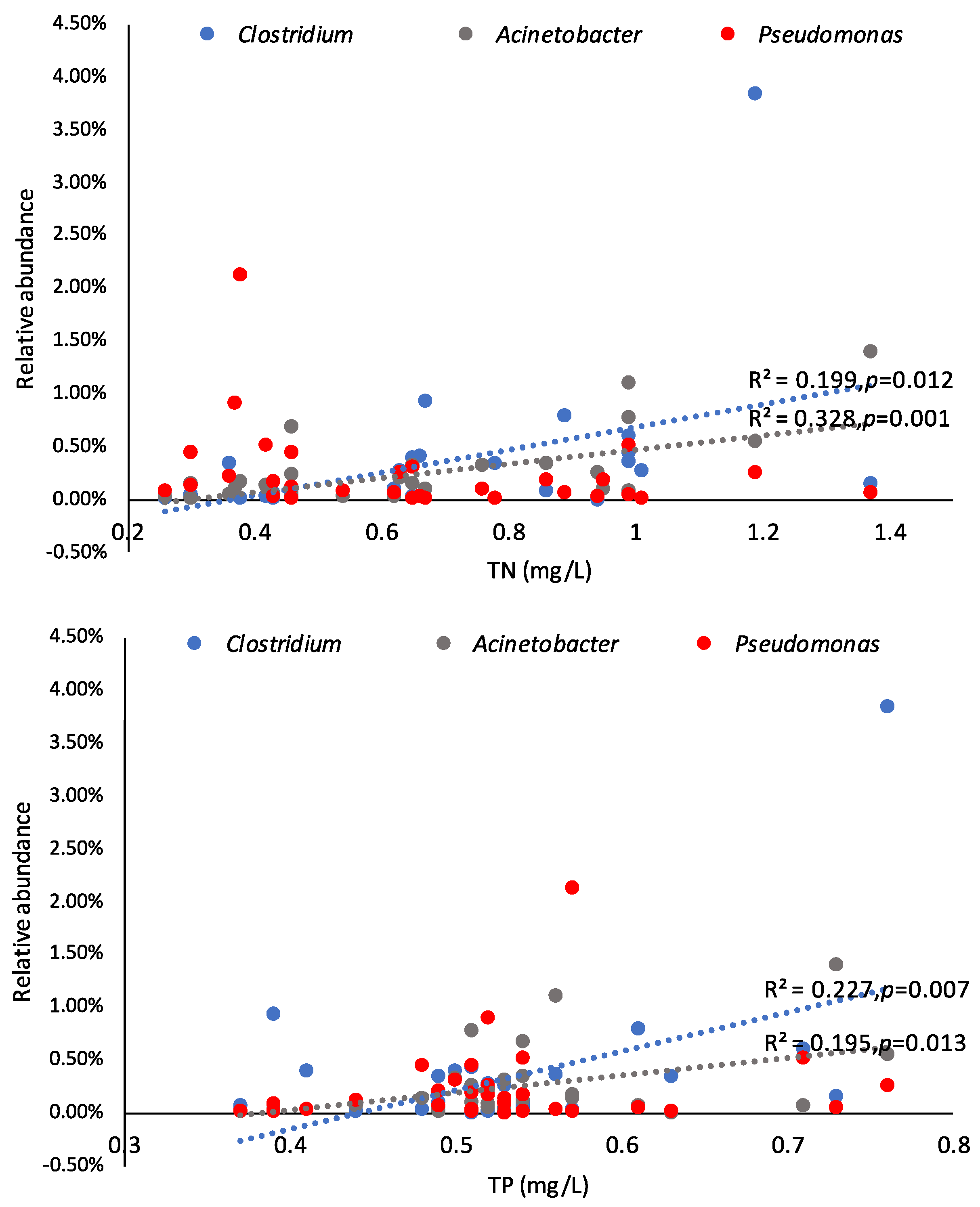
3.3. Environmental Influences on the Microbial Community
4. Discussion
4.1. Adaptive Patterns and Their Driving Factors in Microbial Communities
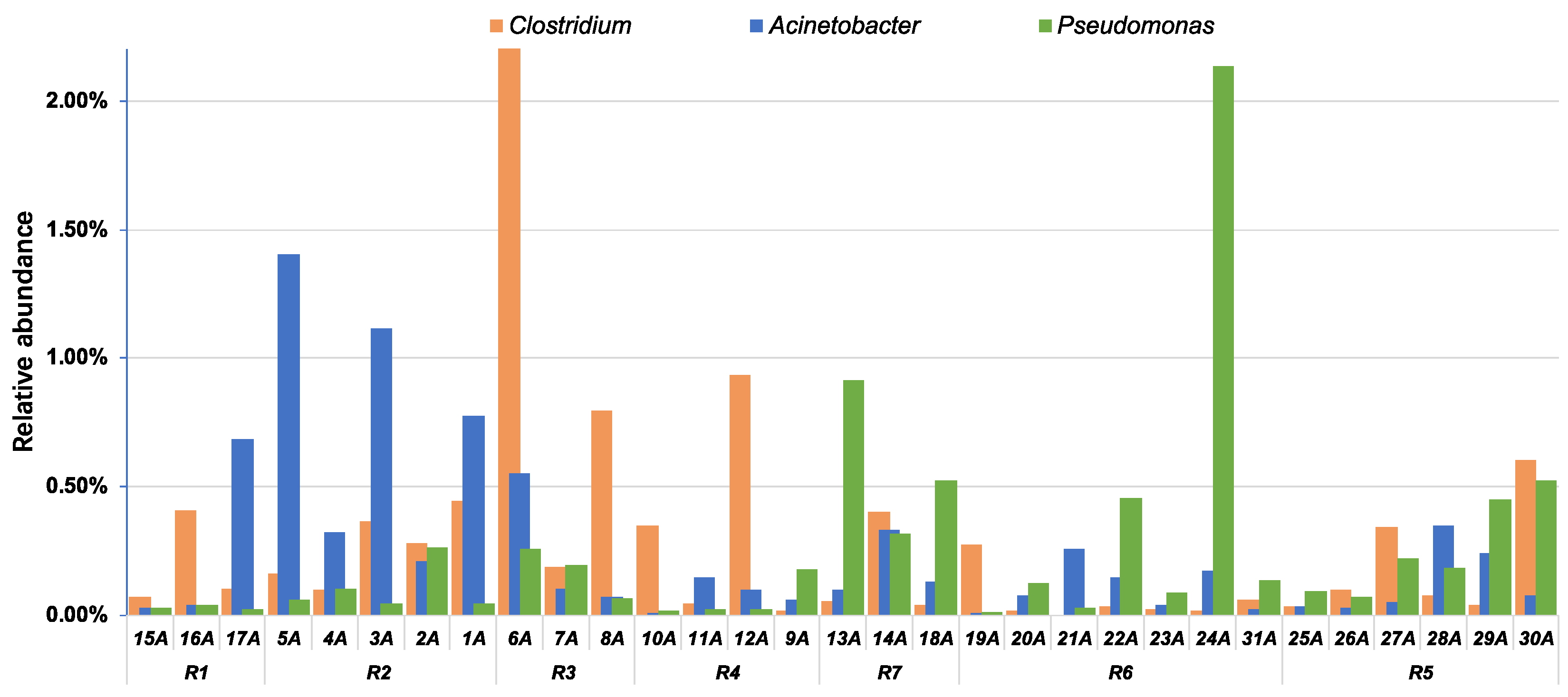
4.2. Implications for the Watershed Management to Sponge City Construction and Indication of Fecal-Associated Bacteria
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Genet. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Findlay, S. Stream microbial ecology. J. N. Am. Benthol. Soc. 2010, 29, 170–181. [Google Scholar] [CrossRef]
- Hosen, J.; Febria, C.M.; Crump, B.C.; Palmer, M.A. Watershed Urbanization Linked to Differences in Stream Bacterial Community Composition. Front. Microbiol. 2017, 8, 1452. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, M.K.; Sanders, N.J.; Wardle, D.A. Community and Ecosystem Responses to Elevational Gradients: Processes, Mechanisms, and Insights for Global Change. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 261–280. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl, D.A.; et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef]
- Hu, Y.; Bai, C.; Cai, J.; Dai, J.; Shao, K.; Tang, X.; Gao, G. Co-occurrence Network Reveals the Higher Fragmentation of the Bacterial Community in Kaidu River Than Its Tributaries in Northwestern China. Microbes Environ. 2018, 33, 127–134. [Google Scholar] [CrossRef]
- Niño-García, J.P.; Ruiz-González, C.; Del Giorgio, P.A. Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J. 2016, 10, 1755–1766. [Google Scholar] [CrossRef]
- Besemer, K.; Singer, G.A.; Quince, C.; Bertuzzo, E.; Sloan, W.T.; Battin, T.J. Headwaters are critical reservoirs of microbial diversity for fluvial networks. Proc. R. Soc. B Biol. Sci. 2013, 280, 20131760. [Google Scholar] [CrossRef]
- Ruiz-González, C.; Niño-García, J.P.; Del Giorgio, P.A. Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol. Lett. 2015, 18, 1198–1206. [Google Scholar] [CrossRef]
- Mykrä, H.; Tolkkinen, M.; Heino, J. Environmental degradation results in contrasting changes in the assembly processes of stream bacterial and fungal communities. Oikos 2017, 126, 1291–1298. [Google Scholar] [CrossRef]
- Allan, J.D. Landscapes and Riverscapes: The Influence of Land Use on Stream Ecosystems. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 257–284. [Google Scholar] [CrossRef]
- Poff, N.L.; Bledsoe, B.P.; Cuhaciyan, C.O. Hydrologic variation with land use across the contiguous United States: Geomorphic and ecological consequences for stream ecosystems. Geomorphology 2006, 79, 264–285. [Google Scholar] [CrossRef]
- Staley, C.; Unno, T.; Gould, T.; Jarvis, B.; Phillips, J.; Cotner, S.; Sadowsky, M.J. Application of Illumina next-generation sequencing to characterize the bacterial community of the Upper Mississippi River. J. Appl. Microbiol. 2013, 115, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wang, J.; Liao, J.; Gao, Z.; Jiang, D.; Sun, J.; Zhao, L.; Huang, Y.; Luan, S. Bacterioplankton community responses to key environmental variables in plateau freshwater lake ecosystems: A structural equation modeling and change point analysis. Sci. Total Environ. 2017, 580, 457–467. [Google Scholar] [CrossRef]
- Warnecke, F.; Sommaruga, R.; Sekar, R.; Hofer, J.S.; Pernthaler, J. Abundances, Identity, and Growth State of Actinobacteria in Mountain Lakes of Different UV Transparency. Appl. Environ. Microbiol. 2005, 71, 5551–5559. [Google Scholar] [CrossRef]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, S.; Sadowsky, M.J. Species sorting and seasonal dynamics primarily shape bacterial communities in the Upper Mississippi River. Sci. Total Environ. 2015, 505, 435–445. [Google Scholar] [CrossRef]
- Haukka, K.; Kolmonen, E.; Hyder, R.; Hietala, J.; Vakkilainen, K.; Kairesalo, T.; Haario, H.; Sivonen, K. Effect of Nutrient Loading on Bacterioplankton Community Composition in Lake Mesocosms. Microb. Ecol. 2006, 51, 137–146. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, S.; Sun, G.; Xu, Z.; Xu, M. Phylogenetic diversity, composition and distribution of bacterioplankton community in the Dongjiang River, China. FEMS Microbiol. Ecol. 2012, 80, 30–44. [Google Scholar] [CrossRef]
- Read, D.S.; Gweon, S.; Bowes, M.; Newbold, L.; Field, D.; Bailey, M.J.; Griffiths, R.I. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 2014, 9, 516–526. [Google Scholar] [CrossRef]
- Hosen, J.; McDonough, O.T.; Febria, C.M.; Palmer, M.A. Dissolved Organic Matter Quality and Bioavailability Changes Across an Urbanization Gradient in Headwater Streams. Environ. Sci. Technol. 2014, 48, 7817–7824. [Google Scholar] [CrossRef]
- Corno, G.; Yang, Y.; Eckert, E.M.; Fontaneto, D.; Fiorentino, A.; Galafassi, S.; Zhang, T.; Di Cesare, A. Effluents of wastewater treatment plants promote the rapid stabilization of the antibiotic resistome in receiving freshwater bodies. Water Res. 2019, 158, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, A.; Di Cesare, A.; Eckert, E.M.; Rizzo, L.; Fontaneto, D.; Yang, Y.; Corno, G. Impact of industrial wastewater on the dynamics of antibiotic resistance genes in a full-scale urban wastewater treatment plant. Sci. Total Environ. 2018, 646, 1204–1210. [Google Scholar] [CrossRef]
- Roguet, A.; Eren, A.M.; Newton, R.J.; McLellan, S.L. Fecal source identification using random forest. Microbiome 2018, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, R.; Zhang, Y.; Wang, G.; Li, K. Impact of nutrient addition on diversity and fate of fecal bacteria. Sci. Total Environ. 2018, 636, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, X.; Wang, C.; Miao, L.; Hou, J.; Yuan, Q. Shift in bacterioplankton diversity and structure: Influence of anthropogenic disturbances along the Yarlung Tsangpo River on the Tibetan Plateau, China. Sci. Rep. 2017, 7, 12529. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Staley, C.; Wang, P.; Dalzell, B.; Chun, C.L.; Sadowsky, M.J. A High-Throughput DNA-Sequencing Approach for Determining Sources of Fecal Bacteria in a Lake Superior Estuary. Environ. Sci. Technol. 2017, 51, 8263–8271. [Google Scholar] [CrossRef] [PubMed]
- Nshimyimana, J.P.; Freedman, A.J.E.; Shanahan, P.; Chua, L.C.H.; Thompson, J.R. Variation of Bacterial Communities with Water Quality in an Urban Tropical Catchment. Environ. Sci. Technol. 2017, 51, 5591–5601. [Google Scholar] [CrossRef]
- Sun, H.; He, X.; Ye, L.; Zhang, X.-X.; Wu, B.; Ren, H. Diversity, abundance, and possible sources of fecal bacteria in the Yangtze River. Appl. Microbiol. Biotechnol. 2016, 101, 2143–2152. [Google Scholar] [CrossRef]
- Wanjugi, P.; Fox, G.; Harwood, V.J. The Interplay Between Predation, Competition, and Nutrient Levels Influences the Survival of Escherichia coli in Aquatic Environments. Microb. Ecol. 2016, 72, 526–537. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z. Estimating groundwater recharge following land-use change using chloride mass balance of soil profiles: A case study at Guyuan and Xifeng in the Loess Plateau of China. Hydrogeol. J. 2010, 19, 177–186. [Google Scholar] [CrossRef]
- Li, C.; Huang, M.; Liu, J.; Ji, S.; Zhao, R.; Zhao, D.; Sun, R. Isotope-based water-use efficiency of major greening plants in a sponge city in northern China. PLoS ONE 2019, 14, e0220083. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.S.; Griffiths†, J.; Higgitt, D.; Xu, S.; Zhu, F.; Tang, Y.-T.; Xu, Y.; Thorne, C. “Sponge City” in China—A breakthrough of planning and flood risk management in the urban context. Land Use Policy 2018, 76, 772–778. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, N.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2010, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.T.; Berg-Lyons, N.; Caporaso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. Examining the global distribution of dominant archaeal populations in soil. ISME J. 2010, 5, 908–917. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the Effects of PCR Amplification and Sequencing Artifacts on 16S rRNA-Based Studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- McLellan, S.L.; Huse, S.M.; Mueller-Spitz, S.; Andreishcheva, E.N.; Sogin, M.L. Diversity and population structure of sewage derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 2009, 12, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Savio, D.; Sinclair, L.; Ijaz, U.Z.; Parajka, J.; Reischer, G.H.; Stadler, P.; Blaschke, A.P.; Blöschl, G.; Mach, R.L.; Kirschner, A.; et al. Bacterial diversity along a 2600 km river continuum. Environ. Microbiol. 2015, 17, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Crump, B.C.; Adams, H.E.; Hobbie, J.E.; Kling, G.W. Biogeography of bacterioplankton in lakes and streams of an arctic tundra catchment. Ecology 2007, 88, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Ibekwe, A.M.; Ma, J.; Murinda, S.E. Bacterial community composition and structure in an Urban River impacted by different pollutant sources. Sci. Total Environ. 2016, 566, 1176–1185. [Google Scholar] [CrossRef]
- Hu, A.; Yang, X.; Chen, N.; Hou, L.; Ma, Y.; Yu, C.-P.; Hu, A. Response of bacterial communities to environmental changes in a mesoscale subtropical watershed, Southeast China. Sci. Total Environ. 2014, 472, 746–756. [Google Scholar] [CrossRef]
- Hu, A.; Wang, H.; Li, J.; Liu, J.; Chen, N.; Yu, C.-P. Archaeal community in a human-disturbed watershed in southeast China: Diversity, distribution, and responses to environmental changes. Appl. Microbiol. Biotechnol. 2016, 100, 4685–4698. [Google Scholar] [CrossRef]
- Fortunato, C.S.; Herfort, L.; Zuber, P.; Baptista, A.M.; Crump, B.C. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J. 2011, 6, 554–563. [Google Scholar] [CrossRef]
- Jackson, C.R.; Millar, J.J.; Payne, J.T.; Ochs, C.A. Free-Living and Particle-Associated Bacterioplankton in Large Rivers of the Mississippi River Basin Demonstrate Biogeographic Patterns. Appl. Environ. Microbiol. 2014, 80, 7186–7195. [Google Scholar] [CrossRef]
- Zinger, L.; Gobet, A.; Pommier, T. Two decades of describing the unseen majority of aquatic microbial diversity. Mol. Ecol. 2011, 21, 1878–1896. [Google Scholar] [CrossRef]
- Zeglin, L.H. Stream microbial diversity in response to environmental changes: Review and synthesis of existing research. Front. Microbiol. 2015, 6, 454. [Google Scholar] [CrossRef]
- Zwart, G.; Crump, B.C.; Agterveld, M.K.-V.; Hagen, F.; Han, S. Typical freshwater bacteria: An analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 2002, 28, 141–155. [Google Scholar] [CrossRef]
- Knittel, K.; Boetius, A. Anaerobic Oxidation of Methane: Progress with an Unknown Process. Annu. Rev. Microbiol. 2009, 63, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; Bootsma, M.J.; Morrison, H.G.; Sogin, M.L.; McLellan, S.L. A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of Lake Michigan. Microb. Ecol. 2013, 65, 1011–1023. [Google Scholar] [CrossRef]
- McLellan, S.L.; Eren, A.M. Discovering new indicators of fecal pollution. Trends Microbiol. 2014, 22, 697–706. [Google Scholar] [CrossRef]
- Ahmed, W.; Staley, C.; Sadowsky, M.J.; Gyawali, P.; Sidhu, J.P.S.; Palmer, A.; Beale, D.J.; Toze, S. Toolbox Approaches Using Molecular Markers and 16S rRNA Gene Amplicon Data Sets for Identification of Fecal Pollution in Surface Water. Appl. Environ. Microbiol. 2015, 81, 7067–7077. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.; Jovanovic, D.; Lintern, A.; Teakle, I.; Barnes, M.; Deletic, A.; Coleman, R.; Rooney, G.; Prosser, T.; Coutts, S.; et al. Source tracking using microbial community fingerprints: Method comparison with hydrodynamic modelling. Water Res. 2017, 109, 253–265. [Google Scholar] [CrossRef]
- Savichtcheva, O.; Okayama, N.; Okabe, S. Relationships between Bacteroides 16S rRNA genetic markers and presence of bacterial enteric pathogens and conventional fecal indicators. Water Res. 2007, 41, 3615–3628. [Google Scholar] [CrossRef]
- Lawes, J.; Neilan, B.A.; Brown, M.V.; Clark, G.F.; Johnston, E. Elevated nutrients change bacterial community composition and connectivity: High throughput sequencing of young marine biofilms. Biofouling 2016, 32, 57–69. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Wang, Y.; Xu, Y.; Duan, G.; Huang, M.; Peng, J. Adaptive Variations of Sediment Microbial Communities and Indication of Fecal-Associated Bacteria to Nutrients in a Regulated Urban River. Water 2020, 12, 1344. https://doi.org/10.3390/w12051344
Cao X, Wang Y, Xu Y, Duan G, Huang M, Peng J. Adaptive Variations of Sediment Microbial Communities and Indication of Fecal-Associated Bacteria to Nutrients in a Regulated Urban River. Water. 2020; 12(5):1344. https://doi.org/10.3390/w12051344
Chicago/Turabian StyleCao, Xiaofeng, Yajun Wang, Yan Xu, Gaoqi Duan, Miansong Huang, and Jianfeng Peng. 2020. "Adaptive Variations of Sediment Microbial Communities and Indication of Fecal-Associated Bacteria to Nutrients in a Regulated Urban River" Water 12, no. 5: 1344. https://doi.org/10.3390/w12051344
APA StyleCao, X., Wang, Y., Xu, Y., Duan, G., Huang, M., & Peng, J. (2020). Adaptive Variations of Sediment Microbial Communities and Indication of Fecal-Associated Bacteria to Nutrients in a Regulated Urban River. Water, 12(5), 1344. https://doi.org/10.3390/w12051344